Impact Factor ISSN: 1837-9664

 Global reach, higher impact
Global reach, higher impactJ Cancer 2026; 17(4):703-711. doi:10.7150/jca.129125 This issue Cite
Research Paper
The Overexpression of Myelin and Lymphocyte Protein (MAL) Downregulates MUC1 and Enhances Cisplatin Sensitivity in Non-Small Cell Lung Cancer Cells
Ana Karina Saldaña-Villa1*, Bismarck Vázquez-Almazán1,2*, Catalina Flores-Maldonado3, Juan Carlos Vizuet-de-Rueda4, Elisa Natalia Oropeza-Durán5, José Bonilla-Delgado6, Enoc Mariano Cortés-Malagón7, Rubén Gerardo Contreras3, Luis Manuel Teran4, Blanca Ortiz-Quintero1, Roberto Lara-Lemus1 ![]()
1. Departamento de Biomedicina Molecular e Investigación Traslacional, Instituto Nacional de Enfermedades Respiratorias Ismael Cosío Villegas, Mexico City 14080, MEXICO.
2. Posgrado en Ciencias Biológicas, Unidad de Posgrado, Edificio D, 1er Piso, Circuito de Posgrados, Ciudad Universitaria, Coyoacán 04510, Mexico City, MEXICO.
3. Departamento de Fisiología, Biofísica y Neurociencias Centro de Investigación y de Estudios Avanzados del Instituto Politécnico Nacional (Cinvestav). Mexico City, MEXICO.
4. Departmento de Inmunogénetica y Alergia, Instituto Nacional de Enfermedades Respiratorias Ismael Cosío Villegas Mexico City, 14080, MEXICO.
5. Departamento de Biotecnología, Facultad de Ciencias de la Salud, Universidad Anáhuac México Campus Norte, Huixquilucan 52786, MEXICO.
6. Laboratorio de la Unidad de Investigación en Oncología Molecular, Hospital de Alta Especialidad de Ixtapaluca, Servicios de Salud del Instituto Mexicano del Seguro Social para el Bienestar, Ixtapaluca 56530, MEXICO.
7. División de Investigación, Hospital Juárez de México 07760, Mexico City, MEXICO.
* These authors contributed equally to this work.
Received 2025-11-28; Accepted 2026-1-20; Published 2026-3-17
Abstract

The epigenetic repression of myelin and lymphocyte protein gene (MAL) and over-expression of MUC1 protein are two well-documented hallmarks of various carcinomas including lung cancer. The purpose of this work was to investigate whether ectopic expression of MAL could modify the proliferative activity of MUC1 in human lung adenocarcinoma HCC827 cells. We generated stable HCC827 cell lines, expressing GFP- or myc-MAL constructs, then, followed the expression of MUC1 by RT-qPCR and Western blot, and tested proliferation and sensitivity of those cells to cisplatin. Our results showed that ectopic expression of MAL nearly eliminated cellular levels of MUC1-C. This effect is primarily due to down-regulation of MUC1 expression, as confirmed by RT-qPCR. Additionally, lysosomal-associated degradation of MUC1 may also contribute, as observed when the cells were treated with ammonium chloride and chloroquine. The reduced quantity of MUC1-C negatively affected both cyclin D1 and c-Myc, protein levels, which are linked to cell-proliferative signals involving MUC1-C. Furthermore, expression of MAL decreased the viability of HCC827 cells, and increased their sensitivity to cisplatin. MAL and MUC1 showed an antagonistic relationship in cancer cells, and these findings provide new insights with respect to the regulation of MUC1 and a better understanding of MAL as a potential tumor suppressor.
Keywords: Myelin and lymphocyte protein, MUC1, lung cancer, cisplatin resistance
Introduction
Myelin and lymphocyte protein (MAL), also named T-lymphocyte maturation-associated protein, is a proteolipid constituent of cholesterol and glycosphingolipid-enriched micro domains (GEMs) that have been implicated in the apical transport of proteins in epithelial cells [1,2]. MAL expression was initially demonstrated to occur during T-cell maturation [3] and was also reported in myelin-forming cells [4,5], as well as polarized epithelial cells [6-8]. Several reports have indicated that silencing of MAL by hyper-methylation of its promoter [9] is found in different kinds of carcinomas, such esophageal [10-13], gastric [14], lung [15], colon [16], head and neck [17,18], cervical [19,20], breast [21,22], and bladder [23]. Conversely, it has also been described that higher MAL expression levels are associated with the development of some types of lymphoma [24-27] and ovarian cancer [28-30]. In this sense, tumor suppressor properties of MAL have been described by at least two well-documented reports; the first one, through the induction of apoptosis via the Fas signaling pathway, and the second, across the inhibition of cell migration and metastasis by blocking the signal transducer and activator of transcription 3 (STAT3) phosphorylation [10,31].
MUC1 (mucin 1), is a heterodimeric membrane-bound mucin, found in the apical region of epithelial cells. However, MUC1 is over-expressed and abnormally glycosylated in several types of adenocarcinomas [32-35]. The oncogenic activity of MUC1 primarily depends on the MUC1-carboxy-terminal subunit (MUC1-C), which interacts with various mediators of cell proliferation signaling pathways [33]. Overexpression of MUC1 and the loss of the plasma membrane (PM) polarity in transformed cells, enable interactions and phosphorylation of MUC1-C by receptor tyrosine kinases (RTKs), such as epidermal growth factor receptor (EGFR) [34,36]. The binding of galectin-3 to the extracellular domains of EGFR and MUC1 stabilizes their interaction [36], and it has been reported that MUC1-C can prevent the lysosomal degradation of EGFR [37], thereby extending EGF´s proliferative activity.
MUC1-C activates various pro-survival pathways, such as Ras, PI3K/Akt, and Wnt [38-41]. In this context, MUC1-C is a potent oncogene that exhibits several features that enhance tumor growth and metastasis. Recently, Rajabi et al. described a new pro-oncogenic activity of MUC1-C, involving the downregulation of tumor-suppressor genes [42]. Therefore, we wondered whether a functional relationship exists between MAL and MUC1. In this sense, Fanayan et al. identified MAL2, another member of the MAL family, as a partner of MUC1 in breast carcinoma cells [43]. They suggested that, in those cells, MAL2 could deliver MUC1 to the basolateral region, thereby promoting the phosphorylation of MUC1-C by EGFR and other tyrosine kinases, such as c-Src. Our laboratory is studying the role of MAL as a tumor suppressor in lung cancer. We hypothesize that MAL, a membrane raft scaffold protein, might interfere with the cellular trafficking of MUC1 and potentially alter its effects on cell proliferation. In this paper, we present evidence indicating that the expression of MAL in the NSCLC cell line HCC827 promotes the downregulation of the MUC1 gene and could facilitate the lysosomal degradation of MUC1-C. Both effects negatively impact the proliferation factors, cyclin D1, and c-Myc.
Materials and Methods
Cells, plasmids, and transfections
The lung adenocarcinoma HCC827 cells were provided by Dr. Jose Sullivan Lopez at the National Institute of Respiratory Diseases, Mexico. Dr. Lourdes Gutierrez supplied HEK293 cells at the National Institute of Public Health, Mexico. Both the HCC827 and HEK293 cells were cultured under standard conditions (5% CO2 and 95% air at 37 °C) in RPMI 1640 high-glucose medium, and DMEM high-glucose medium (Life Technologies), respectively. The media were supplemented with 10% FBS, 50 U/mL of penicillin, and 50 µg/mL of streptomycin (Life Technologies). The constructs pcDNA3.1-Hygromycin/GFP-MAL and pcDNA3.1-Hygromycin/myc-MAL were obtained from Dr. Peter Arvan´s laboratory (University of Michigan). Dr. Rebecca Hughey (University of Pittsburgh) kindly supplied the construct pcDNA3.1-Hygromycin/hMUC1-22r. Transfections were conducted using two µg/mL of plasmid pre-mixed with Lipofectamine 3000 reagent (Invitrogen) at a 2:1 ratio in reduced-serum medium. Six-well plates were seeded with 0.75 × 10^6 cells and transfected after 24 hours. Stable HCC827 and HEK293 cell lines expressing GFP-MAL, myc-MAL and hMUC1-22r were selected and maintained with 200 µg/mL of hygromycin B (Invitrogen). Several clones expressing GFP-MAL were isolated after fluorescence assisted cell sorting (Fig. S1A).
Western blotting (WB)
Sub-confluent cell cultures were washed with ice-cold phosphate-buffered saline (PBS), and whole-cell lysates were prepared using RIPA buffer (Tris-HCl 25 mM pH 7.4, NaCl 150 mM, EDTA 1 mM, sodium deoxycholate 2.5 mM, NP40 1%, sodium dodecyl sulfate 0.1% and protease inhibitor cocktail, Complete C, from Roche). Protein concentration was measured using the bicinchoninic acid method (Thermo Scientific). Proteins were separated by SDS-PAGE in a Mini-PROTEAN Tetra cell electrophoresis chamber (Bio-Rad), according to Laemmli [44]. The samples were transferred to nitrocellulose membranes using a semi-dry transfer system (Bio-Rad). Membranes were incubated with appropriate primary antibodies: anti-MUC1-C antibody, EPR 1023 (Abcam) 1:5000, anti-GFP (Bio-Legend) 1:5000, anti-cMyc (Immunology Consultants Laboratory) 1:5000, anti-cyclin D1 Invitrogen) 1/500, anti-EGFR A-10, and anti-MAL E1 (Santa Cruz Biotech) 1:1000 and 1:50 respectively. Secondary antibodies were from Jackson Immuno Research. Detection was conducted using chemiluminescence (Super Signal Kit, Thermo Scientific). Images were captured and analyzed using a ChemiDoc instrument (Bio-Rad).
Ammonium chloride and chloroquine treatment
HCC827 wild-type (wt) cells, and HCC827-GFP-MAL clone (18P) were seeded at a density of 0.75 × 10^6 cells per well on 6-well plates. After 24 hours, the media were re-placed with fresh medium containing 0 or 5 mM ammonium chloride or 20 μM chloroquine. The cells were then incubated for an additional 24 hours. Whole-cell lysates obtained were immediately processed for immunoblotting.
Immunofluorescence
HCC827-wt and HCC827-GFP-MAL cells were cultured on coverslips, washed twice with ice-cold PBS, and fixed with 4% formaldehyde, then permeabilized with 0.1% Triton X-100 and blocked with 2% ultra-pure BSA in PBS (blocking solution). The primary antibody anti-MUC1-C was diluted 1:100 in blocking solution. The secondary antibody was Alexa 555-tagged (Thermo Fisher). Confocal microscopy images were captured using an SP8 microscope, equipped with a Plan-NeoFluar 63x NA 1.4 objective (Leica Microsystems). We acquired confocal stacks of 40-60 images and created the maximum projection for each.
Cell proliferation and cisplatin assays
HCC827-wt and 18P cells were seeded at a density of 2 × 10^4 cells per well in 96-well plates and incubated for 24 hours with various concentrations of cis diamine- dichloroplatinum (II) (cisplatin) (Sigma-Aldrich). A set of cells without any drug treatment was allowed to grow for 72 hours to assess differences in proliferation due to MAL expression. Cell proliferation was determined using the XTT reagent (Roche) according to the manufacturer´s instructions.
cDNA synthesis and quantitative reverse transcription polymerase chain reaction RT-qPCR) analysis
The SV Total RNA Isolation System Kit (Promega, Cat. No. Z3100) was used to extract total RNA from all samples. For quantitative real-time PCR (RT-qPCR), RNA was treated with DNase I to remove genomic DNA. One microgram of total RNA was reverse transcribed in a 20 µL reaction mixture using SuperScript II-Reverse Transcriptase (Invitrogen) and an oligo(dT) primer. Real-time PCR quantification was performed with a StepOne Real-time PCR system (Applied Biosystems). RT-qPCR was conducted following the Maxima Probe qPCR Master Mix (2X)/ROX protocol (Thermo Scientific). A “no DNA” template control was included in each run. The data from the RT-qPCR experiments were analyzed using the relative quantification method, or 2-ΔΔCT method, where the ΔΔCT value = ((CT1 Target - CT1 Reference) - (CT0 Target - CT0 Reference)) [45]. The mean CT values for both the target and internal reference genes were calculated, and the target gene fold change was normalized to HsGADPH. The relative expression of the target gene was then normalized to the control sample, whit the the control cells' expression set to 1 (i.e., 18P). The primers used included: HsGAPDH (AF261085.1) Forward 5´-GTCTCCTCTGACTTCAACAGCG-3´, Reverse 5´-ACCACCCTGTTGCTGTAGCCAA-3´. HsMAL (NM_002371.4) Forward 5´-GCTGGGTGATGTTCGTGTCTG-3´, Reverse 5´-TGAGGCGCTGAGGTAAAAGA-3´; and HsMUC1 (NM_002456.6) Forward 5´-TCGTAGCCCCTATGAGAAGG-3´, Reverse 5´-CCACTGCTGGGTTTGTGTAA-3´. All experiments were performed in triplicate (technical replicates) from three biological replicates.
Statistical analysis
The statistical analysis used an independent-samples t-test to compare the means between two groups, and the normality of variances was verified using Shapiro-Wilk's test. The t-test was used to determine whether the observed differences were statistically significant (p < 0.05).
Results
The ectopic expression of MAL reduces the levels of MUC1
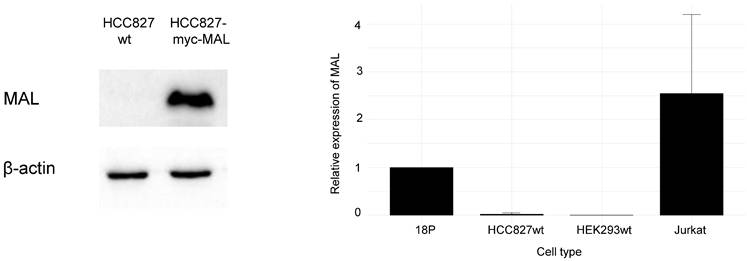
To our knowledge, no published data indicate that human lung carcinoma cells HCC827 express MAL. We began by evaluating endogenous MAL expression using WB and RT-qPCR. As shown in Fig. 1A, comparing the whole-cell lysate of HCC827-wt with that of cells transiently transfected with the myc-MAL construct, the non-transfected cells did not show any protein bands attributable to MAL. Furthermore, RT-qPCR analysis revealed only a negligible amount of MAL-mRNA in wt cells compared to the HCC827-GFP-MAL cells, (18P), as illustrated in Fig. 1B. This result aligns with published data for the other nine lung cancer cell lines [15]. Next, as shown in Fig. 2A, WB analysis revealed that transfection with either GFP-MAL or myc-MAL significantly reduced MUC1-C levels in both HCC827-wt, and HEK293 cells stably transfected with human MUC1 (HEK293-MUC1h22r) [46,47]. This cell line was transiently transfected with the myc-MAL construct, and WB was performed 48 hours after transfection. The result indicates that the phenotype is reproducible in HEK293-MUC1h22r, suggesting that the mechanisms induced by MAL expression are functional in cells that do not endogenously express MAL and MUC1. Fig. 2B displays the indirect immunofluorescence (IIF) results for HCC827 cells transiently transfected with the GFP-MAL construct. A significant reduction in the MUC1-C signal (red) can be observed in MAL-expressing cells, consistent with the WB. To confirm this, HCC827-GFP-MAL cells were sorted by flow cytometry, and several individual clones were isolated. WB analyses of whole-cell lysates revealed a significant decrease in MUC1-C levels in all MAL-expressing clones (see Fig. S1A).
Endogenous expression of MAL in HCC827-wt cells by WB and RT-qPCR. A. The expression of MAL in cell lysates from cells HCC827-wt and HCC827 transiently transfected with the myc-MAL construct, were analyzed by WB. 20 μg of protein were loaded in each case. B. RT-qPCR analysis of HCC827-wt, HCC827-GFP-MAL-expressing (18P clone), HEK293 and Jurkat cells. Relative gene expression in control cells (18P) was defined as 1. HEK293 and Jurkat cells were included as negative and positive controls of the MAL expression respectively.
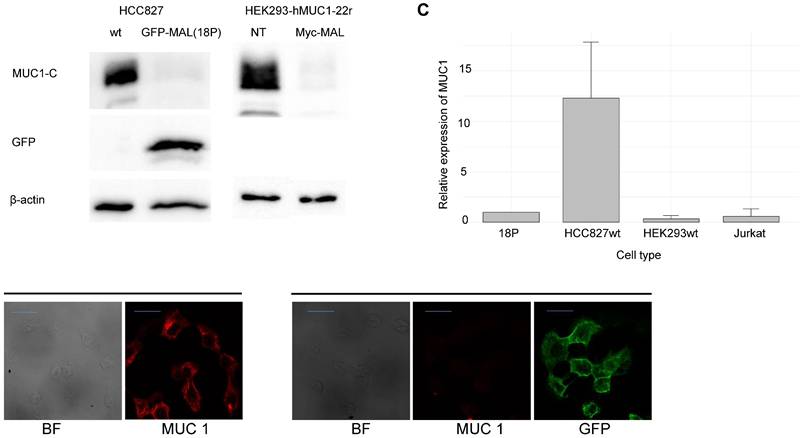
The ectopic expression of MAL affect MUC1 expression. A. WB of cell lysates from HCC827-wt,18P clone and HEK293-hMUC1-22r. This cell line was generated by the stable transfection of the human MUC1 cDNA, having 22 repeats of the variable number tandem repeat sequence (VNTR), described elsewhere [46], then, cells were transiently transfected with the myc-MAL cDNA. 20 μg of protein were loaded in each lane. B. Indirect immunofluorescence of HCC827-wt and HCC827 transiently transfected with GFP-MAL. MUC1, is in red. The decrease in the signal of MUC1-C agrees with the WB results. (bar = 20 μm). C. RT-qPCR analysis of MUC1-mRNA from 18P clone, HCC827-wt, HEK293 and Jurkat cells.
The expression of MAL reduces the amount of MUC1-RNAm and induces lysosomal degradation of the protein
To clarify why MUC1-C levels decrease in HCC827 cells upon MAL expression, we first examined whether MAL affects MUC1 mRNA levels. Fig. 2C shows RT-qPCR results measuring MUC1 mRNA in both HCC827-wt and 18P cells. In the presence of MAL, we observed a significant decrease in MUC1 mRNA levels. While this could explain the substantial reduction in MUC1-C, we also investigated whether MAL is involved in degrading MUC1-C. It is known that MUC1 is primarily broken down in lysosomes, so we treated HCC827-wt and 18P cells with two lysosomal protease inhibitors. As shown in Fig. S1B, lysosomal-associated protein degradation with ammonium chloride or chloroquine partially restored MUC1-C levels in 18P cell lysates, indicating that MAL expression also promoted some degree of lysosomal degradation of MUC1-C.
MAL expression affects the levels of cyclin D1 and cMyc
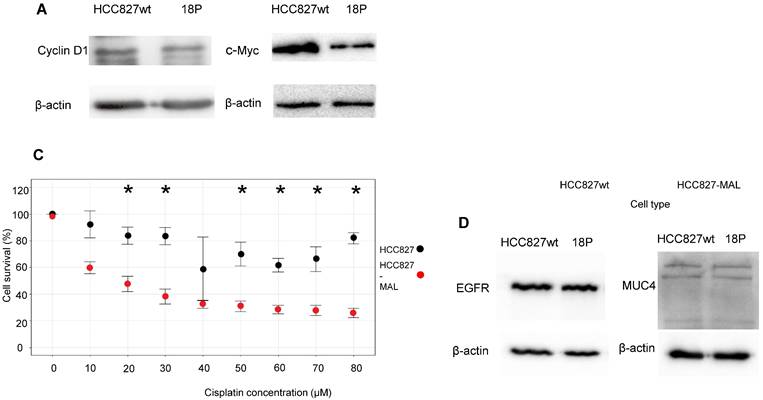
We examined whether MAL expression could affect the Wnt-MUC1-C relationship. β-catenin plays essential roles in processes such as cell-cell adhesion and transcriptional regulation. It binds to the sequence SAGNGGSSLS in the cytoplasmic tail of MUC1-C [48]. In the nucleus, the MUC1-C-β-catenin complex activates the transcription of Wnt target genes [49] including CCND1 and c-Myc [50-52]. Therefore, we hypothesized that the decrease in MUC1-C caused by MAL would lower the expression of cyclin D1 (CD1) and c-Myc. As shown in Fig. 3A, WB analysis revealed a significant decrease in c-Myc and a slight reduction in CD1 levels in 18P cells. These findings support a tumor-suppressor role for MAL by interfering with the pro-oncogenic functions of MUC1-C.
Effects of MAL upon the expression of different proteins, proliferation and cisplatin resistance. A. WB analysis of HCC827wt and 18P clone cell lysates for cyclin D1, and c-Myc; lower expression is observed in both cases when MAL is expressed. B. Proliferation of HCC827wt and 18P cells after 72 hours of culture. C. HCC827-wt (black dots) and 18P (red dots) were treated after 24 hours with different concentrations of cisplatin. Data represent mean ± SD from three independent experiments (n = 3). Statistical significance was determined with an unpaired two-tailed Student's t-test (*p < 0.05). D. EGFR and MUC4 WB of HCC827-wt and 18P cell lysates, no differences in the expression of both proteins were observed when MAL is expressed.
The ectopic MAL expression decreases the viability of HCC827 cells and increases their sensitivity to cisplatin
We hypothesized that decreased levels of CD1 and c-Myc might impact the proliferative capacity of HCC827 cells. To investigate this, we first compared the viability of HCC827-wt and 18P cells. As shown in Fig. 3B, the viability of 18P cells after 72 hours was significantly lower than that of the wt cells. This indicates that ectopic expression of MAL negatively impacts the growth of lung cancer cells. Next, we evaluated the response of HCC827-wt and 18P cells to the genotoxic agent cisplatin. Cells were exposed to increasing concentrations of cisplatin, and as shown in Fig. 3C, MAL-expressing cells exhibited lower survival and statistically significant differences at almost all cisplatin concentrations. The profile displayed by HCC827-wt cells showed a degree of cisplatin resistance, but MAL expression rendered the cells sensitive to the drug. Therefore, MAL expression hinders the proliferation of lung cancer cells.
The effect of MAL seems to be specific to MUC1
To determine whether the MAL-induced phenotype was specific to MUC1, we examined its effects on other proteins. Like MUC1, EGFR is endocytosed through a mechanism involving clathrin-coated vesicles, recycled, and ultimately degraded in lysosomes [53-55]. We investigated whether MAL expression could alter EGFR levels in 18P cells. As shown in Fig. 3D, the levels of EGFR were not affected by the ectopic expression of MAL. Additionally, we tested another membrane-bound mucin, MUC4, and again, no significant change in its cellular levels was observed in response to MAL (Fig. 3D). Therefore, the mechanism by which MAL reduces MUC1-C appears to be specific.
Discussion
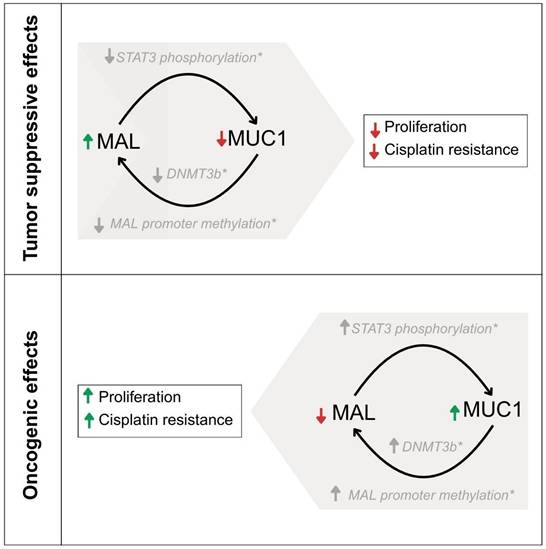
The down-regulation of MAL and over-expression of MUC1 have been observed in similar types of carcinomas [10-23, 32-35]; however, no experimental evidence has previously demonstrated a direct effect of MAL, on the oncogenic activity of MUC1-C [56]. In this study, MAL was undetectable in HCC827 cell-lysates by Western blot analysis (Fig. 1A). Indeed, MAL has been reported to be downregulated in nine other lung carcinoma cell lines through hypermethylation of its promoter, and its expression is restored after DNA demethylation with 5-Aza-2'-deoxycytidine [15]. The results shown here reveal a strong correlation between ectopic MAL-cDNA expression and cellular levels of MUC1-C. Our data showed a significant decrease in MUC1-C levels in the HCC827-wt and HEK293-hMUC1-22r cell lines following transfection with either GFP- or Myc-MAL constructs. To explore the cause of this reduction, we examined both MUC1-C degradation and MUC1-mRNA levels. When lysosomal proteolytic activity was inhibited with ammonium chloride or chloroquine, we observed a slight recovery in cellular MUC1-C levels (Fig. S1B), suggesting that MAL may partially promote lysosomal degradation of MUC1-C. This finding is novel and the underlying mechanisms are unclear. In this regard, Razawi et al. [57] demonstrated that mutation of two specific tyrosine residues in the MUC1-C cytoplasmic tail, (Y20 and 60N) affects MUC1's clathrin-mediated endocytosis and causes the accumulation of MUC1 in endosomes. Our results suggest that MAL disrupts MUC1 recycling by directing it into the lysosomal compartment, even when signals for endocytosis and recycling are intact. Interestingly, it was reported that thyroid and breast cancer cells accumulate MUC1 in cytoplasmic vesicles, likely endosomes [58,59]. From our immunofluorescence images (Fig. 2B), HCC827-wt cells also exhibit a cytoplasmic punctate pattern of MUC1-C, similar to that observed in breast and thyroid cancers. One possible explanation is that MAL may facilitate the transport of “stuck endosomes” loaded with MUC1 into lysosomes for degradation. The notable reduction in MUC1-C at the protein level cannot be explained solely by degradation. Therefore, we investigated MUC1 gene expression. RT-qPCR results showed that MAL-expressing cells (18P) had lower MUC1 mRNA levels (Fig. 2C). This is a significant and interesting finding. The MAL promoter is hypermethylated in several carcinomas [60], and it was demonstrated that MUC1-C activates the expression of DNA methyltransferases 1 and 3b (DNMT1 and 3b) [61,62]. Indeed, the MAL gene is silenced by DNMT3b activity in NSCLC cell lines, including HCC827 [63]. On the other hand, previous reports have shown that ectopic expression of MAL reduces cell motility and tumorigenesis in esophageal carcinoma cells, likely by inducing apoptosis [10]. More recently, Geng et al. reported that MAL interferes with the pro-oncogenic activity of STAT-3 in gastric cancer cells, by blocking its phosphorylation [31]. Additionally, the MUC1 promoter contains a phosphorylated STAT-3 (pSTAT-3) responsive element [64]. Inhibition of STAT-3 expression decreases cell motility and MUC1 expression in breast cancer cells [65]. Although the mechanisms by which MAL interferes with MUC1 expression remain unclear, the reported relationship between MAL and STAT-3 may help explain why ectopic MAL expression downregulates MUC1 in HCC827 cells. Meanwhile, MUC4 is also upregulated by pSTAT-3 [66,67], but we did not observe any decrease in MUC4 protein levels attributable to MAL expression (Fig. 3E). If MAL influences MUC1 expression via pSTAT-3, this should be confirmed through future experiments. In cancer biology, our findings suggest that MAL plasmid expression counteracts the proliferative effects of MUC1-C and enhances cell sensitivity to cisplatin (Fig. 3C and D). Targeting MUC1-C with the cell-penetrating peptide GO-203, has been shown to inhibit cell proliferation, reverse cisplatin resistance, and reduce migration and invasion in esophageal carcinoma cells [68]. In our study, we did not observe significant differences in invasion and cell migration between HCC827-wt and MAL-expressing cells (not shown). Lastly, we could not demonstrate a direct interaction between MAL and MUC1 via co-immunoprecipitation (not show), but Fanayan et al. using the yeast two-hybrid system, found that MUC1 is a partner for MAL-2, and that MAL can bind to MUC1 [43]. Therefore, we cannot dismiss the possibility of direct or indirect interactions between these proteins, which warrants future investigation. To conclude, this study provides the first experimental evidence that MAL reduces the MUC1-C levels in cancer cells, demonstrating an antagonistic relationship between MUC1 and MAL. This relationship results in a decreased proliferative capacity driven by MUC1-C and increased sensitivity to cisplatin. A proposed model explaining this antagonism is shown in Figure 4.
A proposed model suggesting the possible explanation of the antagonist functions of MAL and MUC1-C. The roles of STAT3 and DNMT3b are indicated in grey color as they were published previously by other researchers.
Supplementary Material
Supplementary figure.
Acknowledgements
This work was supported and financed by Instituto Nacional de Enfermedades Respiratorias Ismael Cosío Villegas, project ID: B31-18. BV-A received a postgraduate-student fellowship from CONACYT, México. This paper serves as a fulfillment of BV-A for obtaining a M.Sc. degree in the Posgrado en Ciencias Biológicas UNAM. We extended special thanks to Dr. Miguel A. Alonso for its valuable comments.
Funding
This work was supported and financed by the Instituto Nacional de Enfermedades Respiratorias Ismael Cosío Villegas, project ID: B31-18, and received no external funding.
Author contributions
A.K.S.-V., methodology, investigation, data curation. B.V.-A., methodology, investigation, data curation. C.F.-M., methodology, investigation, validation, review and editing, J.C.V.-R., methodology, software, formal analysis, visualization, resources. E.N.O.-D., methodology, investigation, visualization J.B.-D., methodology, writing-original draft preparation. E.M.C.-M., methodology, writing-original draft preparation. R.G.C., formal analysis, writing review and editing. L.T., formal analysis, review, and editing. B.O.-Q., formal analysis, writing, editing, supervision, and funding acquisition. R.L.-L., conceptualization; methodology, investigation, data curation, validation, formal analysis, writing-original draft preparation and editing, supervision, project administration, funding acquisition. All authors have read and agreed to the published
Competing Interests
The authors have declared that no competing interest exists.
References
1. Millan J, Puertollano R, Fan L. et al. The MAL proteolipid is a component of the detergent-insoluble membrane subdomains of human T-lymphocytes. Biochem J. 1997;321:247-252 10.1042/bj3210247
2. Martin-Belmonte F, Puertollano R, Millan J. et al. The MAL proteolipid is necessary for the overall apical delivery of membrane proteins in the polarized epithelial Madin-Darby canine kidney and Fischer rat thyroid cell lines. Mol Biol Cell. 2000;11:2033-2045 10.1091/mbc.11.6.2033
3. Alonso MA, Weissman SM. cDNA cloning and sequence of MAL, a hydrophobic protein associated with human T-cell differentiation. Proc Natl Acad Sci USA. 1987;84:1997-2001 10.1073/pnas.84.7.1997
4. Kim T, Fiedler K, Madison DL. et al. Cloning and characterization of MVP17: a developmentally regulated myelin protein in oligodendrocytes. J Neurosci Res. 1995;42:413-422 10.1002/jnr.490420316
5. Schaeren-Wiemers N, Valenzuela DM, Frank M. et al. Characterization of a rat gene, rMAL, encoding a protein with four hydrophobic domains in central and peripheral myelin. J Neurosci. 1995;15:5753-5764 10.1523/JNEUROSCI.15-08-05753.1995
6. Zacchetti D, Peranen J, Murata M. et al. VIP17/MAL, a proteolipid in apical transport vesicles. FEBS Lett. 1995;377:465-469 10.1016/0014-5793(95)01396-2
7. Millán J, Puertollano R, Fan L. et al. Caveolin and MAL, two protein components of internal detergent-insoluble membranes, are in distinct lipid microenvironments in MDCK cells. Biochem Biophys Res Commun. 1997;233:707-712 10.1006/bbrc.1997.6530
8. Martín-Belmonte F, Kremer L, Albar JP. et al. Expression of the MAL gene in the thyroid: the MAL proteolipid, a component of glycolipid-enriched membranes, is apically distributed in thyroid follicles. Endocrinology. 1998;139:2077-2084 10.1210/endo.139.4.5875
9. Zorzan E, Elgendy R, Guerra G. et al. Hypermethylation-Mediated silencing of CIDEA, MAL and PCDH17 tumour suppressor genes in canine DLBCL: from multi-omics analyses to mechanistic studies. Int.J. Mol. Sci. 2022;23:4021 10.3390/ijms23074021
10. Mimori K, Shiraishi T, Mashino K. et al. MAL gene expression in esophageal cancer suppresses motility, invasion and tumorigenicity and enhances apoptosis through the Fas pathway. Oncogene. 2003;22:3463-3471 10.1038/sj.onc.1206378
11. Wang Z, Wang M, Xu X. et al. Studies of MAL gene in human esophageal cancer by RNA in situ hybridization. Zhonghua Yixue Yichuanxue Zazhi. 2000;17:329-331
12. Kazemi-Noureini S, Colonna-Romano S, Ziaee AA. et al. Differential gene expression between squamous cell carcinoma of esophageus and its normal epithelium; altered pattern of mal, akr1c2, and rab11a expression. World J Gastroenterol. 2004;10:1716-1721 10.3748/wjgv10.i12.1716
13. Jin Z, Wang L, Zhang Y. et al. MAL hypermethylation is a tissue-specific event that correlates with MAL mRNA expression in esophageal carcinoma. Sci Rep. 2013;3:2838-2843 10.1038/srep02838
14. Buffart TE, Overmeer RM, Steenbergen RDM. et al. MAL promoter hypermethylation as a novel prognostic marker in gastric cancer. Brit J Cancer. 2008;99:1802-1807 10.1038/sj.bjc.6604777
15. Suzuki M, Shiraishi K, Eguchi A. et al. Aberrant methylation of LINE-1, SLIT2, MAL and IGFBP7 in non-small cell lung cancer. Oncol Rep. 2013;29:1308-1314 10.3892/or.2013.2266
16. Lind GE, Ahlquist T, Kolberg M. et al. Hypermethylated MAL gene a silent marker of early colon tumorigenesis. J Trans Med. 2008;6:13 10.1186/1479-5876-6-13
17. Beder LB, Gunduz M, Hotomi M. et al. T-lymphocyte maturation-associated protein gene as a candidate metastasis suppressor for head and neck squamous cell carcinomas. Cancer Sci. 2009;100:873-880 10.1111/j.1349-7006.2009.01132.x
18. Cao W, Zhang Z, Xu Q. et al. Epigenetic silencing of MAL, a putative tumor suppressor gene, can contribute to human epithelium cell carcinoma. Mol Cancer. 2010;9:296-308 10.1186/1476-4598-9-296
19. Hatta M, Nagai H, Okino K. et al. Down-regulation of members of glycolipid-enriched membrane raft gene family, MAL and BENE, in cervical squamous cell cancers. J Obstet Gynaecol Res. 2004;30:53-58 10.1111/j.1341-8076.2004.00156.x
20. Overmeer RM, Henken FE, Bierkens M. et al. Repression of MAL tumor suppressor activity by promoter methylation during cervical carcinogenesis. J Pathol. 2009;219:327-336 10.1002/path.2598
21. Guerrero-Preston R, Hadar T, Laskie-Ostrow K. et al. Differential promoter methylation of kinesin family member 1a in plasma is associated with breast cancer and DNA repair capacity. Oncol Rep. 2014;32:505-512 10.3892/or.2014.3262
22. Horne HN, Lee PS, Murphy SK. et al. Inactivation of the MAL gene in breast cancer is a common event that predicts benefit from adjuvant chemotherapy. Mol Cancer Res. 2009;7:199-209 10.1158/1541-7786.MCR-08-0314
23. Blaveri E, Simko JP, Korkola JE. et al. Bladder cancer outcome and subtype classification by gene expression. Clin Cancer Res. 2005;11:4044-4055 10.1158/1078-0432.CCR-04-2409
24. Hsi ED, Sup SJ, Alemany C. et al. MAL is expressed in a subset of Hodgkin lymphoma and identifies a population of patients with poor prognosis. Am J Clin Pathol. 2006;125:776-782 10.1309/98KL-HRDA-M5CM-DHE2
25. Copie-Bergman C, Gaulard P.; Maouche-Chretien L. et al. The MAL gene is expressed in primary mediastinal large B-cell lymphoma. Blood. 1999;94:3567-3575
26. Copie-Bergman C.; Plonquet A, Alonso MA. et al. MAL expression in lymphoid cells: further evidence for MAL as a distinct molecular marker of primary mediastinal large B-cell lymphomas. Mod Pathol. 2002;11:1172-1180 10.1097/01.MP.0000032534.81894.B3
27. Tracey L, Villuendas R, Ortiz P. et al. Identification of genes involved in resistance to interferon-alpha in cutaneous T-cell lymphoma. Am J Pathol. 2002;161:1825-1837 10.1016/s0002-9440(10)64459-8
28. Schwartz DR, Kardia SLR, Shedden KA. et al. Gene expression in ovarian cancer reflects both morphology and biological behavior, distinguishing clear cell from other poor-prognosis ovarian carcinomas. Cancer Res. 2002;62:4722-4729
29. Berchuck A, Iversen ES, Lancaster JM. et al. Patterns of gene expression that characterize long-term survival in advanced stage serous ovarian cancers. Clin Cancer Res. 2005;11:3686-3696 10.1158/1078-0432.CCR-04-2398
30. Lee PS, Teaberry VS, Bland AE. et al. Elevated MAL expression is accompanied by promoter hypomethylation and platinum resistance in epithelial ovarian cancer. Int J Cancer. 2010;126:1378-1389 10.1002/ijc.24797
31. Geng Z, Li J, Li S. et al. MAL protein suppresses the metastasis and invasion of GC cells by interfering with the phosphorylation of STAT3. J Transl Med. 2022;20:50 doi:10.1186/s12967-022-03254-5. 10.1186/s12967-022-03254-5
32. Sousa AM, Grandgenett PM, David L. et al. Reflections on MUC1 glycoprotein: the hidden potential of isoforms in carcinogenesis. APMIS. 2016;124:913-924 10.1111/apm.12587
33. Kufe DW, MUC1-C oncoprotein as a target in breast cancer. activation of signaling pathways and therapeutic approaches. Oncogene. 2013;32:1073-1081 10.1038/onc.2012.158
34. Bafna S, Kaur S, Batra SK, Membrane-bound mucins. the mechanistic basis for alterations in the growth and survival of cancer cells. Oncogene. 2010;29:2893-2904 10.1038/onc.2010.87
35. Irimura T, Denda K, Iida S. et al. Diverse glycosylation of MUC1 and MUC2: potential significance in tumor immunity. J Biochem. 1999;126:975-985 10.1093/oxfordjournals.jbchem.a022565
36. Kufe D. Mucins in cancer: function, prognosis and therapy. Nat Rev Cancer. 2009;9:874-885 10.1038/nrc2761
37. Pochampalli MR, el Bejjani RM, Schroeder JA. MUC1 is a novel regulator of ErbB1 receptor trafficking. Oncogene. 2007;26:1693-1701 10.1038/sj.onc.1209976
38. Li Y, Kuwahara H, Ren J. et al. The c-Src tyrosine kinase regulates signaling of the human DF3/MUC1 carcinoma-associated antigen with GSK3 beta and beta-catenin. J Biol Chem. 2001;276:6061-6064 10.1074/jbc.C000754200
39. Pandey P, Kharbanda S, Kufe D. Association of the DF3/MUC1 breast cancer antigen with Grb2 and the Sos/Ras ex-change protein. Cancer Res. 1995;55:4000-4003
40. Raina D.; Kharbanda S. and Kufe D, The MUC1 oncoprotein activates the anti-apoptotic phosphoinositide 3-kinase/Akt and Bcl-xL pathways in rat 3Y1 fibroblasts. J Biol Chem. 2004;279:20607-20612 10.1074/jbc.M310538200
41. Wen Y, Caffrey TC, Wheelock MJ. et al. Nuclear association of the cytoplasmic tail of MUC1 and beta-catenin. J Biol Chem. 2003;278:38029-38039 10.1074/jbc.M304333200
42. Rajabi H, Hiraki M, Kufe D. MUC1-C activates polycomb repressive complexes and downregulates tumor suppressor genes in human cancer cells. Oncogene. 2018;37:2079-2088 10.1038/s41388-017-0096-9
43. Fanayan S, Shehata M, Agterof AP. et al. Mucin 1 (MUC1) is a novel partner for MAL2 in breast carcinoma cells. BMC Cell Biology. 2009;10:7 10.1186/1471-2121-10-7
44. Laemmli UK. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature. 1970;227:680-685 10.1038/227680a0
45. Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2-ΔΔCT method. Methods. 2001;25:402-408 10.1006/meth.2001.1262
46. Poland PA, Kinlough CL, Rokaw MD. et al. Differential glycosylation of MUC1 in tumors and transfected epithelial and lymphoblastoid cell lines. Glycoconjugate J. 1997;14:89-96 10.1023/a:1018569100438
47. Lara-Lemus R, Saldaña-Villa AK, Vázquez-Almazán B. Myelin and lymphocyte protein and mucin-1 in a transgenic expression model. Mens Bioquim. 2018;42:48 -
48. Huang L, Ren J, Chen D. et al. MUC1 cytoplasmic domain coactivates Wnt target gene transcription and confers transformation. Cancer Biol Ther. 2003;2:702-706
49. Li Y, Bharti A, Chen D. et al. Interaction of glycogen synthase kinase 3beta with the DF3/MUC1 carcinoma-associated antigen and beta-catenin. Mol Cell Biol. 1998;18:7216-7224 10.1128/MCB.18.12.7216
50. Rajabi H, Ahmad R, Jin C. et al. MUC1-C Oncoprotein Induces TCF7L2 transcription factor activation and promotes cyclin D1 expression in human breast cancer cells. J Biol Chem. 2012;287:10703-10713 10.1074/jbc.M111.323311
51. Liu X, Caffrey TC, Steele MM. et al. MUC1 regulates cyclin D1 gene expression through p120 catenin and β-catenin. Oncogenesis. 2014;3:e107 10.1038/oncsis.2014.19
52. Bouillez A, Rajabi H, Pitroda S. et al. Inhibition of MUC1-C suppresses myc expression and attenuates malignant growth in KRAS mutant lung adenocarcinomas. Cancer Res. 2016;76:1538-1548 10.1158/0008-5472.CAN-15-1804
53. Tomas A, Futter CE, Eden ER, EGF receptor trafficking. consequences for signaling and cancer. Trends Cell Biol. 2014;24:26-34 10.1016/j.tcb.2013.11.002
54. Madshus IH, Stang E. Internalization and intracellular sorting of the EGF receptor: a model for understanding the mechanisms of receptor trafficking. J Cell Sci. 2009;122:3433-3439 10.1242/jcs.050260
55. Chi S, Cao H, Wang Y. et al. A novel form of the clathrin adaptor protein Eps15 mediates recycling of the epidermal growth factor receptor. J Biol Chem. 2011;286:35196-35208 10.1074/jbc.M111.247577
56. Lara-Lemus R, On the role of myelin, lymphocyte protein (MAL) in cancer. a puzzle with two faces. J Cancer. 2019;10:2312-2318 10.7150/jca.30376
57. Razawi H, Kinlough CL, Staubach S. et al. Evidence for core 2 to core 1 O-glycan remodeling during the recycling of MUC1. Glycobiology. 2013;23:935-945 10.1093/glycob/cwt030
58. Ceriani RL, Chan CM, Baratta FS. et al. Levels of expression of breast epithelial mucin detected by monoclonal antibody BrE-3 in breast cancer prognosis. Int J Cancer. 1992;51:343-354 10.1002/ijc.2910510303
59. Bieche I, Ruffet E, Zweibaum A. et al. MUC1 mucin gene, transcripts, and protein in adenomas and papillary carcinomas of the thyroid. Thyroid. 1997;7:725-731 10.1089/thy.1997.7.725
60. Rubio-Ramos A, Labat-de-Hoz L, Correas I. et al. The MAL protein, an integral component of specialized membranes, in normal cells and cancer. Cells. 2021;10:1065 doi.org/10.3390/cells10051065. 10.3390/cells10051065
61. Rajabi H, Tagde A, Kufe D. MUC1-C drives DNA methylation in cancer. Aging. 2016;8:3155-56 10.18632/aging.101153
62. Rajabi H, Tagde A, Alam M. et al. DNA methylation by DNMT1 and DNMT3b methyltransferases is driven by the MUC1-C oncoprotein in human carcinoma cells. Oncogene. 2016;35:6439-6445 10.1038/onc.2016.180
63. Teneng I, Tellez CS, Picchi MA. et al. Global identification of genes targeted by DNMT3b for epigenetic silencing in lung cancer. Oncogene. 2015;34:621-630 10.1038/onc.2013.580
64. Gaemers IC, Vos HL, Volders HH. et al. A stat-responsive element in the promoter of the episialin/MUC1 gene is in-volved in its overexpression in carcinoma cells. J Biol Chem. 2001;276:6191-6199 10.1074/jbc.M009449200
65. Yuan Z.L, Guan YJ, Wang L. et al. Central role of the threonine residue within the p+1 loop of receptor tyrosine kinase in STAT3 constitutive phosphorylation in metastatic cancer cells. Mol Cell Biol. 2004;24:9390-9400 10.1128/MCB.24.21.9390-9400.2004
66. Li G, Zhao L, Li W. et al. Feedback activation of STAT3 mediates trastuzumab resistance via upregulation of MUC1 and MUC4 expression. Oncotarget. 2014;5:8317-8329 10.18632/oncotarget.2135
67. Yang Y, Fang E, Luo J. et al. The antioxidant alphalipoic acid inhibits proliferation and invasion of human gastric cancer cells via suppression of STAT3-mediated MUC4 gene expression. Oxid Med Cell Longev. 2019 3643715. 10.1155/2019/3643715
68. Zhao Y-Q, Wu T, Wang L-F. et al. Targeting MUC1-C reverses the cisplatin resistance of esophageal squamous cell carcinoma in vitro and in vivo. Transl Cancer Res. 2021;10:645-655 10.21037/tcr-20-2495
Author contact
![]() Corresponding author: Roberto Lara-Lemus, Dpto. de Biomedicina Molecular e Investigación Traslacional, Instituto Nacional de Enfermedades Respiratorias “Ismael Cosío Villegas”. Calz. De Tlalpan No. 4502, Col. Sección XVI, Tlalpan, Ciudad de México 14080. Tel. + 52 55 54871700. e-mail: antonio.laragob.mx; alarale.rcom.
Corresponding author: Roberto Lara-Lemus, Dpto. de Biomedicina Molecular e Investigación Traslacional, Instituto Nacional de Enfermedades Respiratorias “Ismael Cosío Villegas”. Calz. De Tlalpan No. 4502, Col. Sección XVI, Tlalpan, Ciudad de México 14080. Tel. + 52 55 54871700. e-mail: antonio.laragob.mx; alarale.rcom.