Impact Factor ISSN: 1837-9664

 Global reach, higher impact
Global reach, higher impactJ Cancer 2026; 17(4):740-758. doi:10.7150/jca.132694 This issue Cite
Research Paper
An integrated bulk and single-cell transcriptomic analysis reveals stemness-driven immune regulation and therapeutic vulnerability in colorectal cancer
I-Hung Chen1,2,3, Kai-Fu Chang4,5, Chien-Cheng Chao4,5, Chung-Hsien Lin4, Chih-Hsuan Chang4,5, Ching-Chung Ko6,7,8, Hui-Ru Lin2,9, Chi-Jen Wu9,10, Chien-Han Yuan2,4,11,12, Sachin Kumar13,14,15, Dahlak Daniel Solomon13,14,16, Do Thi Minh Xuan17, Neethu Palekkode13,14,18, Ayman Fathima14,19, Yung-Kuo Lee2,4,5,20, Chung-Bao Hsieh21 ![]() , Yuen-Jung Wu22
, Yuen-Jung Wu22 ![]()
1. Department of Internal Medicine, Tri-Service General hospital, National Defense Medical University, Taipei City, Taiwan.
2. Institute of Medical Science and Technology, National Sun Yat-Sen University, Kaohsiung 80424, Taiwan.
3. Department of Internal Medicine, Kaohsiung Armed Forces General Hospital, National Defense Medical University, Kaohsiung 80284, Taiwan.
4. Medical Laboratory, Medical Education and Research Center, Kaohsiung Armed Forces General Hospital, National Defense Medical University, Kaohsiung 80284, Taiwan.
5. Division of Experimental Surgery Center, Department of Surgery, Tri-Service General Hospital, National Defense Medical Center, Taipei, Taiwan.
6. Department of Medical Imaging, Chi-Mei Medical Center, Tainan, 710402, Taiwan.
7. Department of Health and Nutrition, Chia Nan University of Pharmacy and Science, Tainan, 71710, Taiwan.
8. School of Medicine, College of Medicine, National Sun Yat-Sen University, Kaohsiung, 80424, Taiwan.
9. Nursing Department, Kaohsiung Armed Forces General Hospital, National Defense Medical University, Kaohsiung, 80284, Taiwan.
10. College of Nursing, Kaohsiung Medical University, Kaohsiung, 80708, Taiwan.
11. Department of Otolaryngology, Kaohsiung Armed Forces General Hospital, National Defense Medical University, Kaohsiung, 80284, Taiwan.
12. Department of Otolaryngology, National Defense Medical Center, Taipei, 11490, Taiwan
13. PhD Program for Cancer Molecular Biology and Drug Discovery, College of Medical Science and Technology, Taipei Medical University, Taipei, 11031, Taiwan.
14. Graduate Institute of Cancer Biology and Drug Discovery, College of Medical Science and Technology, Taipei Medical University, Taipei, 11031, Taiwan.
15. Faculty of Applied Sciences and Biotechnology, Shoolini University of Biotechnology and Management Sciences, Himachal Pradesh, 173229, India.
16. Yogananda School of AI Computers and Data Sciences, Shoolini University, Solan, 173229, India.
17. Faculty of Pharmacy, Van Lang University, 69/68 Dang Thuy Tram Street, Binh Loi Trung Ward, Ho Chi Minh City, 70000, Vietnam
18. Department of Biotechnology, Mother Teresa Women's University, Kodaikanal, Tamil Nadu, 624101, India.
19. Computer Engineering with specialization in Artificial Intelligence and Machine Learning, Presidency University, Yelahanka, Bengaluru, 560064 India.
20. School of Medicine, National Defense Medical University, Taipei, 11490, Taiwan.
21. Taipei City Hospital Zhongxing Branch, Taipei City, 103212, Taiwan.
22. Department of Surgery, Kaohsiung Armed Forces General Hospital, National Defense Medical University, Kaohsiung, 80284, Taiwan.
Received 2026-2-5; Accepted 2026-3-4; Published 2026-3-25
Abstract

Tumor stemness is increasingly recognized as a key contributor to tumor heterogeneity, immune regulation, and therapeutic resistance in colorectal cancer (CRC). In this study, we developed a stemness-based risk model using bulk transcriptomic data from The Cancer Genome Atlas and evaluated its prognostic and therapeutic relevance through integrative analyses. The proposed risk score robustly stratified patients into distinct prognostic groups and remained an independent predictor of overall survival after adjustment for clinicopathological variables. Stemness-high tumors exhibited altered immune infiltration patterns and coordinated upregulation of immune checkpoint-related genes. Although the association between stemness score and immune evasion potential was modest, its clinical relevance was supported by validation in independent immunotherapy-treated cohorts, where low-risk patients demonstrated improved survival and higher response rates. Single-cell RNA sequencing (scRNA-seq) analysis further revealed that enhanced stemness and dedifferentiation were predominantly localized within malignant epithelial cells. Together, these findings establish tumor stemness as a central determinant of prognosis, immune regulation, and therapeutic vulnerability in CRC.
Keywords: colorectal cancer, tumor stemness, immune microenvironment, immunotherapy, single-cell RNA sequencing, prognostic model
Introduction
Colorectal cancer (CRC) remains one of the leading causes of cancer-related morbidity and mortality worldwide, despite substantial advances in surgical techniques, chemotherapy, targeted therapy, and immunotherapy [1-4]. However, patient outcomes remain highly heterogeneous, even among individuals with similar clinicopathological features. This heterogeneity reflects the complex biological diversity of colorectal tumors, which arises from genetic, epigenetic, and microenvironmental factors [5-7]. Consequently, conventional staging systems alone are insufficient to accurately predict prognosis or therapeutic response, underscoring the need for biologically informed biomarkers that capture intrinsic tumor properties [8, 9].
Tumor stemness, defined as the degree to which cancer cells retain stem cell-like characteristics such as self-renewal, plasticity, and dedifferentiation, has emerged as a critical determinant of tumor aggressiveness and therapeutic resistance [10-13]. Accumulating evidence suggests that stemness-associated transcriptional programs contribute to tumor progression, metastasis, and recurrence across multiple cancer types. In CRC, elevated stemness has been linked to poor prognosis and resistance to conventional therapies [14-16]. Nevertheless, the prognostic significance of stemness-related gene signatures and their integration into clinically applicable risk stratification models remain incompletely understood [17-19].
Beyond its role in intrinsic tumor biology, stemness is increasingly recognized as a key modulator of the tumor immune microenvironment [20]. Stemness-high tumors have been reported to exhibit altered immune cell infiltration patterns, dysregulated immune checkpoint signaling, and enhanced immune evasion capabilities [21-23]. These features may critically influence responsiveness to immune checkpoint blockade and other immunotherapeutic strategies [24, 25]. However, the relationship between stemness and immunotherapy response in CRC is complex and context-dependent, particularly when assessed using bulk transcriptomic data that obscure cell-type-specific contributions [18, 26, 27].
In this study, we developed a stemness-based prognostic risk model for CRC using bulk transcriptomic data and systematically evaluated its associations with survival outcomes, immune infiltration, immune checkpoint regulation, immunotherapy response, and drug sensitivity. To overcome the limitations of bulk-level analyses, we further integrated scRNA-seq data from paired normal and tumor colorectal tissues to resolve the cellular origins and differentiation states underlying stemness-associated phenotypes [28, 29]. By combining bulk and single-cell analyses with external validation in immunotherapy-treated cohorts and protein-level verification, our study provides a comprehensive framework linking stemness, tumor heterogeneity, immune regulation, and therapeutic vulnerability in CRC.
Methods
Data acquisition and preprocessing of bulk transcriptomic datasets
Bulk RNA sequencing data and corresponding clinical information for CRC patients were obtained from The Cancer Genome Atlas (TCGA) database. Gene expression data were normalized to transcripts per million values and log2-transformed for downstream analyses [30, 31]. Patients with incomplete survival information were excluded. External validation datasets, including GSE38882 for prognostic validation and IMvigor210 and GSE78820 for immunotherapy response analyses, were downloaded from the Gene Expression Omnibus or publicly available repositories [32]. Batch effects between datasets were addressed using standard normalization procedures where appropriate [33-35].
Stemness score calculation and patient stratification
Stemness scores were calculated for each tumor sample based on predefined stemness-associated gene expression patterns using a single-sample scoring approach (specifically, the ssGSEA algorithm). Patients were stratified into high- and low-stemness groups using the median stemness score as the cutoff for differential expression and survival analyses [36-38]. For selected analyses, patients were further categorized into high, median, and low stemness groups to assess dose-dependent trends [39].
Differential expression analysis and functional enrichment
Differentially expressed genes between high- and low-stemness groups were identified using linear modeling with empirical Bayes moderation [40-42]. Genes with an absolute log2 fold change above 1.0 and adjusted p values below 0.05 were considered significant. Functional enrichment analyses were performed using Gene Ontology and pathway databases to characterize biological processes associated with stemness-related transcriptional changes [43, 44].
Construction of the stemness-based prognostic risk model
Univariate Cox proportional hazards regression was applied to identify stemness-associated genes significantly correlated with overall survival. Candidate genes were subsequently subjected to least absolute shrinkage and selection operator Cox regression to construct a parsimonious prognostic model [19]. The optimal penalty parameter was determined by cross-validation [45-47]. Risk scores were calculated as a weighted sum of gene expression values multiplied by their corresponding regression coefficients, and patients were stratified into high- and low-risk groups based on the median risk score [48].
Survival analysis and independent prognostic evaluation
Kaplan-Meier survival analysis with log-rank tests was used to compare survival outcomes between risk groups [49]. Time-dependent receiver operating characteristic curves were generated to evaluate predictive performance at different time points. Univariate and multivariate Cox regression analyses were conducted to assess the independent prognostic value of the risk score after adjustment for clinicopathological variables, including age, sex, and pathological staging. A nomogram integrating molecular and clinical factors was constructed for comparative performance assessment [50].
Immune infiltration, immune checkpoint, and immune evasion analyses
Tumor immune cell infiltration was estimated using the CIBERSORT algorithm [51]. Differences in immune cell composition between risk groups were evaluated using non-parametric statistical tests. Immune checkpoint gene expression profiles were compared across stemness and risk groups. Stromal, immune, and ESTIMATE scores were calculated to assess overall tumor microenvironment composition. Immune evasion potential was evaluated using Tumor Immune Dysfunction and Exclusion scores, and correlations between stemness scores and TIDE scores were assessed using Spearman correlation analysis [52].
Immunotherapy response validation
The association between the stemness-based risk model and immunotherapy response was evaluated in IMvigor210 and GSE78820 cohorts. Patients were classified according to reported treatment responses, including complete response, partial response, stable disease, and progressive disease. Survival outcomes, risk score distributions, and response proportions were compared between risk groups to assess predictive relevance in immunotherapy-treated populations [29].
Drug sensitivity prediction
In silico drug sensitivity analysis was performed by estimating half-maximal inhibitory concentration values for multiple therapeutic agents using established pharmacogenomic modeling approaches [53]. Estimated IC50 values were compared between high- and low-risk groups [54-56], to identify differential drug susceptibility patterns associated with stemness-based risk stratification [57].
Single-cell RNA sequencing analysis
ScRNA-seq data from paired normal and tumor colorectal tissues were obtained from GSE196964. Quality control filtering was applied to remove low-quality cells based on gene count and mitochondrial gene expression thresholds [58, 59]. Data were normalized, scaled, and subjected to dimensionality reduction using principal component analysis and uniform manifold approximation and projection. Cell clusters were annotated based on canonical marker genes [60-62]. CytoTRACE analysis was performed to infer cellular differentiation states and stemness at single-cell resolution. Functional enrichment analyses were conducted for major cell populations [63, 64].
Immunohistochemical validation
Protein-level validation of selected stemness-associated signature genes was performed using publicly available immunohistochemical staining data from CRC tissue microarrays [65-67]. Representative staining patterns and expression distributions were evaluated to support transcriptomic findings [68].
Statistical analysis
All statistical analyses were conducted using R (4.4.1) software. Survival analyses were performed using Cox proportional hazards models and Kaplan-Meier methods. Group comparisons were conducted using Wilcoxon rank-sum or Kruskal-Wallis tests as appropriate. Correlation analyses were performed using Spearman correlation coefficients. All statistical tests were two-sided, and p values less than 0.05 were considered statistically significant unless otherwise specified.
Results
Stemness-associated transcriptional alterations and prognostic signature construction in TCGA CRC
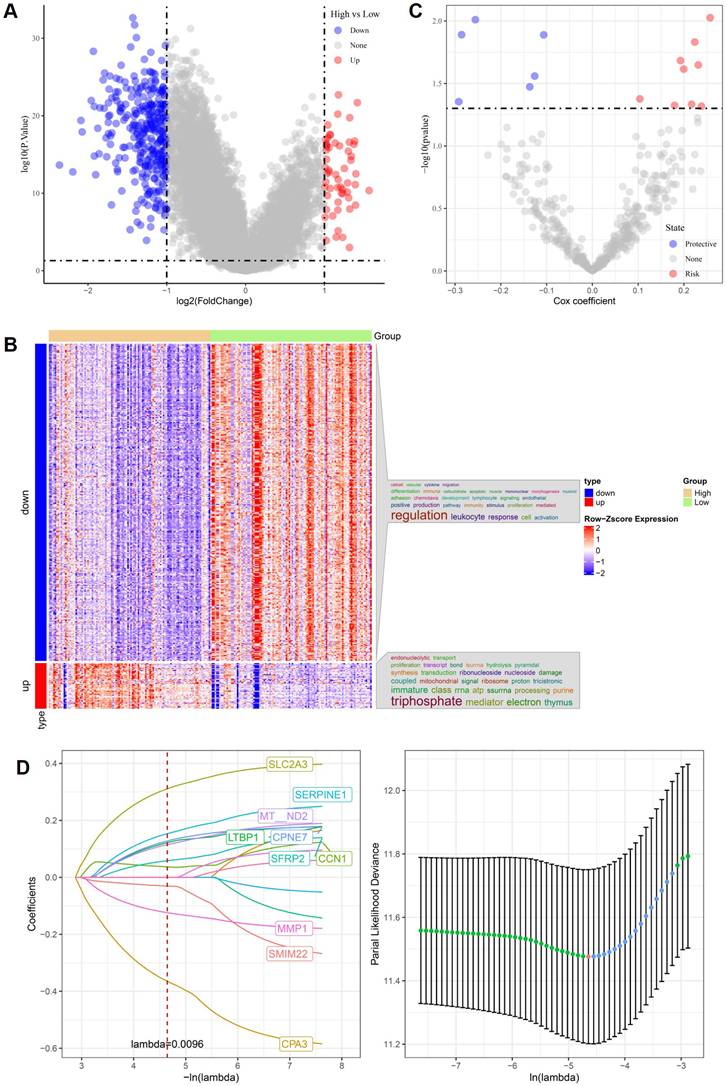
Using bulk RNA-seq data from the TCGA CRC cohort, patients were stratified into high- and low-stemness groups based on transcriptome-derived stemness scores. Differential expression analysis revealed extensive stemness-associated transcriptional reprogramming, as illustrated by the volcano plot in Figure 1A. Genes upregulated in the high-stemness group and those downregulated exhibited clear asymmetrical distributions, indicating distinct molecular states associated with stemness in CRC. Hierarchical clustering of these differentially expressed genes demonstrated robust separation between high- and low-stemness tumors (Figure 1B). Genes enriched in high-stemness tumors were predominantly associated with immune-related and inflammatory processes, including leukocyte response and immune activation, whereas genes downregulated in the high-stemness group were mainly linked to mitochondrial function, ribosomal activity, RNA processing, and oxidative metabolism. This reciprocal pattern suggests that stemness-high CRC are characterized by immune-related transcriptional activation accompanied by metabolic suppression. To assess prognostic relevance, univariate Cox regression analysis was performed and identified multiple stemness-associated genes significantly correlated with overall survival (Figure 1C). Both risk-associated and protective genes were observed, indicating heterogeneous contributions of stemness-related pathways to patient outcomes. Subsequently, LASSO Cox regression was applied to construct a parsimonious prognostic model. An optimal lambda value of 0.0096 was determined by cross-validation, yielding a compact gene signature comprising SLC2A3, SERPINE1, MT-ND2, LTBP1, CPNE7, SFRP2, CCN1, MMP1, SMIM22, and CPA3 (Figure 1D). The stability of the cross-validation curve supported the robustness of this model. Overall, these results demonstrate that stemness-associated transcriptional programs identified from TCGA CRC data are tightly linked to immune regulation, metabolic remodeling, and clinical prognosis, providing a solid foundation for subsequent external validation and single-cell-level mechanistic analyses.
Stemness-associated gene signatures and prognostic modeling in TCGA CRC. (A) Volcano plot of differentially expressed genes between high- and low-stemness groups. (B) Heatmap of stemness-associated genes showing clear separation between stemness-defined tumors. (C) Univariate Cox regression analysis identifying risk-associated and protective genes. (D) LASSO Cox regression analysis demonstrating gene selection and optimal lambda determination.
Prognostic performance and external validation of the stemness-based risk model in CRC
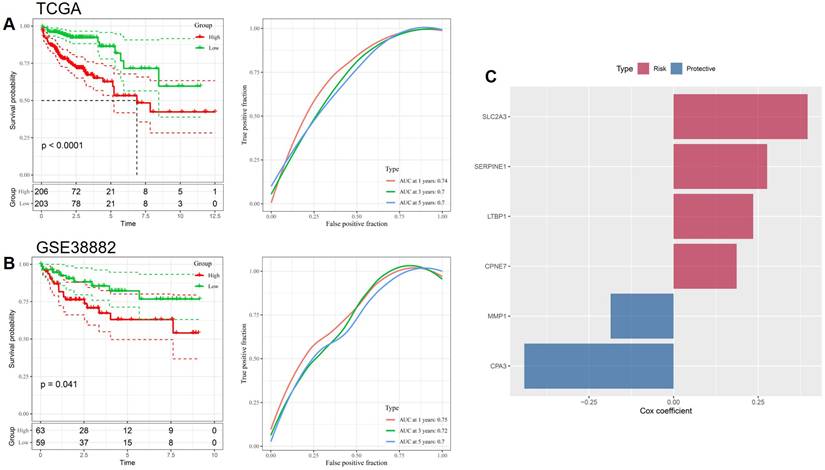
To evaluate the prognostic value of the stemness-based gene signature, patients in the TCGA CRC cohort were stratified into high- and low-risk groups according to the calculated risk score. Kaplan-Meier survival analysis demonstrated that patients in the high-risk group exhibited significantly worse overall survival compared with those in the low-risk group (p < 0.0001, Figure 2A). Time-dependent ROC analysis further confirmed the predictive performance of the model, with AUC values of approximately 0.74, 0.70, and 0.70 at 1, 3, and 5 years, respectively, indicating stable prognostic accuracy over time. The robustness of the risk model was subsequently validated in an independent external cohort, GSE38882. Consistent with the TCGA results, high-risk patients in the validation cohort showed significantly poorer survival outcomes than low-risk patients (p = 0.041, Figure 2B). ROC analysis in GSE38882 yielded comparable AUC values across multiple time points, supporting the generalizability of the stemness-based signature across different patient populations and platforms. The contribution of individual genes to the prognostic model was further examined by Cox regression coefficients (Figure 2C). SLC2A3, SERPINE1, LTBP1, and CPNE7 were identified as risk-associated genes, whereas MMP1 and CPA3 functioned as protective factors. This balanced composition of risk and protective genes underscores the biological heterogeneity of stemness-associated pathways in CRC and supports the interpretability of the proposed prognostic model.
Prognostic validation of the stemness-based risk model in CRC. (A) Kaplan-Meier survival analysis and time-dependent ROC curves in the TCGA cohort. (B) External validation of survival discrimination and predictive performance in the GSE38882 cohort. (C) Cox regression coefficients of genes included in the prognostic signature, classified as risk-associated or protective.
Independent prognostic value and comparative performance of the stemness-based risk score in CRC
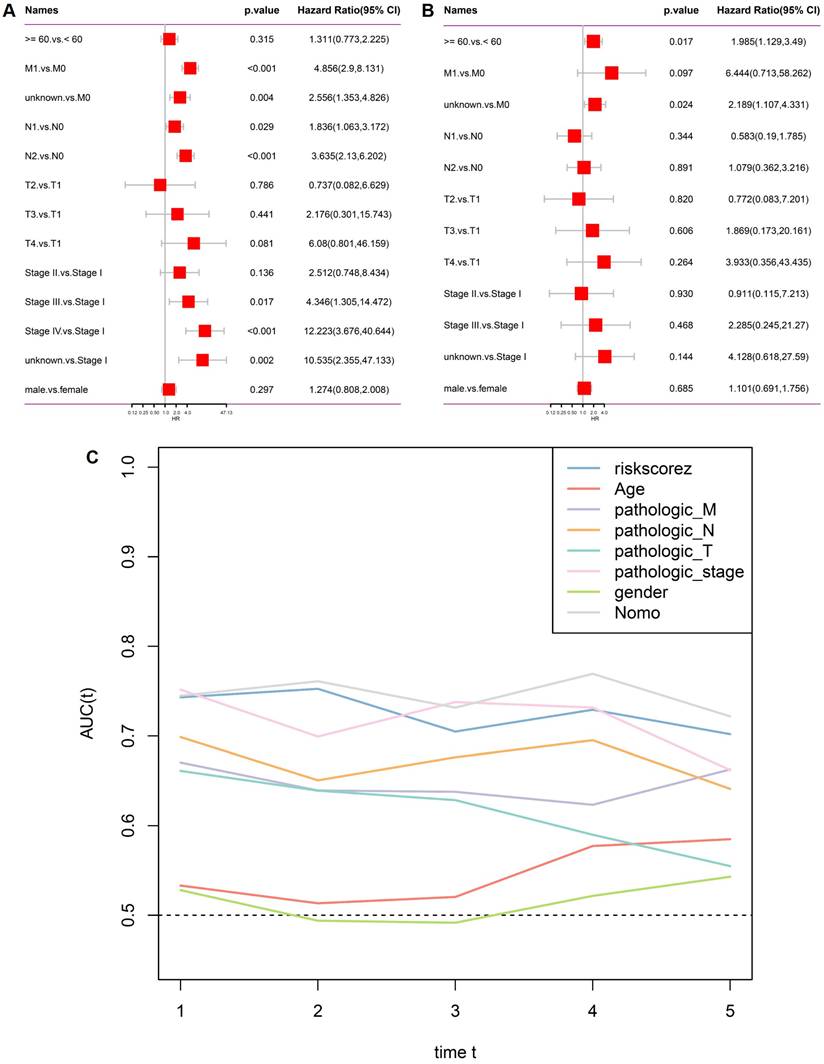
To determine whether the stemness-based risk score serves as an independent prognostic factor, univariate and multivariate Cox regression analyses were performed in the TCGA CRC cohort. In univariate analysis, multiple clinicopathological variables, including metastatic status, nodal involvement, and advanced pathological stage, were significantly associated with overall survival, together with the risk score (Figure 3A). After adjustment for conventional clinical parameters, multivariate Cox regression demonstrated that the risk score remained independently associated with patient survival, whereas most clinicopathological features lost statistical significance (Figure 3B). These results indicate that the stemness-based risk model provides prognostic information beyond standard clinical staging variables. To further compare predictive performance, time-dependent AUC analysis was conducted. As shown in Figure 3C, the risk score consistently outperformed individual clinical factors, including age, pathological T, N, M status, and overall stage, across multiple time points. The integrated nomogram model achieved the highest AUC values, supporting the complementary value of combining molecular and clinical features for survival prediction in CRC.
Independent prognostic analysis and comparative predictive performance of the stemness-based risk model. (A) Univariate Cox regression analysis of clinicopathological variables and risk score in the TCGA cohort. (B) Multivariate Cox regression analysis demonstrating the independent prognostic value of the risk score. (C) Time-dependent AUC comparison of the risk score, clinicopathological variables, and the integrated nomogram model.
Association between the stemness-based risk score and the immune microenvironment in CRC
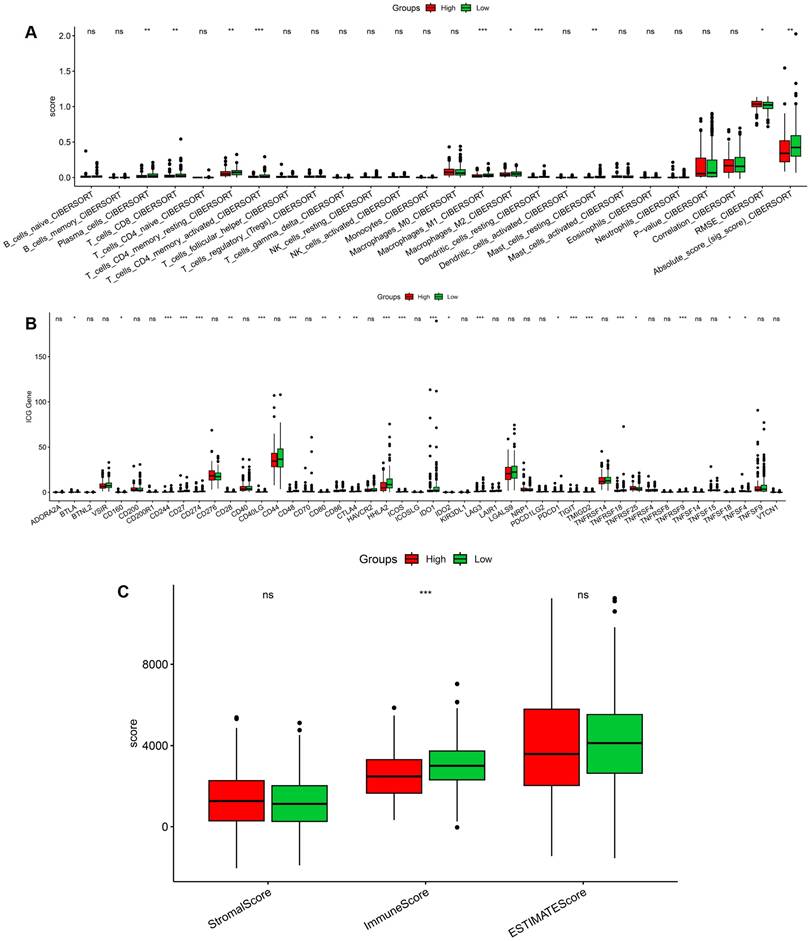
To explore the relationship between the stemness-based risk score and the tumor immune microenvironment, immune cell infiltration was estimated using CIBERSORT. Comparison between high- and low-risk groups revealed significant differences in multiple immune cell subsets, including activated T-cell populations, macrophage subtypes, and dendritic cells (Figure 4A). These findings indicate that the stemness-based risk stratification is accompanied by distinct immune infiltration patterns in CRC. We further examined the expression of immune checkpoint genes between risk groups. Several immune checkpoint-related genes exhibited differential expression between high- and low-risk tumors, suggesting altered immune regulatory states associated with stemness-related risk profiles (Figure 4B). This pattern is consistent with a more immunologically active yet potentially immunosuppressive microenvironment in high-risk CRCs. In addition, ESTIMATE analysis demonstrated that the high-risk group was associated with significantly higher immune scores, whereas stromal scores showed no significant difference between groups (Figure 4C). The ESTIMATE score, reflecting overall non-tumor cellular content, was also elevated in high-risk tumors. Collectively, these results suggest that the stemness-based risk score is closely linked to immune infiltration and immune regulatory activity in the colorectal tumor microenvironment.
Immune microenvironment characteristics associated with the stemness-based risk score in CRC. (A) Comparison of CIBERSORT-estimated immune cell infiltration between high- and low-risk groups. (B) Differential expression of immune checkpoint-related genes between risk groups. (C) Comparison of stromal score, immune score, and ESTIMATE score between high- and low-risk tumors.
Clinical associations of stemness score in CRC
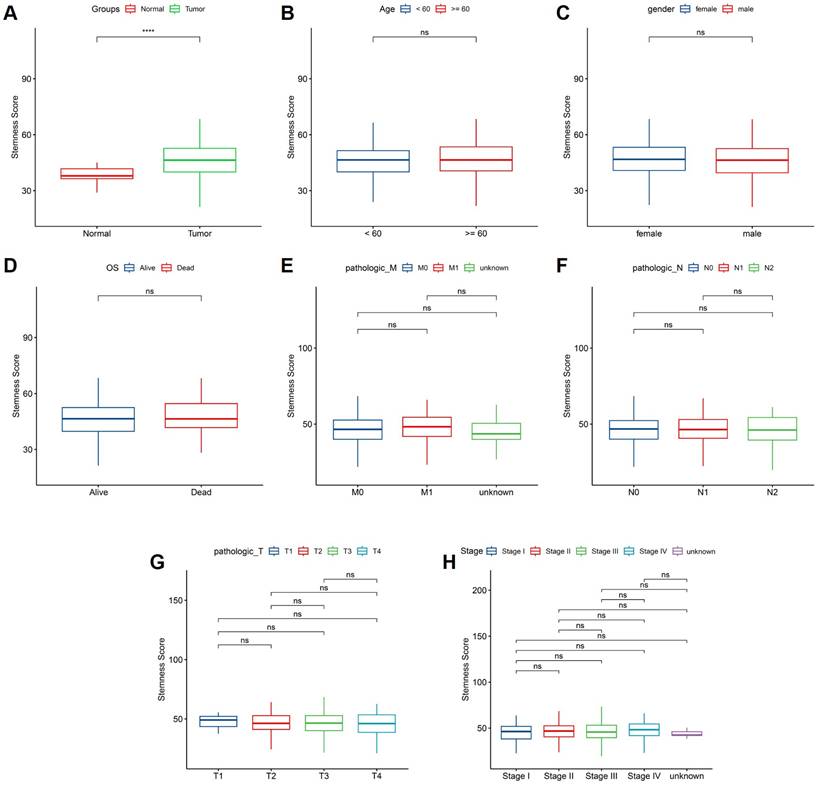
We next investigated the associations between stemness score and clinicopathological characteristics in the TCGA CRC cohort. As shown in Figure 5A, tumor tissues exhibited significantly higher stemness scores than normal tissues, indicating enhanced stemness features in CRC. In contrast, stemness scores did not differ significantly according to age or sex (Figure 5B-C). No significant difference in stemness score was observed between patients who were alive or deceased at the time of follow-up (Figure 5D). Similarly, stemness scores showed no significant associations with metastatic status, lymph node involvement, primary tumor depth, or overall pathological stage (Figure 5E-H). These results suggest that stemness score primarily reflects intrinsic tumor biological properties rather than conventional clinicopathological stratifications.
Association between stemness score and clinicopathological features in CRC. (A) Comparison of stemness score between normal and tumor tissues. (B-C) Stemness score stratified by age and sex. (D) Comparison of stemness score according to overall survival status. (E-H) Distribution of stemness score across pathological M, N, T classifications and tumor stage.
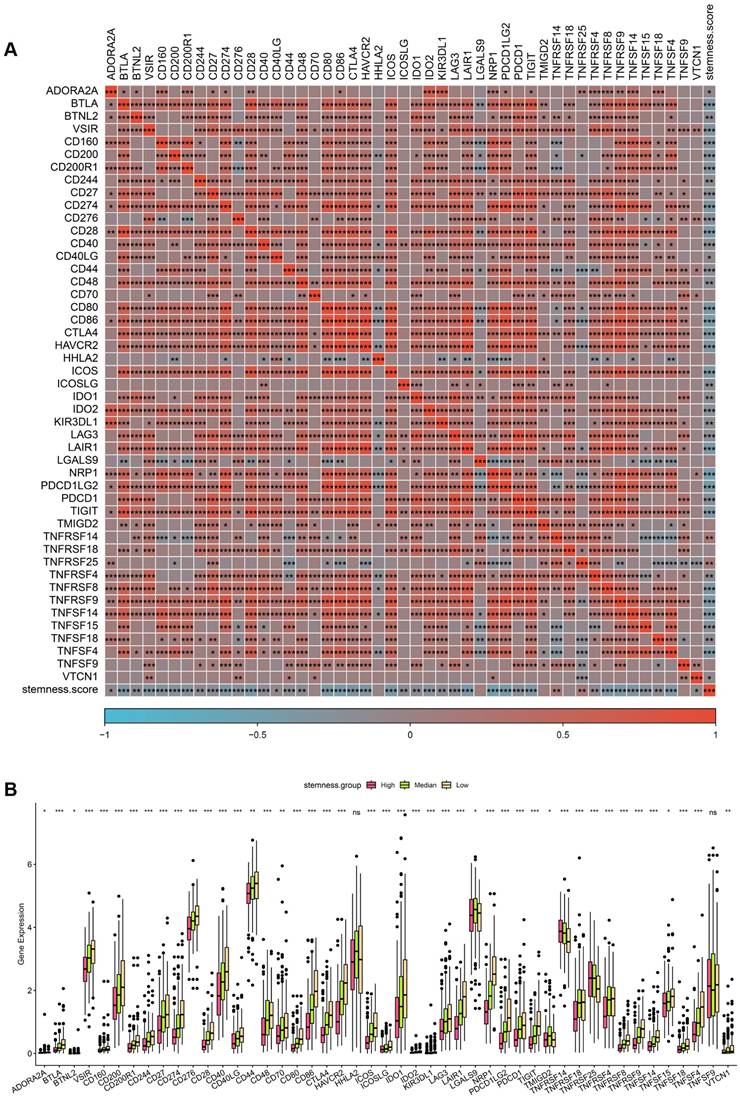
Stemness score is tightly associated with immune checkpoint regulatory networks in CRC
To further investigate the relationship between stemness and immune regulation, correlation analysis was performed between stemness score and immune checkpoint-related genes in the TCGA CRC cohort. As shown in Figure 6A, stemness score exhibited widespread positive correlations with multiple immune checkpoint genes, including PDCD1, CD274, CTLA4, LAG3, TIGIT, HAVCR2, IDO1, and members of the TNF and TNFR superfamilies. These results indicate that increased stemness is accompanied by coordinated activation of immune inhibitory signaling pathways. Patients were subsequently stratified into high, median, and low stemness groups to compare immune checkpoint gene expression levels. Consistent with the correlation analysis, the high-stemness group demonstrated significantly elevated expression of a broad range of immune checkpoint genes compared with the median and low stemness groups (Figure 6B). This stepwise increase in immune checkpoint expression across stemness strata suggests a stemness-dependent immunoregulatory gradient in CRC. Collectively, these findings support a close coupling between stemness-associated transcriptional programs and immune checkpoint activation, implying that stemness-high CRCs may adopt immune-evasive phenotypes through enhanced inhibitory immune signaling.
Association between stemness score and immune checkpoint gene expression in CRC. (A) Correlation heatmap showing relationships between stemness score and immune checkpoint-related genes in the TCGA cohort. (B) Comparison of immune checkpoint gene expression across high, median, and low stemness groups.
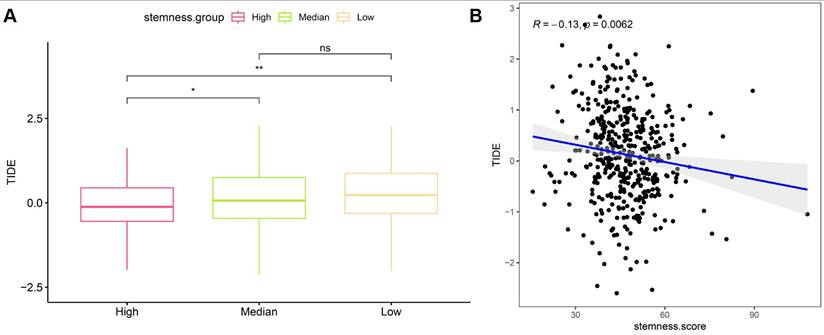
Association between stemness score and immune evasion potential in CRC
To evaluate the relationship between stemness and immune evasion, Tumor Immune Dysfunction and Exclusion (TIDE) scores were analyzed in the TCGA CRC cohort. Patients stratified by stemness levels showed significant differences in TIDE scores, with the high-stemness group exhibiting lower TIDE values compared with the median group, while no significant difference was observed between the median and low stemness groups (Figure 7A). Correlation analysis further demonstrated a modest but significant negative association between stemness score and TIDE score (R = -0.13, p = 0.0062), indicating that tumors with higher stemness tended to display reduced predicted immune escape potential (Figure 7B). These findings suggest that stemness-associated transcriptional programs are linked to immune modulation and may influence immunotherapy-related response patterns in CRC.
Relationship between stemness score and immune evasion potential in CRC. (A) Comparison of TIDE scores across high, median, and low stemness groups. (B) Correlation between stemness score and TIDE score in the TCGA cohort.
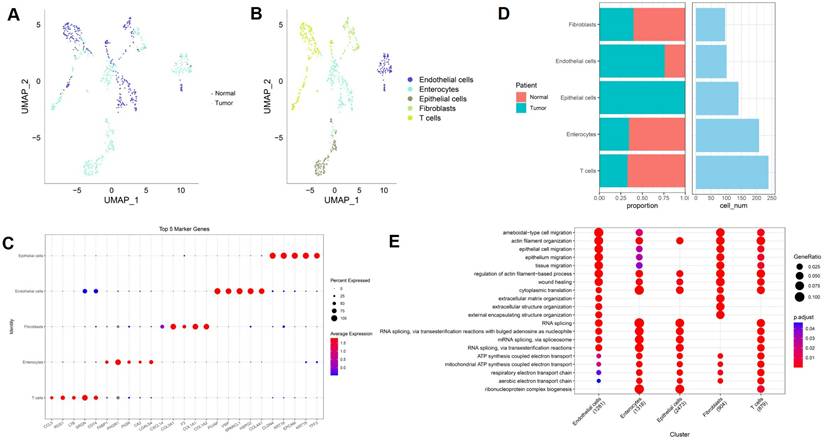
Single-cell landscape of paired normal and tumor colorectal tissues reveals cell-type-specific transcriptional programs
ScRNA-seq data from GSE196964 were analyzed to characterize the cellular composition of paired normal colon and colorectal tumor tissues. UMAP visualization demonstrated clear separation between normal and tumor cells, indicating distinct transcriptional states associated with malignant transformation (Figure 8A). Subsequent cell-type annotation identified five major cell populations, including epithelial cells, enterocytes, endothelial cells, fibroblasts, and T cells (Figure 8B). Marker gene analysis confirmed robust cell-type-specific expression patterns, with epithelial cells expressing EPCAM and KRT family genes, fibroblasts enriched for COL1A1 and COL1A2, endothelial cells expressing PECAM1 and VWF, enterocytes characterized by FABP1 and APOA1, and T cells marked by CD3D and CD3E (Figure 8C). These results validate the accuracy of cell-type annotation in this dataset. Quantitative comparison revealed marked differences in cellular composition between normal and tumor samples (Figure 8D). Tumor tissues exhibited an increased proportion of epithelial cells and fibroblasts, accompanied by a relative reduction in enterocytes and T cells, highlighting substantial remodeling of the tumor microenvironment in CRC. Functional enrichment analysis further revealed cell-type-specific biological processes (Figure 8E). Epithelial cells were predominantly enriched for RNA processing and metabolic pathways, fibroblasts showed strong enrichment in extracellular matrix organization and wound healing processes, endothelial cells were associated with migration and angiogenesis-related functions, and T cells displayed enrichment in immune activation-related pathways. Together, these findings delineate the multicellular architecture and functional heterogeneity of colorectal tumors at single-cell resolution.
Single-cell transcriptomic landscape of paired normal and tumor colorectal tissues in GSE196964. (A) UMAP plot showing separation of normal and tumor cells. (B) UMAP visualization of annotated cell types. (C) Dot plot of representative marker genes across major cell populations. (D) Comparison of cell-type proportions and cell numbers between normal and tumor samples. (E) Functional enrichment analysis of major cell populations.
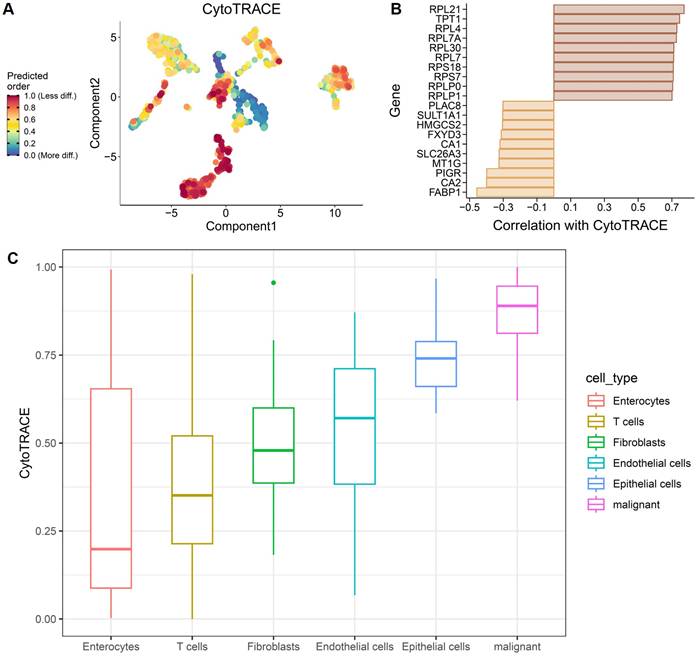
Single-cell CytoTRACE analysis reveals enhanced stemness and dedifferentiation in malignant colorectal cells
To characterize cellular differentiation states at single-cell resolution, CytoTRACE analysis was applied to the GSE196964 dataset. Projection of CytoTRACE scores onto the low-dimensional embedding revealed a continuous gradient of predicted differentiation states across cell populations, with malignant epithelial cells exhibiting the highest CytoTRACE scores, indicative of reduced differentiation and enhanced stemness (Figure 9A). Correlation analysis identified genes positively and negatively associated with CytoTRACE scores (Figure 9B). Genes positively correlated with CytoTRACE were predominantly ribosomal and translational regulators, consistent with a progenitor-like transcriptional program, whereas genes negatively correlated with CytoTRACE included enterocyte differentiation markers such as FABP1, CA1, CA2, and SLC26A3, reflecting loss of mature intestinal epithelial features in stemness-high cells. Comparison of CytoTRACE scores across annotated cell types further confirmed a hierarchical differentiation landscape (Figure 9C). Malignant cells exhibited the highest CytoTRACE scores, followed by epithelial cells, endothelial cells, and fibroblasts, whereas enterocytes and T cells showed the lowest scores. These results demonstrate that colorectal tumor cells adopt a dedifferentiated, stemness-enriched state relative to normal epithelial and stromal compartments.
CytoTRACE-based inference of differentiation states in CRC single-cell data. (A) Projection of CytoTRACE scores onto low-dimensional embedding, indicating predicted differentiation states. (B) Genes positively and negatively correlated with CytoTRACE scores. (C) Distribution of CytoTRACE scores across major cell types.
Validation of the stemness-based risk model in independent immunotherapy cohorts
To validate the predictive value of the stemness-based risk model in the context of immunotherapy, two independent immunotherapy-treated cohorts, IMvigor210 and GSE78820, were analyzed. In the IMvigor210 cohort, Kaplan-Meier survival analysis demonstrated significantly poorer survival outcomes in patients classified into the high-risk group compared with those in the low-risk group (Figure 10A). Risk score distributions further revealed that patients with progressive disease or stable disease exhibited significantly higher risk scores than those achieving complete or partial response (Figure 10B). Consistently, response proportion analysis showed that the low-risk group was enriched for complete or partial responders, whereas the high-risk group was predominantly composed of patients with progressive or stable disease (Figure 10C).
Validation of the stemness-based risk model in immunotherapy datasets (IMvigor210 and GSE78820). (A) Kaplan-Meier survival analysis of the IMvigor210 cohort showing significantly poorer survival in the high-risk group compared with the low-risk group. (B) Boxplot analysis in IMvigor210 demonstrating higher risk scores in patients with progressive disease or stable disease compared with those achieving complete or partial response. (C) Distribution of treatment responses in IMvigor210, with the low-risk group enriched for complete or partial response and the high-risk group enriched for progressive or stable disease. (D) Kaplan-Meier survival analysis of the GSE78820 cohort showing inferior survival outcomes in high-risk patients. (E) Subgroup Kaplan-Meier analysis in GSE78820 confirming a consistent trend toward reduced survival in the high-risk group. (F) Additional survival validation in a smaller GSE78820 subset further demonstrating poorer outcomes in high-risk patients. (G) Boxplot analysis in GSE78820 showing significantly higher risk scores in non-responders compared with responders. (H) Distribution of treatment responses in GSE78820 indicating that low-risk patients predominantly derived clinical benefit, whereas high-risk patients were mainly non-responders.
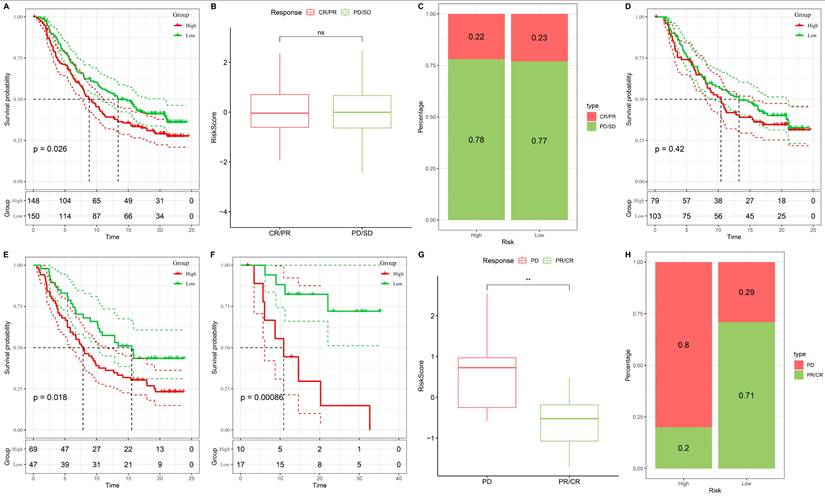
These findings were independently confirmed in the GSE78820 cohort. High-risk patients displayed inferior survival outcomes compared with low-risk patients (Figure 10D), and subgroup survival analyses consistently demonstrated a trend toward reduced survival in the high-risk group across different validation settings (Figure 10E-F). In addition, non-responders in GSE78820 exhibited significantly higher risk scores than responders (Figure 10G). Response distribution analysis further indicated that most low-risk patients derived clinical benefit from immunotherapy, whereas high-risk patients were largely non-responders (Figure 10H). Collectively, these results demonstrate that the stemness-based risk model robustly predicts immunotherapy response and survival outcomes across independent cohorts, supporting its potential utility as a biomarker for immunotherapy stratification in CRC.
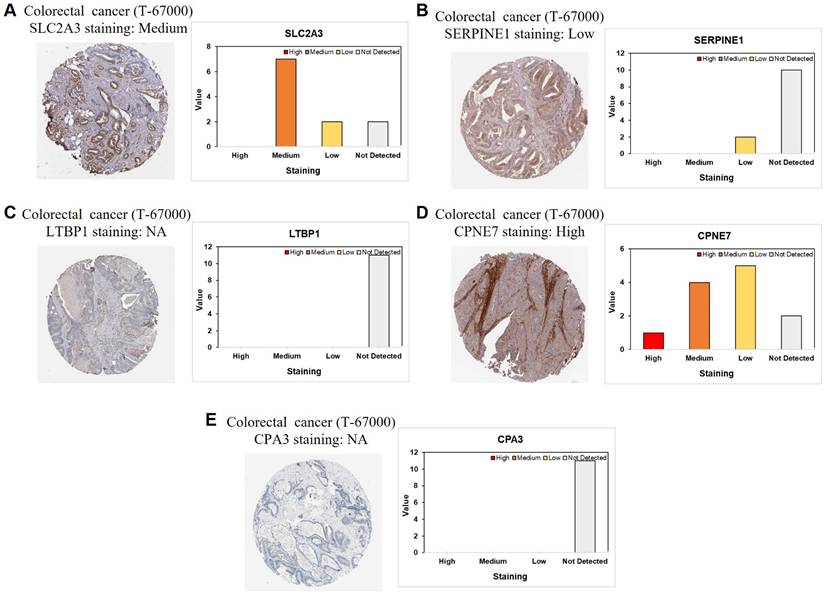
Protein-level validation of stemness-associated signature genes in CRC tissues
To further validate the stemness-associated risk signature at the protein level, immunohistochemical staining data from CRC tissue microarrays were examined. Representative staining patterns for SLC2A3, SERPINE1, LTBP1, CPNE7, and CPA3 are shown in Figure 11A-E. SLC2A3 protein expression exhibited predominantly medium staining intensity in CRC tissues, supporting its transcriptional upregulation observed in stemness-high tumors (Figure 11A). In contrast, SERPINE1 showed generally low or undetectable protein expression, suggesting potential post-transcriptional regulation or tumor heterogeneity affecting protein abundance (Figure 11B). LTBP1 and CPA3 proteins were largely undetectable in the analyzed samples, consistent with their variable or low expression patterns inferred from bulk and single-cell analyses (Figure 11C and 11E). Notably, CPNE7 demonstrated high protein expression in CRC tissues, aligning with its identification as a risk-associated gene in the prognostic model (Figure 11D). The differential protein expression patterns among these signature genes highlight heterogeneous regulatory mechanisms at the protein level and provide orthogonal validation for selected components of the stemness-based risk model. Collectively, these immunohistochemical findings support the biological relevance of the stemness-associated gene signature and reinforce its translational potential in CRC.
Immunohistochemical validation of stemness-associated signature genes in CRC. Representative immunohistochemical staining and corresponding intensity distribution of (A) SLC2A3, (B) SERPINE1, (C) LTBP1, (D) CPNE7, and (E) CPA3 in CRC tissue microarrays.
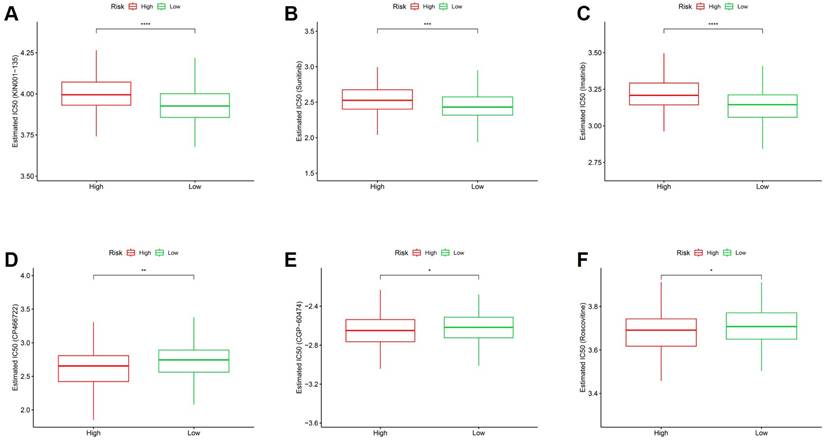
Stemness-based risk score is associated with differential drug sensitivity in CRC
These in silico IC50 estimations provide a global susceptibility profile, though they should be interpreted alongside the high degree of cellular heterogeneity. To explore the potential therapeutic implications of the stemness-based risk model, in silico drug sensitivity analysis was performed by comparing estimated IC50 values between high- and low-risk groups. As shown in Figure 12A-C, high-risk tumors exhibited significantly higher estimated IC50 values for multiple agents, including KIN001-135, Sunitinib, and Imatinib, indicating reduced drug sensitivity in stemness-high CRC. Conversely, low-risk tumors demonstrated significantly lower estimated IC50 values for several compounds, including CP466722, CGP-60474, and Roscovitine (Figure 12D-F), suggesting increased susceptibility to these agents. These differential drug response patterns indicate that stemness-associated risk stratification is closely linked to therapeutic vulnerability profiles. Collectively, these findings suggest that the stemness-based risk score may inform personalized treatment strategies by identifying subsets of CRC patients with distinct drug sensitivity landscapes. While providing a global profile, these estimations should be interpreted considering the inherent cellular heterogeneity identified in our single-cell analysis.
Association between stemness-based risk groups and predicted drug sensitivity in CRC. (A-C) Comparison of estimated IC50 values for KIN001-135, Sunitinib, and Imatinib between high- and low-risk groups. (D-F) Comparison of estimated IC50 values for CP466722, CGP-60474, and Roscovitine between risk groups.
Discussion
In this study, we systematically characterized the prognostic, immunological, and therapeutic relevance of tumor stemness in CRC by integrating bulk transcriptomic analyses, scRNA-seq, immunotherapy validation cohorts, and protein-level evidence. We developed a stemness-based risk model that robustly stratified patients into distinct prognostic groups across internal and external cohorts [17, 18]. Beyond survival prediction, this model captured fundamental biological differences associated with stemness, including transcriptional reprogramming, immune microenvironment remodeling, and differential therapeutic vulnerabilities. These findings highlight tumor stemness as a central axis linking intrinsic tumor biology with clinical outcomes in CRC [69-71].
At the bulk transcriptomic level, stemness-associated gene expression patterns were strongly associated with adverse prognosis and remained independently predictive after adjustment for conventional clinicopathological variables. Importantly, the prognostic value of the stemness-based risk score exceeded that of individual clinical features and was further enhanced when integrated into a nomogram framework [72, 73]. Immune profiling revealed that stemness-high tumors exhibited distinct immune infiltration patterns and coordinated upregulation of immune checkpoint-related genes, suggesting a complex immune regulatory state rather than simple immune exclusion [17, 74]. Although the correlation between stemness score and TIDE score was modest, it was statistically significant and directionally consistent, which is expected given the biological heterogeneity of bulk data and the composite nature of immune evasion metrics. Crucially, the immunotherapy relevance of the stemness-based risk model was substantiated in two independent immunotherapy-treated cohorts, supporting its translational significance beyond correlation-based analyses [18, 75, 76].
By incorporating scRNA-seq data from paired normal and tumor colorectal tissues, we further resolved the cellular origins and differentiation states underlying stemness-associated phenotypes [26]. Single-cell analysis demonstrated that malignant epithelial cells exhibited the highest stemness and dedifferentiation states, as inferred by CytoTRACE, while normal epithelial and immune cell populations showed more differentiated profiles [77]. Functional enrichment analyses revealed that stemness-high malignant cells were characterized by transcriptional programs related to RNA processing, metabolic remodeling, and cellular plasticity, whereas stromal and endothelial compartments displayed distinct extracellular matrix and migratory signatures. These findings provide mechanistic support for the bulk-level observations and underscore the importance of cellular heterogeneity in shaping stemness-driven tumor behavior [78, 79].
From a therapeutic perspective, the stemness-based risk model was associated with differential sensitivity to multiple anticancer agents, suggesting potential utility for guiding personalized treatment strategies [69]. In addition, protein-level validation using immunohistochemical data confirmed heterogeneous expression patterns of selected signature genes, reinforcing the biological relevance of the proposed model while also highlighting post-transcriptional and microenvironmental regulatory complexity [80]. Nevertheless, several limitations should be acknowledged. The stemness score and risk model were derived primarily from retrospective datasets, and prospective clinical validation is warranted [17]. Furthermore, while single-cell analyses provided valuable mechanistic insights, functional experiments will be required to establish causal relationships between stemness programs, immune modulation, and therapeutic response [69, 81].
In conclusion, our study presents an integrative framework that links tumor stemness with prognosis, immune regulation, and therapeutic vulnerability in CRC. By combining bulk and single-cell transcriptomics with immunotherapy validation and protein-level evidence, we provide a comprehensive view of stemness as a key determinant of tumor heterogeneity and clinical outcome. These findings lay the groundwork for future studies aimed at targeting stemness-associated pathways and optimizing precision oncology strategies in CRC. This independence suggests that stemness features may represent an intrinsic, early-stage molecular event that precedes anatomical progression.
Conclusions
In this study, we established a stemness-based risk model that effectively stratifies CRC patients according to prognosis and therapeutic relevance. By integrating bulk transcriptomic analyses with scRNA-seq, immunotherapy validation cohorts, and protein-level evidence, we demonstrate that tumor stemness is a key determinant of tumor heterogeneity, immune regulation, and clinical outcome in CRC. Our findings indicate that stemness-associated transcriptional programs are linked to adverse survival, distinct immune microenvironment characteristics, and differential sensitivity to immunotherapy and anticancer agents. Single-cell analyses further revealed that enhanced stemness and dedifferentiation are predominantly localized within malignant epithelial cells, providing mechanistic support for bulk-level observations. Collectively, this integrative framework highlights the potential of stemness-based biomarkers to inform prognostic assessment and therapeutic stratification in CRC, and provides a foundation for future studies aimed at translating stemness-targeted strategies into precision oncology applications.
Abbreviations
CRC: colorectal cancer
TCGA: The Cancer Genome Atlas
GEO: Gene Expression Omnibus
OS: overall survival
ROC: receiver operating characteristic
AUC: area under the curve
HR: hazard ratio
CI: confidence interval
LASSO: least absolute shrinkage and selection operator
ssGSEA: single-sample gene set enrichment analysis
TME: tumor microenvironment
IHC: immunohistochemistry
scRNA-seq: single-cell RNA sequencing
UMAP: uniform manifold approximation and projection
PCA: principal component analysis
DEGs: differentially expressed genes
GO: Gene Ontology
KEGG: Kyoto Encyclopedia of Genes and Genomes
CIBERSORT: cell-type identification by estimating relative subsets of RNA transcripts
ESTIMATE: estimation of stromal and immune cells in malignant tumor tissues using expression data
TIDE: tumor immune dysfunction and exclusion
IC50: half-maximal inhibitory concentration
PR: partial response
CR: complete response
SD: stable disease
PD: progressive disease
KM: Kaplan-Meier
TMA: tissue microarray
CytoTRACE: cellular trajectory reconstruction analysis using gene counts and expression
Acknowledgements
We would like to acknowledge Yi-Ting Wu, Yun-Yu Lin, and Yueh-Yuan Shieh for their excellent technical support at the Laboratory of Research and Medical Education and Research Center, Kaohsiung Armed Forces General Hospital, Kaohsiung, Taiwan. This research was funded by the Kaohsiung Armed Forces General Hospital grant nos. MND-MAB-C15-114063, MND-MAB-C01-115065 and KAFGH_D_115029. This work was also funded by National Science and Technology Council (NSTC), Taiwan (grant no. NSTC 114-2320-B-393-003). The APC was funded by Kaohsiung Armed Forces General Hospital. Furthermore, this work received financial support from the Healthy Taiwan Cultivation Plan of the Ministry of Health and Welfare (MOHW), Taiwan. (grant no. A20016).
Availability of data and materials
All datasets and materials generated in this study can be provided by the corresponding author upon reasonable request.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Morgan E, Arnold M, Gini A, Lorenzoni V, Cabasag CJ, Laversanne M. et al. Global burden of colorectal cancer in 2020 and 2040: incidence and mortality estimates from GLOBOCAN. Gut. 2023;72:338-44
2. Bray F, Laversanne M, Sung H, Ferlay J, Siegel RL, Soerjomataram I. et al. Global cancer statistics 2022: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2024;74:229-63
3. Abedizadeh R, Majidi F, Khorasani HR, Abedi H, Sabour D. Colorectal cancer: a comprehensive review of carcinogenesis, diagnosis, and novel strategies for classified treatments. Cancer Metastasis Rev. 2024;43:729-53
4. Siegel RL, Wagle NS, Cercek A, Smith RA, Jemal A. Colorectal cancer statistics, 2023. CA Cancer J Clin. 2023;73:233-54
5. Saoudi González N, Salvà F, Ros J, Baraibar I, Rodríguez-Castells M, García A. et al. Unravelling the Complexity of Colorectal Cancer: Heterogeneity, Clonal Evolution, and Clinical Implications. Cancers (Basel). 2023 15
6. Toghey S, Harvey-Jones EJ, Towler JD, Hafkamp CJH, Chong IY. Advances in Colorectal Cancer Cell Biology and Clonal Evolution. Int J Mol Sci. 2026 27
7. Househam J, Heide T, Cresswell GD, Spiteri I, Kimberley C, Zapata L. et al. Phenotypic plasticity and genetic control in colorectal cancer evolution. Nature. 2022;611:744-53
8. Mondal S, Preetam S, Deshwal RK, Thapliyal S, Rustagi S, Alghamdi S. et al. Advances in prognostic and predictive biomarkers for breast cancer: Integrating multigene assays, hormone receptors, and emerging circulating biomarkers. Clin Chim Acta. 2026;578:120513
9. Batis N, Brooks JM, Payne K, Sharma N, Nankivell P, Mehanna H. Lack of predictive tools for conventional and targeted cancer therapy: Barriers to biomarker development and clinical translation. Adv Drug Deliv Rev. 2021;176:113854
10. Nwokolo GC, Ganesan TS, Pors K, Falconer RA, Smarakan S. Cancer stem cells in focus: Deciphering the dynamic functional landscape of stemness in cancer. Biochim Biophys Acta Rev Cancer. 2025;1880:189440
11. Leck LYW, Abd El-Aziz YS, McKelvey KJ, Park KC, Sahni S, Lane DJR. et al. Cancer stem cells: Masters of all traits. Biochim Biophys Acta Mol Basis Dis. 2025;1871:167549
12. Paul R, Dorsey JF, Fan Y. Cell plasticity, senescence, and quiescence in cancer stem cells: Biological and therapeutic implications. Pharmacol Ther. 2022;231:107985
13. Aramini B, Masciale V, Grisendi G, Bertolini F, Maur M, Guaitoli G. et al. Dissecting Tumor Growth: The Role of Cancer Stem Cells in Drug Resistance and Recurrence. Cancers (Basel). 2022 14
14. Angius A, Scanu AM, Arru C, Muroni MR, Rallo V, Deiana G. et al. Portrait of Cancer Stem Cells on Colorectal Cancer: Molecular Biomarkers, Signaling Pathways and miRNAome. Int J Mol Sci. 2021 22
15. Frank MH, Wilson BJ, Gold JS, Frank NY. Clinical Implications of Colorectal Cancer Stem Cells in the Age of Single-Cell Omics and Targeted Therapies. Gastroenterology. 2021;160:1947-60
16. Zhan Y, Sun D, Gao J, Gao Q, Lv Y, Du T. et al. Single-cell transcriptomics reveals intratumor heterogeneity and the potential roles of cancer stem cells and myCAFs in colorectal cancer liver metastasis and recurrence. Cancer Lett. 2025;612:217452
17. Liu Z, Xu H, Weng S, Ren Y, Han X. Stemness Refines the Classification of Colorectal Cancer with Stratified Prognosis, Multi-Omics Landscape, Potential Mechanisms, and Treatment Options. Front Immunol. 2022;13:828330
18. Zheng H, Liu H, Li H, Dou W, Wang J, Zhang J. et al. Characterization of stem cell landscape and identification of stemness-relevant prognostic gene signature to aid immunotherapy in colorectal cancer. Stem Cell Res Ther. 2022;13:244
19. Wei R, Quan J, Li S, Liu H, Guan X, Jiang Z. et al. Integrative Analysis of Biomarkers Through Machine Learning Identifies Stemness Features in Colorectal Cancer. Front Cell Dev Biol. 2021;9:724860
20. Dianat-Moghadam H, Sharifi M, Salehi R, Keshavarz M, Shahgolzari M, Amoozgar Z. Engaging stemness improves cancer immunotherapy. Cancer Lett. 2023;554:216007
21. Hussein OJ, Rayan M, Matarid TR, Elkhalifa D, Abunada HH, Therachiyil L. et al. The role of immune checkpoints in modulating cancer stem cells anti-tumor immune responses: implications and perspectives in cancer therapy. J Exp Clin Cancer Res. 2025;44:305
22. Tomei S, Ibnaof O, Ravindran S, Ferrone S, Maccalli C. Cancer Stem Cells Are Possible Key Players in Regulating Anti-Tumor Immune Responses: The Role of Immunomodulating Molecules and MicroRNAs. Cancers (Basel). 2021 13
23. Nallasamy P, Nimmakayala RK, Parte S, Are AC, Batra SK, Ponnusamy MP. Tumor microenvironment enriches the stemness features: the architectural event of therapy resistance and metastasis. Mol Cancer. 2022;21:225
24. Meric-Bernstam F, Larkin J, Tabernero J, Bonini C. Enhancing anti-tumour efficacy with immunotherapy combinations. Lancet. 2021;397:1010-22
25. Ursino C, Mouric C, Gros L, Bonnefoy N, Faget J. Intrinsic features of the cancer cell as drivers of immune checkpoint blockade response and refractoriness. Front Immunol. 2023;14:1170321
26. Lin K, Chowdhury S, Zeineddine MA, Zeineddine FA, Hornstein NJ, Villarreal OE. et al. Identification of Colorectal Cancer Cell Stemness from Single-Cell RNA Sequencing. Mol Cancer Res. 2024;22:337-46
27. Dai Y, Luo X, Zhang L, Yan J. Roles of immunosuppressive myeloid states in colorectal cancer checkpoint inhibitor non-response: single-cell and spatial proteomics, and reprogramming approaches. Front Immunol. 2025;16:1742654
28. Duan Y, Li A, Miao D, Tan D, Wang K, Shi J. Colorectal cancer stem cells crosstalk in tumor immune microenvironment and targeted therapeutic strategies. Front Immunol. 2025;16:1714954
29. Cai L, Guo X, Zhang Y, Xie H, Liu Y, Zhou J. et al. Integrated analysis of single-cell and bulk RNA-sequencing to predict prognosis and therapeutic response for colorectal cancer. Sci Rep. 2025;15:7986
30. Nunes L, Li F, Wu M, Luo T, Hammarström K, Torell E. et al. Prognostic genome and transcriptome signatures in colorectal cancers. Nature. 2024;633:137-46
31. Holmström S, Hautaniemi S, Häkkinen A. POIBM: batch correction of heterogeneous RNA-seq datasets through latent sample matching. Bioinformatics. 2022;38:2474-80
32. Cao L, Li T, Ba Y, Chen E, Yang J, Zhang H. Exploring Immune-Related Prognostic Signatures in the Tumor Microenvironment of Colon Cancer. Front Genet. 2022;13:801484
33. Chiang YC, Wang CY, Kumar S, Hsieh CB, Chang KF, Ko CC. et al. Metal ion transporter SLC39A14-mediated ferroptosis and glycosylation modulate the tumor immune microenvironment: pan-cancer multi-omics exploration of therapeutic potential. Cancer Cell Int. 2025;25:363
34. Su BH, Kumar S, Cheng LH, Chang WJ, Solomon DD, Ko CC. et al. Multi-omics profiling reveals PLEKHA6 as a modulator of β-catenin signaling and therapeutic vulnerability in lung adenocarcinoma. Am J Cancer Res. 2025;15:3106-27
35. Xuan DTM, Yeh IJ, Liu HL, Su CY, Ko CC, Ta HDK. et al. A comparative analysis of Marburg virus-infected bat and human models from public high-throughput sequencing data. Int J Med Sci. 2025;22:1-16
36. Kumar S, Wu CC, Wulandari FS, Chiao CC, Ko CC, Lin HY. et al. Integration of multi-omics and single-cell transcriptome reveals mitochondrial outer membrane protein-2 (MTX-2) as a prognostic biomarker and characterizes ubiquinone metabolism in lung adenocarcinoma. J Cancer. 2025;16:2401-20
37. Xuan DTM, Wu CC, Wang WJ, Hsu HP, Ta HDK, Anuraga G. et al. Glutamine synthetase regulates the immune microenvironment and cancer development through the inflammatory pathway. Int J Med Sci. 2023;20:35-49
38. Xuan DTM, Yeh IJ, Su CY, Liu HL, Ta HDK, Anuraga G. et al. Prognostic and Immune Infiltration Value of Proteasome Assembly Chaperone (PSMG) Family Genes in Lung Adenocarcinoma. Int J Med Sci. 2023;20:87-101
39. Abdelrahman Z, Abdelatty A, Luo J, McKnight AJ, Wang X. Stratification of glioma based on stemness scores in bulk and single-cell transcriptomes. Comput Biol Med. 2024;175:108304
40. Wang CY, Chao YJ, Chen YL, Wang TW, Phan NN, Hsu HP. et al. Upregulation of peroxisome proliferator-activated receptor-α and the lipid metabolism pathway promotes carcinogenesis of ampullary cancer. Int J Med Sci. 2021;18:256-69
41. Liu HL, Yeh IJ, Phan NN, Wu YH, Yen MC, Hung JH. et al. Gene signatures of SARS-CoV/SARS-CoV-2-infected ferret lungs in short- and long-term models. Infect Genet Evol. 2020;85:104438
42. Wu YH, Yeh IJ, Phan NN, Yen MC, Hung JH, Chiao CC. et al. Gene signatures and potential therapeutic targets of Middle East respiratory syndrome coronavirus (MERS-CoV)-infected human lung adenocarcinoma epithelial cells. J Microbiol Immunol Infect. 2021;54:845-57
43. Lu J, Zhang H, Gu X, Liu Y, Zhao C, Wang X. Identification of key genes regulating colorectal cancer stem cell characteristics by bioinformatics analysis. Medicine (Baltimore). 2025;104:e40910
44. Ban C, Yang F, Wei M, Liu Q, Wang J, Chen L. et al. Integrative Analysis of Gene Expression Through One-Class Logistic Regression Machine Learning Identifies Stemness Features in Multiple Myeloma. Front Genet. 2021;12:666561
45. Tang D, Chen M, Huang X, Zhang G, Zeng L, Zhang G. et al. SRplot: A free online platform for data visualization and graphing. PLoS One. 2023;18:e0294236
46. Wickham H, Sievert C. ggplot2: elegant graphics for data analysis: springer New York; 2009
47. Chen PS, Hsu HP, Phan NN, Yen MC, Chen FW, Liu YW. et al. CCDC167 as a potential therapeutic target and regulator of cell cycle-related networks in breast cancer. Aging (Albany NY). 2021;13:4157-81
48. Dunias ZS, Van Calster B, Timmerman D, Boulesteix AL, van Smeden M. A comparison of hyperparameter tuning procedures for clinical prediction models: A simulation study. Stat Med. 2024;43:1119-34
49. Chu T, Meng Y, Wu P, Li Z, Wen H, Ren F. et al. The prognosis of patients with small cell carcinoma of the cervix: a retrospective study of the SEER database and a Chinese multicentre registry. Lancet Oncol. 2023;24:701-8
50. Chen X, Wang X, Chen K, Zheng Y, Chappell RJ, Dey J. Comparison of survival distributions in clinical trials: A practical guidance. Clin Trials. 2020;17:507-21
51. Chen B, Khodadoust MS, Liu CL, Newman AM, Alizadeh AA. Profiling Tumor Infiltrating Immune Cells with CIBERSORT. Methods Mol Biol. 2018;1711:243-59
52. Jiang P, Gu S, Pan D, Fu J, Sahu A, Hu X. et al. Signatures of T cell dysfunction and exclusion predict cancer immunotherapy response. Nat Med. 2018;24:1550-8
53. Chen Y, Juan L, Lv X, Shi L. Bioinformatics Research on Drug Sensitivity Prediction. Front Pharmacol. 2021;12:799712
54. Wu YJ, Chiao CC, Chuang PK, Hsieh CB, Ko CY, Ko CC. et al. Comprehensive analysis of bulk and single-cell RNA sequencing data reveals Schlafen-5 (SLFN5) as a novel prognosis and immunity. Int J Med Sci. 2024;21:2348-64
55. Anuraga G, Lang J, Xuan DTM, Ta HDK, Jiang JZ, Sun Z. et al. Integrated bioinformatics approaches to investigate alterations in transcriptomic profiles of monkeypox infected human cell line model. J Infect Public Health. 2024;17:60-9
56. Wang CY, Xuan DTM, Ye PH, Li CY, Anuraga G, Ta HDK. et al. Synergistic suppressive effects on triple-negative breast cancer by the combination of JTC-801 and sodium oxamate. Am J Cancer Res. 2023;13:4661-77
57. Park A, Joo M, Kim K, Son WJ, Lim G, Lee J. et al. A comprehensive evaluation of regression-based drug responsiveness prediction models, using cell viability inhibitory concentrations (IC50 values). Bioinformatics. 2022;38:2810-7
58. Hippen AA, Falco MM, Weber LM, Erkan EP, Zhang K, Doherty JA. et al. miQC: An adaptive probabilistic framework for quality control of single-cell RNA-sequencing data. PLoS Comput Biol. 2021;17:e1009290
59. Osorio D, Cai JJ. Systematic determination of the mitochondrial proportion in human and mice tissues for single-cell RNA-sequencing data quality control. Bioinformatics. 2021;37:963-7
60. Solomon DD, Ko CC, Chen HY, Kumar S, Wulandari FS, Xuan DTM. et al. A machine learning framework using urinary biomarkers for pancreatic ductal adenocarcinoma prediction with post hoc validation via single-cell transcriptomics. Brief Bioinform. 2025 26
61. Li CY, Anuraga G, Chang CP, Weng TY, Hsu HP, Ta HDK. et al. Repurposing nitric oxide donating drugs in cancer therapy through immune modulation. J Exp Clin Cancer Res. 2023;42:22
62. Hagerling C, Gonzalez H, Salari K, Wang CY, Lin C, Robles I. et al. Immune effector monocyte-neutrophil cooperation induced by the primary tumor prevents metastatic progression of breast cancer. Proc Natl Acad Sci U S A. 2019;116:21704-14
63. Kang M, Gulati GS, Brown EL, Qi Z, Avagyan S, Armenteros JJA. et al. Improved reconstruction of single-cell developmental potential with CytoTRACE 2. Nat Methods. 2025;22:2258-63
64. Gulati GS, Sikandar SS, Wesche DJ, Manjunath A, Bharadwaj A, Berger MJ. et al. Single-cell transcriptional diversity is a hallmark of developmental potential. Science. 2020;367:405-11
65. Chiao CC, Liu YH, Phan NN, An Ton NT, Ta HDK, Anuraga G. et al. Prognostic and Genomic Analysis of Proteasome 20S Subunit Alpha (PSMA) Family Members in Breast Cancer. Diagnostics (Basel). 2021 11
66. Anuraga G, Wang WJ, Phan NN, An Ton NT, Ta HDK, Berenice Prayugo F. et al. Potential Prognostic Biomarkers of NIMA (Never in Mitosis, Gene A)-Related Kinase (NEK) Family Members in Breast Cancer. J Pers Med. 2021 11
67. Ta HDK, Wang WJ, Phan NN, An Ton NT, Anuraga G, Ku SC. et al. Potential Therapeutic and Prognostic Values of LSM Family Genes in Breast Cancer. Cancers (Basel). 2021 13
68. Walker BS, Zarour LR, Wieghard N, Gallagher AC, Swain JR, Weinmann S. et al. Stem Cell Marker Expression in Early Stage Colorectal Cancer is Associated with Recurrent Intestinal Neoplasia. World J Surg. 2020;44:3501-9
69. Chen J, Wu S, Peng Y, Zhao Y, Dong Y, Ran F. et al. Constructing a cancer stem cell related prognostic model for predicting immune landscape and drug sensitivity in colorectal cancer. Front Pharmacol. 2023;14:1200017
70. Ye ML, Li SQ, Yin YX, Li KZ, Li JL, Hu BL. Integrative Analysis Revealed Stemness Features and a Novel Stemness-Related Classification in Colorectal Cancer Patients. Front Cell Dev Biol. 2022;10:817509
71. Omran MM, Fouda MS, Mekkawy SA, Tabll AA, Abdelaziz AG, Omran AM. et al. Molecular Biomarkers and Signaling Pathways of Cancer Stem Cells in Colorectal Cancer. Technol Cancer Res Treat. 2024;23:15330338241254061
72. Wang X, Chen X, Zhao M, Li G, Cai D, Yan F. et al. Integration of scRNA-seq and bulk RNA-seq constructs a stemness-related signature for predicting prognosis and immunotherapy responses in hepatocellular carcinoma. J Cancer Res Clin Oncol. 2023;149:13823-39
73. Liu L, Zhang M, Cui N, Liu W, Di G, Wang Y. et al. Integration of single-cell RNA-seq and bulk RNA-seq to construct liver hepatocellular carcinoma stem cell signatures to explore their impact on patient prognosis and treatment. PLoS One. 2024;19:e0298004
74. Basak U, Mukherjee S, Chakraborty S, Sa G, Dastidar SG, Das T. In-silico analysis unveiling the role of cancer stem cells in immunotherapy resistance of immune checkpoint-high pancreatic adenocarcinoma. Sci Rep. 2025;15:10355
75. Ye W, Su W, Lei C, Huang C, Du M. Single-cell analysis identifies a stemness-associated tumor cell subpopulation and develops a prognostic scoring model in esophageal squamous cell carcinoma. Transl Oncol. 2026;64:102653
76. Wang C, Chen Y, Zhou R, Yang Y, Fang Y. Systematic Analysis of Tumor Stem Cell-related Gene Characteristics to Predict the PD-L1 Immunotherapy and Prognosis of Gastric Cancer. Curr Med Chem. 2024;31:2467-82
77. Becker WR, Nevins SA, Chen DC, Chiu R, Horning AM, Guha TK. et al. Single-cell analyses define a continuum of cell state and composition changes in the malignant transformation of polyps to colorectal cancer. Nat Genet. 2022;54:985-95
78. Deng S, Yang Y, Gao D, Gao J, Xiong Y. Targeting Pan-Cancer Stemness: Core Regulatory lncRNAs as Novel Therapeutic Vulnerabilities. Int J Mol Sci. 2025 26
79. Cheng J, Cao J, Yang Y, Wang Y, Hu X, Liu Z. et al. Multi-omics analysis unraveling stemness features associated with oncogenic dedifferentiation in 12 cancers. Cancer Lett. 2025;625:217816
80. Chen E, Zou Z, Wang R, Liu J, Peng Z, Gan Z. et al. Predictive value of a stemness-based classifier for prognosis and immunotherapy response of hepatocellular carcinoma based on bioinformatics and machine-learning strategies. Front Immunol. 2024;15:1244392
81. Lai J, Lin X, Zheng H, Xie B, Fu D. Characterization of stemness features and construction of a stemness subtype classifier to predict survival and treatment responses in lung squamous cell carcinoma. BMC Cancer. 2023;23:525
Author contact
![]() Corresponding authors: albert0920com.tw (Chung-Bao Hsieh), yungkuonsysu.edu.tw (Yuen-Jung Wu).
Corresponding authors: albert0920com.tw (Chung-Bao Hsieh), yungkuonsysu.edu.tw (Yuen-Jung Wu).