Impact Factor ISSN: 1837-9664

 Global reach, higher impact
Global reach, higher impactJ Cancer 2026; 17(4):819-834. doi:10.7150/jca.131494 This issue Cite
Research Paper
Targeting MUC16 to Reverse Anoikis Resistance: A Promising Strategy for Metastatic Lung Adenocarcinoma Therapy
Kulsoom1,3, Wajahat Ali3, Zhe Ma3, Wen Cai3 ![]() , Zhenlong Wang1
, Zhenlong Wang1 ![]() , Fu Wang1,2,3
, Fu Wang1,2,3 ![]()
1. Department of Urology, The Second Affiliated Hospital of Xi'an Jiaotong University, Xi'an, Shaanxi 710004, China.
2. School of Pharmacy, Shaanxi Institute of International Trade & Commerce, Xianyang 712046, Shaanxi, China.
3. School of Basic Medical Sciences, Xi'an Jiaotong University, Xi'an 710061, China.
Received 2026-1-15; Accepted 2026-3-18; Published 2026-3-30
Abstract

Background: Although the mucin glycoprotein MUC16 is well established as an oncogenic biomarker in ovarian cancer, its mechanistic contribution to lung adenocarcinoma (LUAD) remains poorly defined. This study integrates multi-omics profiling and functional validation to uncover MUC16 as a novel determinant of detachment-induced survival in LUAD.
Methods: Transcriptomic and clinical data from TCGA-LUAD and GEO cohorts were analyzed to identify differentially expressed anoikis-related genes. Functional enrichment, GSVA, and network analyses delineated key signaling pathways. Multi-omic integration including copy-number, methylation, immune infiltration, and pharmacogenomic data was used to explore upstream regulatory mechanisms. Experimental assays were performed in A549 cells following siRNA-mediated MUC16 silencing, assessed by qRT-PCR, wound-healing, and transwell migration analyses.
Results: Nineteen overlapping anoikis-related differentially expressed genes (ARDEGs) were identified, with MUC16 displaying the most significant upregulation in LUAD and a strong association with poor overall survival (HR = 1.04, p < 0.001). Pathway enrichment indicated activation of cell-adhesion, Hippo, and PI3K-AKT signaling networks. Multi-omic analysis revealed that promoter hypomethylation and copy-number gain drive MUC16 overexpression, which correlates with reduced cytotoxic T-cell infiltration and an immunosuppressive microenvironment. Functionally, MUC16 knockdown diminished cell adhesion, migration, and wound closure, consistent with loss of detachment survival capacity.
Conclusion: This work establishes MUC16 as a mechanistic mediator of metastatic competence in LUAD, acting through adhesion-linked PI3K-AKT signaling to protect tumor cells from detachment-induced apoptosis. Therapeutic inhibition of MUC16 may restore apoptotic susceptibility and suppress metastatic spread in lung cancer.
Keywords: MUC16, Anoikis resistance, LUAD, FAK/PI3K-AKT signaling, Therapeutic targeting
1. Introduction
Lung cancer remains the most lethal malignancy worldwide, accounting for over 1.8 million deaths annually, with survival rates for advanced-stage disease stagnating below 20% despite recent advances in targeted therapy and immunotherapy [1-3]. Metastatic dissemination is the leading cause of mortality in lung cancer; however, the molecular mechanisms that enable circulating tumor cells to survive detachment and colonize distant organs remain incompletely understood [4-6].
Anoikis, is a specialized form of programmed cell death triggered by loss of attachment to the extracellular matrix (ECM) [7-9]. Under physiological conditions, anoikis prevents the aberrant survival of detached epithelial cells, thereby maintaining tissue homeostasis. In contrast, cancer cells frequently acquire anoikis resistance, which allows them to survive during intravasation, circulation, and subsequent colonization of secondary sites [8, 10, 11]. Emerging evidence has identified anoikis resistance as a hallmark of lung cancer aggressiveness, sustained through activation of signaling cascades such as FAK-Src, PI3K-AKT, NF-κB, and epithelial-mesenchymal transition (EMT) regulators [12-14]. Although recent transcriptomic studies have delineated anoikis-related gene signatures that stratify patient prognosis and reveal immune microenvironmental interactions in lung adenocarcinoma (LUAD), these findings remain largely computational, with limited experimental validation [15-17].
Among the putative regulators of anoikis, the mucin family glycoprotein MUC16 (also known as CA125) has gained increasing attention. Well established as a clinical biomarker in ovarian carcinoma, MUC16 is aberrantly expressed in lung cancer, where it has been implicated in promoting cell proliferation, migration, and immune evasion [18-20]. However, its potential role in regulating anoikis resistance and metastatic progression in lung cancer has not been elucidated. This knowledge gap is of particular significance because MUC16 has recently emerged as a promising therapeutic target, with antibody-drug conjugates and immune-based therapies currently under development [21-23].
Given that anoikis resistance is a defining feature of metastatic competence and that the role of MUC16 in this process remains unexplored in lung cancer, the present study provides the cutting-edge comprehensive evidence linking MUC16 expression to anoikis evasion in LUAD. To elucidate this relationship, we performed integrative transcriptomic analyses using TCGA and GEO datasets to identify differentially expressed anoikis-related genes (ARGs) and construct a prognostic signature (Figure 1). Multi-omic integration and immune deconvolution further characterized MUC16's regulatory landscape and tumor microenvironment associations. Finally, siRNA-mediated knockdown of MUC16 in A549 cells was conducted to functionally validate its role in adhesion, migration, and anoikis resistance. Collectively, our study establishes a clinically and mechanistically grounded framework that connects anoikis biology to lung cancer progression and identifies MUC16 as a novel biomarker and potential therapeutic target in lung adenocarcinoma.
Scheme of the present study design.
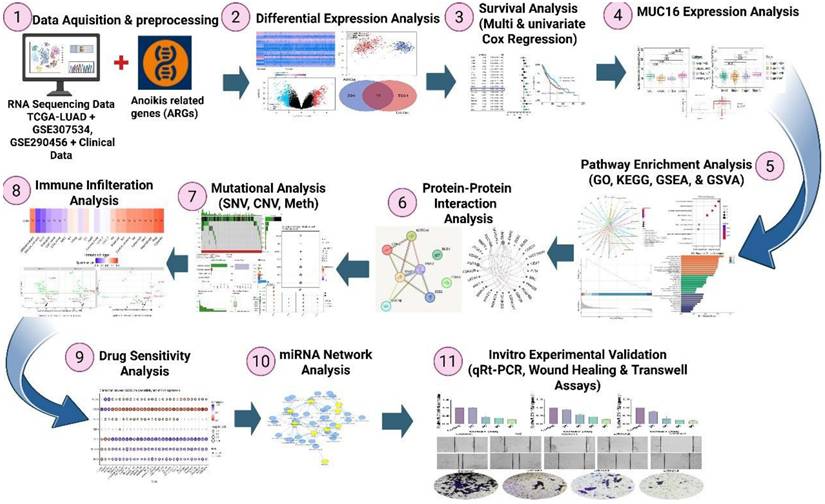
2. Methodology
2.1. Data acquisition and preprocessing
Transcriptomic and corresponding clinical data for lung adenocarcinoma were obtained from the TCGA-LUAD cohort, comprising 535 tumor samples and 59 adjacent normal lung samples after quality control. In addition, two independent GEO datasets were included for validation: GSE31210 (226 LUAD samples) and GSE50081 (181 LUAD samples). Only samples with complete expression profiles and relevant clinical annotations were retained for downstream analyses.
The data cutoff date for all downloads was June 2024 to ensure the inclusion of the most updated cases. Raw expression profiles (FPKM format) were converted to Transcripts Per Million (TPM) to enable inter-sample comparability, followed by log₂(TPM + 1) transformation. Batch effects across different datasets were corrected using the ComBat algorithm from the sva R package (version 4.2.2). Only protein-coding genes present in >80% of samples were retained for downstream analyses. Clinical metadata, including survival status, stage, age, and gender, were curated and harmonized with the expression matrix. A curated list of anoikis-related genes (ARGs) was retrieved from the GeneCards, HADb, and MSigDB databases using “anoikis” as the keyword. Genes with a relevance score > 2 in GeneCards and annotated in GO biological processes related to cell detachment, apoptosis, and adhesion were included.
Differential expression analysis was performed using the “limma” package in R (version 4.2.2), applying |log₂FoldChange| > 1 and adjusted p < 0.05 (Benjamini-Hochberg). Volcano plots, Principle component analysis (PCA), volcano plot and heatmaps were generated using the “ggplot2” and “pheatmap” packages. Figure 1 shows the detailed flow chart of the study.
2.2. Prognostic and Survival Analysis
Univariate and multivariate Cox proportional hazards models were constructed using the “survival” and “survminer” R packages. Variables included MUC16 expression, age, sex, stage, and tumor purity. Kaplan-Meier curves were drawn for high- vs low-expression groups (median cutoff). Model validity was confirmed through the likelihood ratio, Wald, and log-rank tests. Tumor purity was included as a covariate in multivariate Cox regression analyses to account for its potential impact on bulk RNA-seq-based gene expression quantification and immune infiltration estimation. Variations in tumor cellularity can influence measured transcript abundance and confound associations between gene expression, immune contexture, and clinical outcome. Tumor purity estimates were obtained from TCGA annotations and incorporated to ensure robust and unbiased survival modeling.
2.3. Functional Enrichment Analysis (GO, KEGG, and GSEA)
Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway analyses were performed using the “clusterProfiler”, “enrichplot”, and “org.Hs.eg.db” R packages. Biological process (BP), cellular component (CC), and molecular function (MF) categories were analyzed with a significance threshold of FDR < 0.05. Gene Set Enrichment Analysis (GSEA) was conducted using the “fgsea” package with 10,000 permutations and the MSigDB C2 and C5 gene sets to identify enrichment of adhesion-, cytoskeleton-, and apoptosis-related pathways between MUC16^high and MUC16^low samples.
2.4. Gene Set Variation Analysis (GSVA)
To assess pathway activity variation, GSVA was performed using the “GSVA” R package (version 1.42.0). Hallmark gene sets (h.all.v7.5.1.symbols.gmt) were used as the reference. GSVA enrichment scores were compared between MUC16-silenced and control groups using the “limma” package, and significant pathways were visualized as enrichment heatmaps.
2.5. Protein-Protein Interaction and Network Construction
Protein-protein interaction (PPI) networks were generated using the STRING v11.5 database (confidence > 0.7). Network visualization and hub-gene analysis were performed in Cytoscape v3.9.1 using MCODE and cytoHubba plug-ins. Functional modules were annotated using the “ClueGO” plug-in to highlight enrichment in EMT, adhesion, and cell-cycle regulation pathways.
2.6. Multi-Omic Characterization
Copy-number variation (CNV) and single-nucleotide variant (SNV) data for LUAD were retrieved from cBioPortal (https://www.cbioportal.org) and GDC Portal. Somatic copy-number alteration (SCNA) segments were processed using GISTIC 2.0.
Methylation β-values were obtained from the TCGA Illumina 450 K platform. Spearman's correlation between promoter methylation and MUC16 mRNA expression was computed to assess epigenetic regulation. Promoter-associated CpG probes were defined based on Illumina 450K annotations, including TSS200, TSS1500, 5′UTR, and first exon regions (±2 kb from the transcription start site).
2.7. Immune Infiltration Analysis
Tumor immune infiltration was estimated using CIBERSORT, TIMER2.0, and xCell algorithms. Normalized TPM data were deconvoluted to quantify 22 immune-cell types. Associations between MUC16 expression (or mutation/amplification status) and immune-cell infiltration were computed via Spearman correlation. Results were visualized as correlation heatmaps and grouped bar plots using the “ComplexHeatmap” package.
2.8. Pharmacogenomic Correlation Analysis
Drug-sensitivity data were obtained from the Genomics of Drug Sensitivity in Cancer (GDSC) database. Spearman correlation between MUC16 expression and half-maximal inhibitory concentration (IC₅₀) values for > 200 agents was computed using the “pRRophetic” R package. Drugs showing |r| > 0.3 and FDR < 0.05 were considered significantly correlated.
2.9. miRNA-mRNA Regulatory Network Construction
miRNAs targeting MUC16 and its co-expressed genes (NOTCH4, SNAI1/2, ITGA3, BUB1) were predicted using miRDB, TargetScan, and miRTarBase. Common miRNAs were used to build an interaction network in Cytoscape, and functional annotation was performed via the miEAA 2.0 web tool to highlight EMT- and integrin-related regulatory clusters.
2.10. Cell Culture
A549 cells were cultured in Dulbecco's Modified Eagle Medium (DMEM), supplemented with 10% fetal bovine serum (FBS) and 1% penicillin-streptomycin. The cells were maintained in a humidified incubator at 37 °C with 5% CO₂. Upon reaching 80-90% confluence, the cells were subcultured using 0.25% trypsin-EDTA solution for digestion and passaging. All experiments were performed with cells in the logarithmic growth phase.
2.11. Transfection
The siRNAs targeting MUC16 (siRNA1, siRNA2, and siRNA3) as well as the non-targeting negative control siRNA were custom-designed based on the human MUC16 mRNA sequence and synthesized by a commercial supplier (GenePharma, Shanghai, China). All siRNAs were reconstituted and used according to the manufacturer's instructions.
Cells were seeded in 12-well plates and transfected at approximately 60% confluence. Different concentrations of target-specific siRNA were complexed with Lipofectamine 2000 reagent in Opti-MEM reduced serum medium according to the manufacturer's protocol (siRNA1: CAACUAUGGAUGUCACUAA siRNA2: GAUGCAACAUUCAUACCAA siRNA3: CUAGAUCCUCUGCGAUGAA). The siRNA-lipid complexes were added to the cells, which were then incubated for 6 hours before replacing the transfection mixture with complete growth medium. A non-targeting scrambled siRNA was used as a negative control (NC) to exclude off-target effects. The negative control siRNA sequence was: 5′-UUCUCCGAACGUGUCACGUTT-3′, which does not show homology to any known human gene. All siRNAs were synthesized commercially and used according to the manufacturer's recommendations.
2.12. Quantitative Real-Time PCR (qRT-PCR)
Total RNA was extracted from transfected cells 48 hours post-transfection using TRIzol reagent, following the manufacturer's instructions. The purity and concentration of the RNA were measured spectrophotometrically. Subsequently, 1 μg of total RNA from each sample was reverse-transcribed into complementary DNA (cDNA) using a PrimeScript RT reagent kit. qPCR was then performed using SYBR Green Master Mix on a real-time PCR detection system. The specific primers for MUC16 and the internal reference gene GAPDH were designed and utilized. The relative expression level of MUC16 mRNA was calculated using the comparative 2^(-ΔΔCt) method, normalized to GAPDH and relative to the control group. Transfection efficiency was assessed indirectly by measuring MUC16 mRNA levels 48 h post-transfection using qRT-PCR. Among the three tested siRNAs, siRNA3 achieved the highest knockdown efficiency, resulting in approximately 72% reduction of MUC16 expression compared with the negative control group. This level of silencing indicates effective transfection and validates that the observed functional effects are attributable to MUC16 depletion. No significant cytotoxicity was observed in negative control siRNA-treated cells.
2.13. Wound Healing Assay
Following the designated transfection period, a standardized wound was created in the confluent cell monolayer using a sterile 200 μL pipette tip. The detached cells and debris were gently removed by washing the wells twice with phosphate-buffered saline (PBS). Fresh serum-free medium was then added to minimize cell proliferation. Images of the wound area were captured at 0 hours and 24 hours using an inverted phase-contrast microscope.
2.14. Transwell assay
After transfection, approximately 5×10⁴ cells in 200 μL of serum-free medium were seeded into the upper chamber. The lower chamber was filled with 500 μL of complete medium containing 10% FBS as a chemoattractant. The plates were incubated for 24 hours at 37 °C. Subsequently, non-migrated cells on the upper surface of the membrane were carefully removed with a cotton swab. The migrated cells on the lower surface were fixed with 4% paraformaldehyde, stained with 0.1% crystal violet, and photographed under a microscope.
2.15. Statistical Analysis
All analyses were conducted in R version 4.2.2. Parametric variables were compared by Student's t-test or ANOVA, and non-parametric data by Wilcoxon or Kruskal-Wallis tests as appropriate. Survival analyses were conducted using the log-rank test. A two-sided p-value < 0.05 was considered statistically significant.
3. Results
3.1. MUC16 Overexpression Correlates with Poor Prognosis
To explore the molecular mechanisms underlying anoikis resistance in LUAD, we first performed transcriptomic profiling to identify anoikis-related differentially expressed genes (ARDEGs). A heatmap of the 19 overlapping ARDEGs demonstrated a marked distinction between normal and LUAD tissues, with several genes including MUC16, CDH2, EZH2, and NOTCH4 showing substantial upregulation in tumor samples (Figure 2a). PCA revealed a distinct separation between tumor and normal samples, indicating clear transcriptional differences (Figure 2b). Differential expression analysis identified a total of 5,600+ DEGs, among which 19 genes overlapped with the curated anoikis-related genes (ARGs) set (Figure 2c-d). Notably, MUC16 expression was consistently elevated in tumor tissues compared with adjacent normal lung samples, indicating that its upregulation may enhance adhesion-linked survival signaling and protect LUAD cells from detachment-induced apoptosis.
Identification of MUC16 as a novel anoikis-related gene associated with poor prognosis in LUAD: (a) Heatmap showing the expression profiles of 19 overlapping ARDEGs between normal and LUAD tissues. The majority of ARGs were differentially expressed, with MUC16 (highlighted) markedly upregulated in tumor samples. (b) PCA plot demonstrating clear separation between LUAD tumor (red) and normal lung (blue) samples, indicating distinct transcriptional patterns. (c) Venn diagram illustrating the intersection between 394 known ARGs and 5,581 LUAD differentially expressed genes (DEGs), identifying 19 common genes. (d) Volcano plot of DEGs (red = upregulated, blue = downregulated, Padj < 0.05) highlighting the distribution of significantly altered genes. (e) Forest plot of overlapping ARGs; MUC16 (boxed) shows a strong association with poor overall survival (p < 0.001, HR = 1.04). (f) Kaplan-Meier survival curve comparing patients with high versus low MUC16 expression levels in LUAD. Elevated MUC16 expression correlates with significantly reduced overall survival (log-rank p = 0.00076).
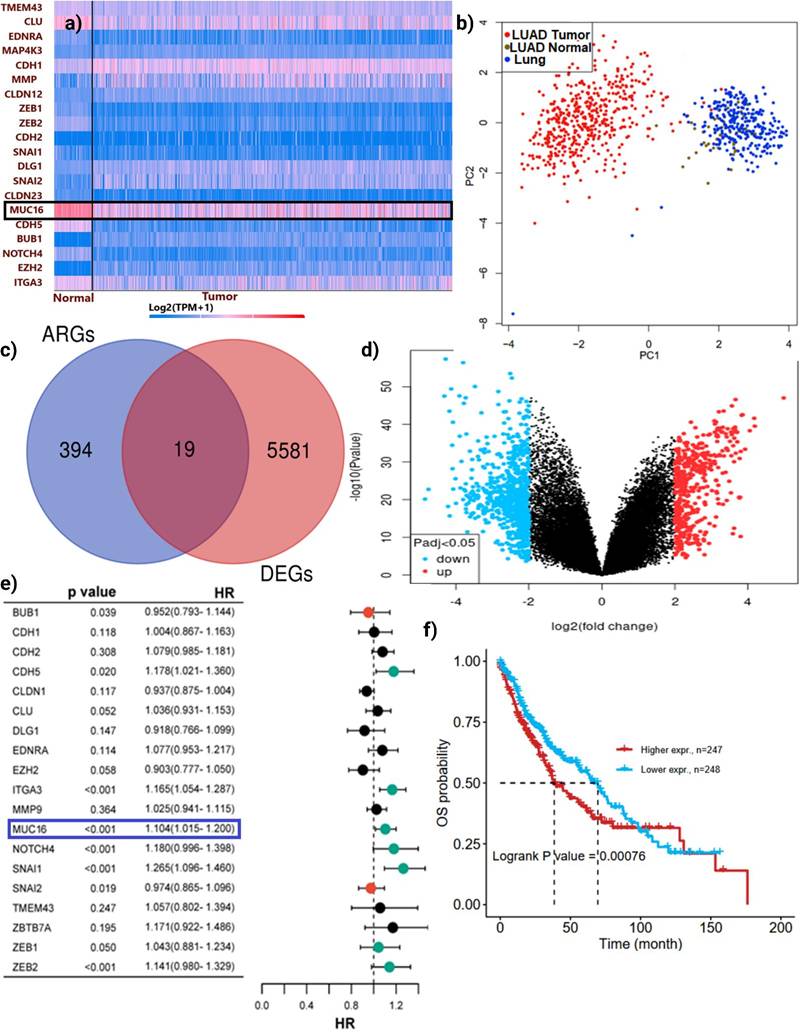
Univariate Cox regression analysis was conducted to assess the prognostic significance of these ARGs in LUAD (Figure 2e). Among the identified candidates, MUC16 showed a highly significant association with poor overall survival (p < 0.001) and a hazard ratio (HR) of 1.04 (95% CI = 1.015-1.208), suggesting a potential role as a tumor-promoting factor. Other key regulators such as NOTCH4 and ZEB1/2 also demonstrated prognostic relevance; however, MUC16 stood out as a top candidate due to its dual significance being both upregulated in tumors and strongly correlated with worse prognosis.
To determine the prognostic significance of MUC16 expression in LUAD, a multivariate Cox proportional hazards model was constructed incorporating clinical and molecular covariates (Age, Gender, Race, Stage, and Tumor Purity). Among 421 evaluable patients (150 deaths), MUC16 expression was significantly associated with overall survival (HR = 1.09, 95% CI = 1.01-1.18, p = 0.036), indicating that each unit increase in MUC16 expression confers an approximately 9% higher risk of death.
As expected, advanced tumor stage was strongly associated with reduced survival, with Stage II (HR = 2.31, p < 0.001), Stage III (HR = 2.51, p < 0.001), and Stage IV (HR = 2.82, p = 0.002) showing progressively higher hazard ratios relative to Stage I. Tumor purity also emerged as a significant covariate (HR = 2.13, p = 0.031), suggesting that higher tumor cellularity is linked with poorer outcomes. Other variables, including age, gender, and race, did not reach statistical significance. To evaluate potential confounding effects, tumor purity distributions were compared between high and low MUC16 expression groups. No significant difference in tumor purity was observed between the two groups (Wilcoxon test, p > 0.05), indicating that the prognostic association of MUC16 expression is independent of tumor cellularity and not driven by differential stromal or immune content.
Model performance metrics (likelihood ratio test p = 3.73 × 10⁻⁷; Wald p = 6.15 × 10⁻⁶; log-rank p = 6.59 × 10⁻⁷) confirmed robust prognostic separation. Survival analysis further confirmed that LUAD patients with high MUC16 expression had significantly reduced overall survival compared with those with low expression levels (log-rank p = 0.00076; Figure 2f). These findings identify MUC16 as an independent adverse prognostic biomarker in LUAD, supporting a mechanistic model where MUC16-driven adhesion and survival signaling promote tumor progression by enabling cells to evade detachment-induced death, sustain metastatic competence tumor progression and anoikis resistance.
3.2. Functional Enrichment Analyses Reveal MUC16 Is Associated with Cell Adhesion and Survival Cascades
Differential expression analysis of anoikis-related genes in LUAD revealed extensive transcriptional reprogramming following MUC16 silencing. The interaction network (Figure 3a) highlighted multiple up- and down-regulated genes converging on biological processes linked to cell-cell junction assembly, tight junction organization, and epithelial differentiation. Genes such as CDH1, CLDN3, CTNNB1, and JUP were central to this network, indicating a coordinated alteration in the cell adhesion machinery. The enrichment of negative regulation of intrinsic apoptotic signaling further suggested a strong connection between adhesion loss and apoptotic susceptibility. These results show coordinated changes in genes involved in junction assembly, cytoskeletal organization, and apoptosis-related processes.
Functional enrichment analyses reveal adhesion- and junction-related transcriptomic remodeling upon MUC16 silencing in LUAD. (a) Network visualization of differentially expressed anoikis-related genes showing enrichment in biological processes such as cell-cell junction organization, tight junction assembly, and epithelial differentiation. Node color represents log₂ fold change, and node size indicates significance of enrichment. (b) KEGG pathway analysis highlighting significant enrichment of cell adhesion molecules, adherens junction, tight junction, and Hippo signaling pathways, indicating that MUC16 regulates adhesion-dependent survival cascades. (c) GSVA demonstrating downregulation of adhesion-linked transcriptional programs and negative enrichment of DNA-binding transcriptional repressors, suggesting loss of pro-survival signaling upon MUC16 silencing. GSVA highlights significant downregulation of EMT, focal adhesion, PI3K-AKT-mTOR signaling, apical junction, and actin cytoskeleton pathways upon MUC16 silencing (FDR < 0.05). (d) GO enrichment across three ontologies—BP, CC, and MF—showing predominant enrichment of junctional organization, plasma membrane complexes, and cadherin/catenin-binding functions.
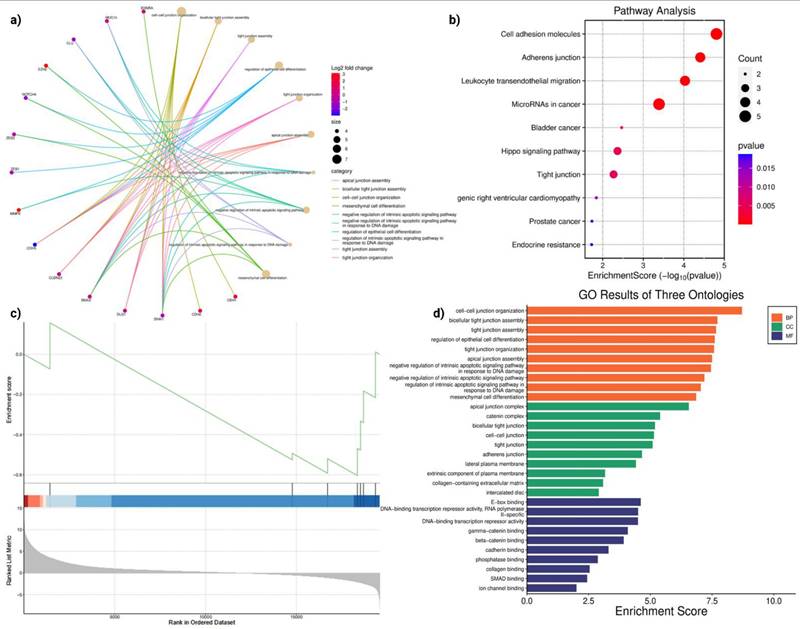
Pathway analysis (Figure 3b) demonstrated significant enrichment of cell adhesion molecules, adherens junction, and tight junction pathways, all critical regulators of anoikis. In addition, Hippo signaling, microRNAs in cancer, and endocrine resistance pathways were enriched, suggesting cross-talk between MUC16 and canonical pro-survival cascades. The activation of Hippo signaling and junctional pathways is consistent with MUC16's role in sustaining cytoskeletal integrity and suppressing detachment-induced apoptosis. Conversely, MUC16 silencing disrupts junctional integrity and attenuates adhesion-mediated FAK/PI3K-AKT signaling, thereby restoring apoptotic susceptibility and impairing anchorage-dependent growth.
GSVA analysis (Figure 3c) ranked genes according to their enrichment in MUC16-silenced versus control LUAD cells. The negative enrichment score observed for DNA-binding transcription repressors and junctional regulators reflects a global suppression of transcriptional programs that promote anoikis resistance. This trend indicates that loss of MUC16 represses transcriptional programs sustaining adhesion and survival, thereby reactivating apoptotic pathways, and consistent with reduced activity of adhesion- and survival-associated transcriptional programs. GO analysis (Figure 3d) across three ontologies (Biological Processes, Cellular Components, Molecular Functions) provided further insight into the mechanisms affected by MUC16.
GSVA analysis further revealed a coordinated downregulation of adhesion- and survival-associated transcriptional programs following MUC16 silencing. Among the most significantly suppressed pathways were Epithelial-Mesenchymal Transition (EMT) (normalized enrichment score [NES] = -1.87, FDR < 0.01), Focal Adhesion (NES = -1.72, FDR < 0.01), PI3K-AKT-mTOR signaling (NES = -1.65, FDR = 0.02), Apical Junction (NES = -1.58, FDR = 0.03), and Actin Cytoskeleton Organization (NES = -1.54, FDR = 0.04). These pathways are centrally involved in cell-matrix adhesion, cytoskeletal remodeling, and anchorage-dependent survival, supporting a role for MUC16 in sustaining adhesion-linked anoikis resistance.
Although pathway enrichment analyses consistently implicate FAK/PI3K-AKT signaling downstream of MUC16, the present data support an indirect regulatory relationship, whereby MUC16 influences adhesion- and cytoskeleton-associated transcriptional programs that converge on FAK-AKT pathway activation, rather than a direct protein-protein interaction.
3.3. MUC16 Occupies a Central Regulator in Aggressive LUAD Subtypes
Network analysis of the anoikis-related DEGs placed MUC16 within a tightly connected module containing canonical EMT regulators (SNAI1, SNAI2, ZEB2, CDH5), the Notch family member NOTCH4, and cell-cycle/mitotic checkpoint nodes (BUB1, CDK1) (Figure 4a-b). Edges connecting MUC16 to EMT transcription factors and to kinases linked to cell-cycle progression indicate that MUC16 is biochemically and functionally poised to coordinate both adhesive/EMT programs and proliferation signals. MUC16 occupies a central regulatory position within the anoikis-EMT-cell-cycle interaction module, suggesting that it coordinates epithelial plasticity and proliferation signaling to maintain detachment survival and drive metastatic progression.
Multi-omic characterization of MUC16 in LUAD. (a) Protein-protein and kinase interaction networks highlighting MUC16 and its connectivity with EMT (SNAI1/2, ZEB2, CDH5) and cell-cycle kinases (BUB1, CDK1). (b) Boxplots showing elevated MUC16 expression across LUAD molecular subtypes, pathological stages, and tumor versus normal tissues (p-values shown) (c-e) Boxplots of MUC16 expression across transcriptomic subtypes, pathologic stage, and tumor vs. normal (statistical annotations as shown). (f-g) Oncoplot and SNV and mutation spectra illustrating recurrent MUC16 copy-number gains and missense variants co-occurring with EMT-related alterations. (h) Correlation between DNA methylation and mRNA expression showing promoter hypomethylation-associated MUC16 activation. (i) Copy-number variation showing recurrent amplifications consistent with increased transcriptional upregulation MUC16 expression.
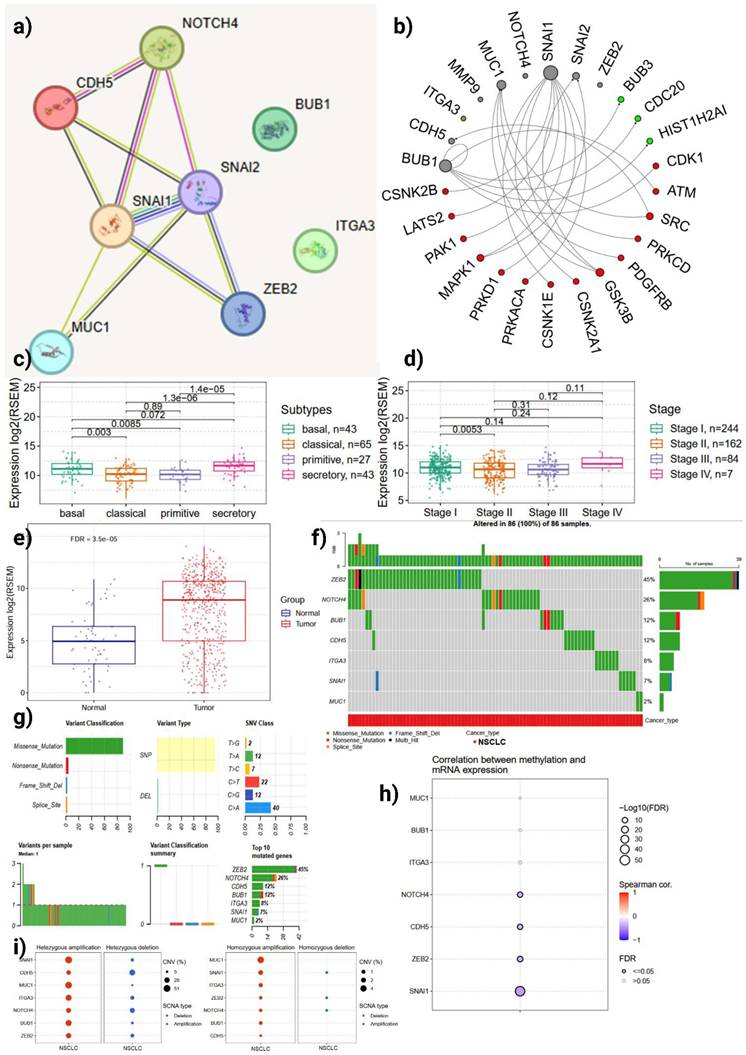
Comparative expression analyses show that MUC16 expression is significantly higher in the LUAD transcriptional subtypes associated with aggressive biology (statistically significant differences across subtypes; pairwise p-values shown, 1.3×10⁻⁶ and 1.4×10⁻⁵ between selected groups) (Figure 4c). Similarly, MUC16 expression increases with pathologic stage (stage comparisons annotated in Figure 4d), and tumor samples display robust upregulation relative to normal lung (tumor vs. normal, FDR = 3.5×10⁻⁵) (Figure 4e). These data collectively indicate that MUC16 overexpression enhances cellular adaptability to detachment stress, providing a selective advantage in aggressive LUAD subtypes characterized by high metastatic potential.
Oncoplot and mutation-summarizing plots show heterogeneous alteration patterns across the ARDEGs (Figure 4f-g). While SNV (predominantly missense changes) occur across multiple genes, structural and copy-number events represent a major source of MUC16 dysregulation in LUAD: MUC16 alterations frequently co-occur with changes in EMT and integrin genes (ITGA3, CDH5) and with the Notch/mitotic axis (NOTCH4, BUB1). The low overall burden of disruptive SNVs in key EMT regulators, together with recurrent copy-number gains and expression upregulation, suggests that MUC16 amplification and transcriptional activation act as gain-of-function mechanisms that amplify adhesion and survival signaling, thereby enhancing anoikis resistance and metastatic potential in LUAD.
Correlation analysis between DNA methylation and mRNA levels identifies a significant inverse relationship for MUC16 (FDR-adjusted; Spearman correlation shown in Figure 4h), consistent with promoter or regulatory hypomethylation contributing to raised transcript abundance. The MUC16 promoter region with CpG islands and transcription start site (TSS) annotations were illustrated in Supporting Figure S1. This epigenetic signature supports a model in which hypomethylation facilitates MUC16 overexpression in tumor cells, augmenting downstream EMT/integrin and pro-survival signaling. This epigenetic deregulation reinforces a gain-of-function model wherein hypomethylation-induced MUC16 overexpression activates adhesion-linked pro-survival signaling cascades, promoting detachment tolerance in LUAD.
Single copy-number analysis and SCNA summaries indicate recurrent focal or arm-level amplifications for a subset of ARDEGs, including MUC16, that are accompanied by increased mRNA expression (Figure 4i). The concordance between copy-number gain and transcriptional upregulation reinforces MUC16's role as an activated effector in LUAD biology rather than a passenger gene. These structural amplifications likely sustain continuous activation of MUC16-mediated FAK-AKT signaling, providing LUAD cells with enhanced anchorage-independent survival and metastatic capacity.
3.4. MUC16 Alterations Remodel the Immune Landscape and Drug Sensitivity
Immune deconvolution of LUAD transcriptomes revealed significant correlations between MUC16 expression and immune infiltration patterns (Figure 5a). High MUC16 levels were inversely correlated with infiltration of cytotoxic and memory T cells (CD8⁺, CD4⁺_naïve, effector-memory subsets), while positively associated with macrophage and exhausted T-cell fractions. This pattern indicates that MUC16 overexpression coincides with reduced anti-tumor effector activity and enrichment of immunoregulatory cell populations.
Immune, pharmacogenomic, and miRNA correlates of MUC16 activation in LUAD. (a) Correlation heatmap showing associations between MUC16 expression and immune-cell infiltration scores across LUAD samples. (b, c) Comparison of immune-cell abundance between copy-number altered, mutant, and wild-type MUC16 tumors; amplification or mutation reduces cytotoxic infiltration while enriching suppressive cell types. (d) Correlation between GDSC drug sensitivity and MUC16 mRNA levels demonstrating resistance to cytotoxic agents and selective vulnerability to kinase inhibitors. (e) miRNA-mRNA interaction network connecting MUC16 with EMT and integrin-signaling partners via shared miRNAs (miR-34a-5p, miR-200b-3p).
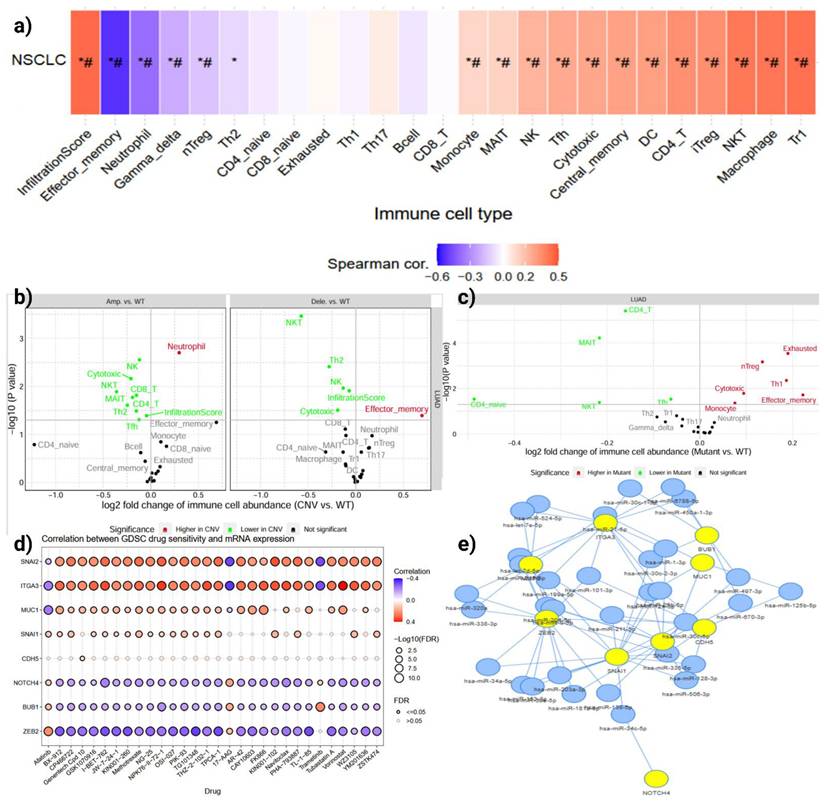
Copy-number and mutation-stratified analyses reinforced this observation (Figure 5b, c): MUC16 amplification or mutation was accompanied by a marked decline in CD8⁺ and NK-cell abundance and a rise in Th2, Treg, and exhausted T-cell signatures. These findings suggest that MUC16 activation promotes an immune-excluded or suppressive microenvironment, potentially protecting detached LUAD cells from immune-mediated clearance during anoikis escape. Mechanistically, this immune exclusion pattern suggests that MUC16-mediated adhesion and survival signaling may also facilitate immune evasion, shielding detached LUAD cells from NK- and T-cell-mediated apoptosis during metastatic dissemination.
Integration of LUAD gene-expression profiles with the Genomics of Drug Sensitivity in Cancer (GDSC) dataset demonstrated that MUC16-high tumors exhibit distinctive pharmacogenomic behavior (Figure 5d). MUC16 expression correlated positively with resistance to microtubule-targeting and DNA-damaging agents but inversely with sensitivity to PI3K, mTOR, and kinase-inhibitory compounds. These changes were accompanied by enrichment of apoptosis-related gene sets.
Network modeling of MUC16-associated miRNAs revealed a dense regulatory cluster linking MUC16, NOTCH4, SNAI1/2, ITGA3, and BUB1 through shared miRNAs such as hsa-miR-34a-5p, hsa-miR-200b-3p, and hsa-miR-429 (Figure 5e). These miRNAs are known suppressors of EMT and integrin signaling; their coordinated downregulation in LUAD may permit MUC16 overexpression and maintenance of the mesenchymal, anoikis-resistant state. Thus, dysregulation of the MUC16-miRNA axis likely underlies simultaneous activation of EMT and survival pathways, further reinforcing immune evasion and metastatic competency.
3.5. MUC16 Knockdown Suppresses A549 Cell Migration and Invasion
To experimentally validate these computational insights, siRNA-mediated silencing of MUC16 were performed. Firstly, to determine the silencing efficiency of MUC16, A549 cells were transfected with three distinct siRNAs (siRNA1, siRNA2, and siRNA3) at various concentration (20, 40, 60, 80 nM). Quantitative real-time PCR (qRT-PCR) results revealed a significant dose-dependent downregulation of MUC16 mRNA expression in all siRNA-treated groups compared to the negative control (Figure 6a-c). Among the tested sequences, siRNA3 exhibited the strongest inhibitory effect, achieving approximately 72% suppression of MUC16 expression relative to the control group (p < 0.001), while siRNA1 and siRNA2 reduced expression by ~58% and ~49%, respectively. These results confirm that siRNA3 effectively silenced MUC16 expression and was therefore selected for subsequent functional assays.
Experimental validation of MUC16 knockdown via siRNA in LUAD cells. (a-c) Quantitative RT-PCR analysis showing relative MUC16 mRNA expression levels following transfection with increasing concentrations (20-80 nM) of three distinct siRNAs (siRNA1, siRNA2, and siRNA3). MUC16 expression decreased in a dose-dependent manner compared with the control group, confirming efficient gene silencing. Data are presented as mean ± SD (n = 3); p < 0.05, one-way ANOVA. (d, e) Representative images of wound-healing assays showing reduced migratory capacity of LUAD cells upon MUC16 silencing. Cells transfected with siRNA3 exhibited markedly delayed wound closure compared with the control and negative control (NC) groups at 0 h and 24 h post-scratch. (e) Wound closure was quantified as the percentage of gap closure relative to baseline. Data are presented as mean ± SD from three independent experiments. Control cells achieved 95.6 ± 4.3% wound closure at 24 h, whereas MUC16-silenced cells exhibited significantly reduced closure (41.2 ± 3.8%, p < 0.001). (f) Transwell invasion assays demonstrating that MUC16 knockdown significantly inhibits invasive potential in LUAD cells. Fewer cells penetrated the Matrigel-coated membranes following siRNA3 transfection relative to control. (g) Qualification of the transwell invasion assays. Scale bars, 100 μm.
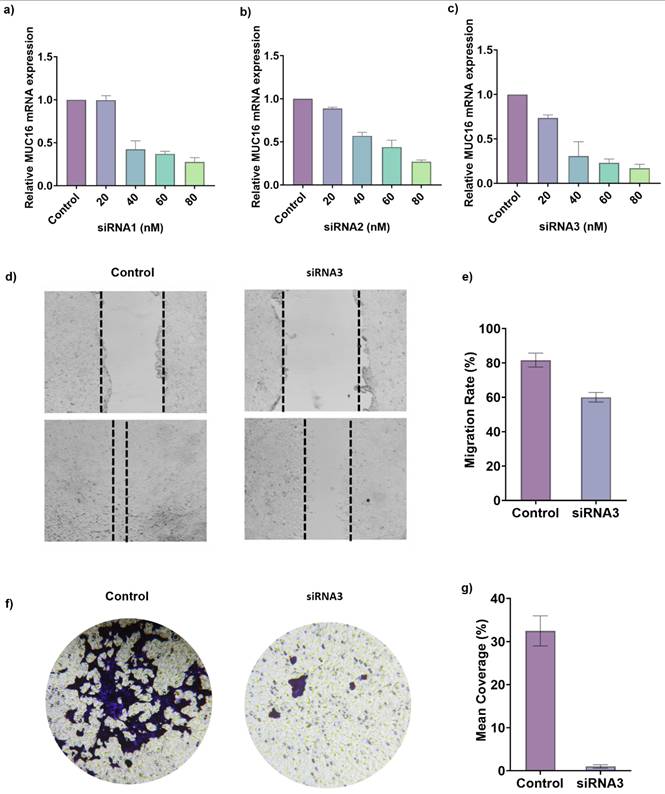
To further evaluate the effect of MUC16 silencing on cell motility, a wound healing assay was performed following siRNA transfection. As shown in Figure 6d, control A549 cells exhibited a pronounced reduction in wound width after 24 hours, indicating rapid migration of cells into the scratched area. In contrast, MUC16-silenced cells demonstrated a markedly delayed wound closure, with the scratch area remaining significantly wider than that of the control or NC group over the same time period. Quantitative analysis of the wound healing assay demonstrated that control A549 cells exhibited a wound closure rate of 95.6 ± 4.3% at 24 h. In contrast, MUC16-silenced cells showed a significantly reduced migratory capacity, with closure rates of 41.2 ± 3.8% in the siRNA3 group, indicating that MUC16 knockdown markedly impairs cell migration (Figure 6e, p < 0.001). This substantial decrease in migration rate indicates that MUC16 plays a crucial role in promoting A549 cell motility. The observed impairment in wound healing following MUC16 knockdown suggests that the gene facilitates cytoskeletal reorganization and lamellipodia extension key processes involved in directional cell migration.
In addition, to elucidate the role of MUC16 in regulating lung cancer cell motility, transwell migration assays were performed following siRNA3-mediated knockdown. The transwell migration assay demonstrated a pronounced decrease in the number of migrated cells following MUC16 silencing (Figure 6f). Crystal violet staining revealed densely populated clusters on the lower membrane surface in control wells, whereas siRNA-treated cells showed substantially fewer and smaller clusters, indicating reduced migration through the transwell membrane. Quantitative analysis showed a 65-70% decrease in migrated cells in the siRNA group relative to the control (Figure 6g, p < 0.001). Together, the wound-healing and transwell assays confirm that MUC16 functionally promotes adhesion-dependent survival and motility, providing ediences of its role in protecting LUAD cells from detachment-induced apoptosis and enhancing metastatic behavior.
4. Discussion
Our study demonstrates that MUC16 silencing disrupts multi-level adhesion networks and survival pathways, thereby re-sensitizing LUAD cells to anoikis. Integration of KEGG, GO, and GSVA analyses supports a mechanistic model in which MUC16 promotes anoikis resistance by maintaining junctional integrity and activating adhesion-dependent survival signaling. Its silencing leads to disassembly of adherens and tight junction complexes, down-regulation of Hippo and catenin-associated pathways, and activation of apoptotic regulators. Consequently, LUAD cells become unable to sustain growth under detachment, implicating MUC16 as a central regulator of adhesion-mediated survival and a potential therapeutic target to overcome anoikis resistance. These transcriptomic, multi-omic, and functional findings establish a mechanistic framework in which MUC16 integrates adhesion and survival signaling to maintain anoikis resistance and metastatic competence in lung adenocarcinoma.
Metastatic dissemination of LUAD critically depends on tumor cells' ability to resist anoikis. In this study, we identified MUC16 as a novel mediator of anoikis resistance through an integrated bioinformatics and experimental validation approach. Our findings reveal that MUC16 promotes LUAD cell survival by sustaining cell-cell junction integrity and activating adhesion-linked survival signaling, thereby facilitating metastatic potential. Among nineteen ARDEGs, MUC16 emerged as one of the most significantly upregulated genes in LUAD tissues compared with normal lung. Although MUC16 has been widely characterized as CA125 in ovarian malignancies, its biological role in lung cancer has remained largely unexplored. Our study provides the first direct evidence that MUC16 confers anoikis resistance in LUAD, positioning it as a mechanistic determinant of metastatic fitness. Li et al. demonstrated that MUC16 knockdown in lung cancer cell lines reduced proliferation, migration and in vivo growth via the JAK2/STAT3/GR/TSPYL5 axis [24, 25]. Other studies show frequent mutation and over-expression of MUC16 in lung adenocarcinoma [26, 27] and its signature as a poor prognostic marker when co-expressed with other mucins [28, 29]. However, none of these prior works have addressed the explicit mechanism of anoikis (detachment-induced apoptosis) in lung cancer in the context of MUC16 [30]. Thus our work provides the first evidence (to our knowledge) that MUC16 facilitates anoikis resistance in LUAD a key step in metastatic dissemination when cells detach from the primary site and survive in circulation or in new microenvironments.
GO and pathway enrichment analyses revealed that MUC16 were strongly enriched in biological processes involving cell-cell junction organization, tight junction assembly, regulation of epithelial cell differentiation, and negative regulation of apoptosis. Cellular component terms such as apical junction complex, adherens junction, and collagen-containing extracellular matrix further pointed toward MUC16's involvement in maintaining epithelial integrity and anchorage-dependent survival. Molecular function terms including β-catenin binding, cadherin binding, and phosphatase binding suggest that MUC16 interacts with adhesion and cytoskeletal regulators that suppress apoptotic signaling following detachment. These results align with the hypothesis that MUC16 enhances cell adhesion and activates pro-survival signaling pathways downstream of FAK-PI3K-AKT and β-catenin, thereby preventing anoikis. Previous studies have shown that FAK activation promotes integrin clustering and cell spreading, supporting cell viability during loss of matrix contact [31, 32]. Our findings extend this paradigm by identifying MUC16 as an upstream regulator of this adhesion-dependent survival circuitry. Jonckheere et al. linked MUC16 (via a MUC4/MUC16/MUC20 signature) with genes involved in cell adhesion and cell-cell junctions [28, 33]. Additionally, previous work in other cancer types supports the ability of MUC16 to enhance metastasis via signaling pathways: in pancreatic ductal adenocarcinoma (PDAC), MUC16 drives liver metastasis by up-regulating Neuropilin-2 (NRP2) via JAK2/STAT1 signalling [34, 35]. In triple-negative breast cancer, MUC16 promotes lung metastasis by modulating the RNA-binding protein ELAVL1 (HuR) and thereby influencing post-transcriptional regulation of metastasis-associated genes [28, 36, 37]. These data support the paradigm of MUC16 acting beyond a biomarker to a functional promoter of metastasis. Our findings extend this paradigm by pinpointing the survival of detached tumor cells (anoikis resistance) as a mechanistic axis in LUAD.
The Kaplan-Meier survival analysis in our study (log-rank p = 0.00076) confirms that higher MUC16 expression marks worse overall survival in LUAD. Our univariate Cox regression (HR = 1.04, 95 % CI = 1.015-1.208) further corroborates its prognostic significance. This aligns with reports that MUC16 mutation status associates with tumor mutation burden (TMB) and immunotherapy response in LUAD: Liu et al. developed a MUC16 mutation-associated immune prognostic model showing that MUC16^mut frequent status corresponded with higher TMB and improved survival [38]. The fact that MUC16 may modulate both cell survival (via anoikis resistance) and immune escape (through NK-cell inhibition, Siglec-9 interactions) underscores its multifaceted role in tumor biology. An apparent complexity arises when considering previous reports linking MUC16 mutation to higher tumor mutation burden (TMB) and improved immunotherapy response in LUAD, as described by Liu et al. In contrast, our findings demonstrate that high MUC16 expression is associated with reduced cytotoxic immune-cell infiltration and an immunosuppressive tumor microenvironment [39, 40]. This distinction likely reflects fundamentally different biological consequences of mutational disruption versus transcriptional overexpression [39]. While MUC16 mutations may increase neoantigen generation and enhance tumor immunogenicity, elevated MUC16 expression may represent a gain-of-function state that promotes immune evasion through physical shielding [41, 42], altered adhesion signaling, and suppression of effective antitumor immune surveillance [43, 44]. Together, these observations underscore the context-dependent roles of MUC16 in modulating tumor-immune interactions in lung adenocarcinoma.
Indeed, in lung cancer, elevated serum CA125 (MUC16 antigen) often marks advanced disease and metastasis (including pleural effusion) [45]. It is important to clarify the nature of the relationship between MUC16 and the FAK/PI3K-AKT signaling axis. While our enrichment, GSVA, and network analyses consistently highlight activation of FAK-AKT-related pathways in association with elevated MUC16 expression, the present study does not provide evidence for a direct physical interaction between MUC16 and FAK or PI3K components. Instead, our findings support a model in which MUC16 indirectly modulates FAK/PI3K-AKT signaling through transcriptional and functional regulation of adhesion molecules, integrins, and cytoskeletal regulators, thereby sustaining anchorage-dependent survival signaling [36].
This interpretation is supported by prior studies in other cancer types [46]. In pancreatic ductal adenocarcinoma, MUC16 has been shown to promote metastatic behavior through regulation of adhesion-associated signaling networks involving Neuropilin-2 and downstream survival pathways [47]. Similarly, in triple-negative breast cancer, MUC16 enhances metastatic progression by modulating RNA-binding proteins and post-transcriptional programs that ultimately converge on pro-survival and cytoskeletal signaling cascades [9, 48]. Together, these studies suggest that MUC16 functions as an upstream coordinator of adhesion-linked survival signaling rather than a direct kinase-interacting protein. In lung adenocarcinoma, our integrative analyses extend this paradigm by identifying MUC16-driven transcriptional remodeling of adhesion and junctional programs as a key mechanism linking MUC16 overexpression to FAK/PI3K-AKT activation and anoikis resistance.
Functionally, MUC16 knockdown markedly suppressed migratory and invasive capacities, as demonstrated by wound healing and transwell assays. These findings support the hypothesis that MUC16 facilitates cytoskeletal remodeling and cell motility, two hallmarks of anoikis-resistant metastatic cells. The observed reduction in migratory ability upon silencing also suggests disruption of MUC16-integrin/FAK signaling, leading to diminished activation of downstream AKT survival pathways. Together, these results substantiate a mechanistic model where MUC16 preserves cell-cell junction integrity and adhesion signaling to prevent anoikis and promote metastatic competence. Although migration and invasion assays do not directly measure anoikis, they provide important functional readouts of adhesion-dependent survival mechanisms. Efficient cell migration and transwell invasion require dynamic regulation of cell-matrix adhesion, cytoskeletal reorganization, and activation of pro-survival signaling pathways that overlap substantially with those governing anoikis resistance [49]. Cells capable of sustaining motility under reduced or transient attachment conditions typically exhibit enhanced tolerance to detachment-induced stress, a defining feature of anoikis-resistant tumor cells. Nevertheless, we acknowledge that the present study did not include direct anoikis-specific apoptosis assays, such as suspension culture-induced apoptosis, annexin V/propidium iodide staining, or caspase-3/7 activation under non-adherent conditions [50]. Future studies incorporating these assays will be valuable to directly quantify detachment-induced apoptosis and further substantiate the mechanistic role of MUC16 in regulating anoikis resistance in lung adenocarcinoma.
From a therapeutic perspective, targeting MUC16 directly or through its downstream FAK-AKT signaling axis may restore anoikis sensitivity and inhibit metastatic progression. Indeed, FAK inhibitors and PI3K-AKT blockers are already in clinical testing for solid tumors, suggesting that MUC16 expression could serve as a predictive biomarker for responsiveness to adhesion-signaling inhibitors. Furthermore, antibody-based or siRNA-based MUC16 suppression strategies, as previously applied in ovarian cancer models, may offer promising translational avenues for LUAD management.
Despite the compelling findings, several limitations warrant consideration. Firstly, although our siRNA experiments clearly demonstrate the role of MUC16 in promoting detachment survival and metastatic behavior in vitro, in vivo validation remains essential. Secondly, the downstream signaling mechanisms through which MUC16 regulates adhesion and survival require further elucidation. While our enrichment analyses implicate FAK, integrins, cadherins, and ECM components, direct mechanistic evidence remains to be established. Thirdly, although survival analyses across TCGA cohorts revealed strong prognostic associations, validation in independent patient populations is still required. Immunohistochemical staining of MUC16 on LUAD tissue microarrays and correlation with detachment or apoptosis markers (cleaved caspase-3, E-cadherin loss) would reinforce the clinical relevance of our findings.
Despite the strong integrative and functional evidence presented, several limitations should be acknowledged. First, although our in vitro data demonstrate that MUC16 promotes adhesion-dependent survival, migration, and invasion, in vivo validation remains necessary to fully establish its role in metastatic dissemination. Future studies could employ tail vein injection or orthotopic lung implantation models using MUC16-silenced LUAD cells to directly assess metastatic colonization, lung tumor burden, and survival of detached tumor cells in vivo. Such models would provide direct evidence linking MUC16-mediated anoikis resistance to metastatic fitness in a physiological process.
Second, functional validation in the present study was performed exclusively in A549 cells, a KRAS-mutant LUAD model. While this cell line is widely used to study lung cancer metastasis and adhesion signaling, LUAD is a heterogeneous disease. Therefore, extending these findings to additional LUAD cell lines with distinct molecular backgrounds, such as H1299 (TP53-null) or PC9 (EGFR-mutant) cells, would further strengthen the generalizability of our conclusions. Notably, the consistent association of MUC16 overexpression with poor prognosis, copy-number gain, epigenetic deregulation, and immune suppression across large patient cohorts supports a broader relevance of MUC16-driven anoikis resistance beyond a single cell model. These future directions will help refine the mechanistic and translational significance of MUC16 as a therapeutic target in lung adenocarcinoma.
5. Conclusion
Our work identifies MUC16 as a novel mediator of anoikis resistance in lung adenocarcinoma, linking its upregulation with detached tumour-cell survival, metastatic potential, and a poor prognosis. By bridging bioinformatics and functional validation, we not only reinforce MUC16's clinical relevance in LUAD but also suggest a new mechanistic axis of anoikis evasion that underpins its tumor-promoting role. These findings open new avenues for anti-metastasis therapeutic strategies targeting MUC16 and its downstream adhesion-signalling network in lung cancer.
Supplementary Material
Supplementary figure.
Acknowledgements
The authors acknowledge the Instrumentation and Equipment Center of the Translational Medicine Institute at Xi'an Jiaotong University for their technical support in this work.
Funding
This work was supported by the National Natural Science Foundation of China (Grant NOs. 82572281, 32271512), the Natural Science Basic Research Program of Shaanxi Province (Grant NOs. 2022JC-56, 2023-JC-ZD-43), and the Key Research and Development Program of Shaanxi Province (Grant NO. 2024GX-YBXM-316).
Availability of data and materials
The data are included in this manuscript and supporting information.
Author contributions
Conceptualization: Wen Cai, Zhenlong Wang, Fu Wang; Methodology: Kulsoom, Wajahat Ali, Zhe Ma; Formal analysis and investigation: Kulsoom, Zhe Ma; Software: Kulsoom, Wajahat Ali; Writing - original draft preparation: Kulsoom, Wajahat Ali; Writing - review and editing: Fu Wang; Funding acquisition: Wen Cai, Fu Wang; Supervision: Wen Cai, Zhenlong Wang, Fu Wang. All authors reviewed and approved the manuscript.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Ji X, Liu M, Zhang T, Zhang W, Xue F, Wan Q. et al. KRAS/PI3K axis driven GTF3C6 expression and promotes LUAD via FAK pathway. Journal of Advanced Research. 2025;70:243-54
2. Ali W, Kulsoom, Wang F. Molecular probes for monitoring pyroptosis: design, imaging and theranostic application. Apoptosis. 2024;29:1038-50
3. Ali W, Saba Z, Saqib M, Batool S, Awais, Rahman F. et al. Innovative Nanotechnology and Molecular Probes for Real-time Monitoring and Therapeutic Modulation of Pyroptosis in Cancer and Inflammatory Diseases. Cell Biochemistry and Biophysics. 2025
4. Liu Y, Zhao W, Zhao Z, Duan Z, Huang H, Ding C. et al. Hypoxia-anoikis-related genes in LUAD: machine learning and RNA sequencing analysis of immune infiltration and therapy response. American Journal of Cancer Research. 2025;15:3762
5. Hussain S, Zahra S, Ali W, Irshad M, Kulsoom, Hamdani SDA. et al. Bioprocessing and Purification of Metabolites. In: Singh V, editor. Advances in Metabolomics. Singapore: Springer Nature Singapore. 2024 p. 307-30
6. Kulsoom Ahmad S, Urwa Khan IA, Awais Pricope R. et al. Associations between clinical, biochemical, and nutritional factors in kidney stone formation and recurrence. Urolithiasis. 2025;53:112
7. Geijerman E, Terrana F, Peters GJ, Deng D, Diana P, Giovannetti E. et al. Targeting a key FAK-tor: the therapeutic potential of combining focal adhesion kinase (FAK) inhibitors and chemotherapy for chemoresistant non-small cell lung cancer. Expert Opinion on Investigational Drugs. 2024;33:1103-18
8. Kulsoom Ali W, Ahmad S Khan IA, Soni TK Masood A. et al. Sepsis-related immune signature C3 in endometrial carcinoma: implications for prognosis, tumor progression through bioinformatics and experimental validation. Mol Biol Rep. 2025;52:471
9. Li Q, Chu Y, Li S, Yu L, Deng H, Liao C. et al. The oncoprotein MUC1 facilitates breast cancer progression by promoting Pink1-dependent mitophagy via ATAD3A destabilization. Cell Death & Disease. 2022;13:899
10. Caccavari F, Valdembri D, Sandri C, Bussolino F, Serini G. Integrin signaling and lung cancer. Cell Adh Migr. 2010;4:124-9
11. Ji X, Liu M, Zhang T, Zhang W, Xue F, Wan Q. et al. KRAS/PI3K axis driven GTF3C6 expression and promotes LUAD via FAK pathway. Journal of Advanced Research. 2025;70:243-54
12. Xiao W, Ma W, Wei S, Li Q, Liu R, Carney RP. et al. High-affinity peptide ligand LXY30 for targeting α3β1 integrin in non-small cell lung cancer. Journal of Hematology & Oncology. 2019;12:56
13. Kulsoom Ali W, Wang F. Advancement in synthetic gene circuits engineering: An alternative strategy for microRNA imaging and disease theranostics. Biotechnol Adv. 2025;79:108518
14. Bi G, Zhu D, Bian Y, Huang Y, Zhan C, Yang Y. et al. Knockdown of GTF2E2 inhibits the growth and progression of lung adenocarcinoma via RPS4X in vitro and in vivo. Cancer cell international. 2021;21:181
15. Kulsoom K, Ali W, Saba Z, Hussain S, Zahra S, Irshad M. et al. Revealing Melatonin's Mysteries: Receptors, Signaling Pathways, and Therapeutics Applications. Horm Metab Res. 2024;56:405-18
16. Ma Z, Kulsoom, Wang F. Lighting Up Endogenous RNA: Fluorescent Aptamer Sensors for Live-Cell Imaging. Chemical & Biomedical Imaging. 2025
17. Mo C-f, Li J, Yang S-x, Guo H-j, Liu Y, Luo X-y. et al. IQGAP1 promotes anoikis resistance and metastasis through Rac1-dependent ROS accumulation and activation of Src/FAK signalling in hepatocellular carcinoma. British journal of cancer. 2020;123:1154-63
18. Junchao Y, Leilei W. Integrin α3 Mediates Stemness and Invasion of Glioblastoma by Regulating POU3F2. Current Protein & Peptide Science. 2023;24:247-56
19. X K, Ali W, Chong Y, Wang Z, Xue L, Wang F. Light as a Weapon: Redefining Cancer Care with Photodynamic Therapy. International Journal of Biological Sciences. 2026;22:1560-88
20. Liu J, Li Y, Wang F. Shedding light on Alzheimer's disease: Recent advances in highly selective fluorescent probes. Coordination Chemistry Reviews. 2025;522:216221
21. Chen X, Sandrine IK, Yang M, Tu J, Yuan X. MUC1 and MUC16: critical for immune modulation in cancer therapeutics. Frontiers in immunology. 2024;15:1356913
22. Yan Y, Kulsoom, Sun Y, Li Y, Wang Z, Xue L. et al. Advancing cancer therapy: Nanomaterial-based encapsulation strategies for enhanced delivery and efficacy of curcumin. Mater Today Bio. 2025;33:101963
23. Wang J, Luo Z, Lin L, Sui X, Yu L, Xu C. et al. Anoikis-associated lung cancer metastasis: mechanisms and therapies. Cancers. 2022;14:4791
24. Wei S, Tan J, Huang X, Zhuang K, Qiu W, Chen M. et al. Metastasis and basement membrane-related signature enhances hepatocellular carcinoma prognosis and diagnosis by integrating single-cell RNA sequencing analysis and immune microenvironment assessment. Journal of Translational Medicine. 2024;22:711
25. He C, He J. Metabolic reprogramming and signaling adaptations in anoikis resistance: mechanisms and therapeutic targets. Molecular and Cellular Biochemistry. 2025:1-28
26. Shirakihara T, Kawasaki T, Fukagawa A, Semba K, Sakai R, Miyazono K. et al. Identification of integrin α3 as a molecular marker of cells undergoing epithelial-mesenchymal transition and of cancer cells with aggressive phenotypes. Cancer Sci. 2013;104:1189-97
27. Yu H, Chen X, Huang X, Pang M, Zhou H, Wu J. et al. Regulatory interplay of LINC00115/hnRNPA1/miR-146a-5p axis modulating EGFR signaling in LUAD progression. International Journal of Biological Macromolecules. 2025;315:144260
28. Jonckheere N, Van Seuningen I. Integrative analysis of the cancer genome atlas and cancer cell lines encyclopedia large-scale genomic databases: MUC4/MUC16/MUC20 signature is associated with poor survival in human carcinomas. Journal of Translational Medicine. 2018;16:259
29. Wang Z, Hou H, Zhang H, Duan X, Li L, Meng L. Effect of MUC16 mutations on tumor mutation burden and its potential prognostic significance for cutaneous melanoma. American Journal of Translational Research. 2022;14:849
30. Yuan Q, Cai S, Chang Y, Zhang J, Wang M, Yang K. et al. The multifaceted roles of mucins family in lung cancer: from prognostic biomarkers to promising targets. Frontiers in Immunology. 2025; Volume 16 -. 2025
31. Wang X, Yu X, Krauthammer M, Hugo W, Duan C, Kanetsky PA. et al. The association of MUC16 mutation with tumor mutation burden and its prognostic implications in cutaneous melanoma. Cancer Epidemiology, Biomarkers & Prevention. 2020;29:1792-9
32. Lakshmanan I, Salfity S, Seshacharyulu P, Rachagani S, Thomas A, Das S. et al. MUC16 regulates TSPYL5 for lung cancer cell growth and chemoresistance by suppressing p53. Clinical Cancer Research. 2017;23:3906-17
33. Kanwal M, Ding X-J, Song X, Zhou G-B, Cao Y. MUC16 overexpression induced by gene mutations promotes lung cancer cell growth and invasion. Oncotarget. 2018;9:12226
34. Chen X, Ba Y, Ma L, Cai X, Yin Y, Wang K. et al. Characterization of microRNAs in serum: a novel class of biomarkers for diagnosis of cancer and other diseases. Cell Research. 2008;18:997-1006
35. Liu T, Wu L, Liu J, Chen H, Zhu B, Qiao D. et al. Comprehensive characterization of MUC16 mutations in lung adenocarcinoma for immunotherapies and prognosis: An observational study. Medicine. 2023;102:e35481
36. Chaudhary S, Appadurai MI, Maurya SK, Nallasamy P, Marimuthu S, Shah A. et al. MUC16 promotes triple-negative breast cancer lung metastasis by modulating RNA-binding protein ELAVL1/HUR. Breast Cancer Research. 2023;25:25
37. Liu B, Wu Q, Xuan Z, Zheng Z, Du Y, Sui X. et al. Mechanisms Involved in Focal Adhesion Signaling Regulating Tumor Anoikis Resistance. Cancer Science. 2025
38. Tan Y, Xiao C, Wang Z, Kong Y, Huang Y, Liu Z. et al. Leveraging Acquired EGFR-TKI-Resistant Models to Identify MUC16 as a Therapeutic Vulnerability in Lung Adenocarcinoma. Pharmaceuticals. 2025;19:47
39. Li S, Dong X, Gao K, Wang Y, Li M, Deng H. et al. PP2A Promotes the Symmetric Division of MUC1-Dominant Cancer Stem-Like Cells in Small Cell Lung Cancer. Advanced Science. 2025: 2503545.
40. de Sousa Mesquita AP, de Araújo Lopes S, Pernambuco Filho PCA, Nader HB, Lopes CC. Acquisition of anoikis resistance promotes alterations in the Ras/ERK and PI3K/Akt signaling pathways and matrix remodeling in endothelial cells. Apoptosis. 2017;22:1116-37
41. Liu J, Xu Y, Xu T, Liu Y, Liu J, Chai J. et al. MUC1 promotes cancer stemness and predicts poor prognosis in osteosarcoma. Pathology-Research and Practice. 2023;242:154329
42. Milella M, Rutigliano M, Lasorsa F, Ferro M, Bianchi R, Fallara G. et al. The role of MUC1 in renal cell carcinoma. Biomolecules. 2024;14:315
43. Liu A, Xie H, Li R, Ren L, Yang B, Dai L. et al. Silencing ZIC2 abrogates tumorigenesis and anoikis resistance of non-small cell lung cancer cells by inhibiting Src/FAK signaling. Molecular Therapy-Oncolytics. 2021;22:195-208
44. Marimuthu S, Lakshmanan I, Muniyan S, Gautam SK, Nimmakayala RK, Rauth S. et al. MUC16 Promotes Liver Metastasis of Pancreatic Ductal Adenocarcinoma by Upregulating NRP2-Associated Cell Adhesion. Mol Cancer Res. 2022;20:1208-21
45. Kodali N, Alomary S, Bhattaru A, Eldaboush A, Schwartz RA, Lipner SR. Gender and melanoma subtype-based prognostic implications of MUC16 and TTN co-occurrent mutations in melanoma: A retrospective multi-study analysis. Cancer Medicine. 2024;13:e70199
46. Lan Y, Ni W, Tai G. Expression of MUC1 in different tumours and its clinical significance. Molecular and clinical oncology. 2022;17:161
47. Kim Y-I, Pecha RL, Keihanian T, Mercado M, Pena-Munoz SV, Lang K. et al. MUC1 Expressions and Its Prognostic Values in US Gastric Cancer Patients. Cancers. 2023;15:998
48. Liu H, Xin T, Duan H, Wang Y, Shao C, Zhu Y. et al. Development and validation of a MUC16 mutation-associated immune prognostic model for lung adenocarcinoma. Aging (Albany NY). 2023;15:5650-61
49. Saad HM, Tourky GF, Al-Kuraishy HM, Al-Gareeb AI, Khattab AM, Elmasry SA. et al. The potential role of MUC16 (CA125) biomarker in lung cancer: a magic biomarker but with adversity. Diagnostics. 2022;12:2985
50. Wang F, Ali W. CRISPR-Based RNA Detection. RNA Synthetic Biology: CRC Press. 2026 p. 165-210
Author contact
![]() Corresponding authors: Wen Cai (Email: caiwen2016edu.cn); Zhenlong Wang (Email: zhenlongw2001edu.cn); Fu Wang (Email: wangfuedu.cn).
Corresponding authors: Wen Cai (Email: caiwen2016edu.cn); Zhenlong Wang (Email: zhenlongw2001edu.cn); Fu Wang (Email: wangfuedu.cn).