Impact Factor ISSN: 1837-9664
- Issue 8; 2026
- Issue 7; 2026
- Issue 6; 2026
- Issue 5; 2026
- Issue 4; 2026
- Volume 17; 2026
- Past Issues
- Editorial Board
- Cover Images
- Index & Coverage
- Special Issues
Advantages and disadvantages of...
Cell surface molecules and...
Cell surface proteins and cancer...
Strategies for improving cancer...
Antigen pollution associated...
Antigen pollution associated...
Proof-of-concept studies using...
Proof-of-concept studies using...
Authentication of cell surface...
Safety of cell surface antigens
Conclusions
Acknowledgements
References

 Global reach, higher impact
Global reach, higher impactJ Cancer 2010; 1:230-241. doi:10.7150/jca.1.230 This volume Cite
Review
Cellular Cancer Vaccines: an Update on the Development of Vaccines Generated from Cell Surface Antigens
Petr G. Lokhov1,2 ![]() , Elena E. Balashova1,3
, Elena E. Balashova1,3
1. ZAO BioBohemia, Moscow, Russia
2. Institute of Biomedical Chemistry RAMS, Moscow, Russia
3. Cardiology Research Center, Moscow, Russia
Received 2010-9-27; Accepted 2010-11-29; Published 2010-11-29
Abstract
A recent advance in anti-cancer therapies has been the use of cancer cells to develop vaccines. However, immunization with cancer cell-based vaccines has not resulted in significant long-term therapeutic benefits. A possible reason for this is that while cancer cells provide surface antigens that are targets for a desired immune response, they also contain a high abundance of housekeeping proteins, carbohydrates, nucleic acids, lipids, and other intracellular contents that are ubiquitous in all mammalian cells. These ubiquitous molecules are not the intended targets of this therapy approach, and thus, the immune response generated is not sufficient to eliminate the cancer cells present. In this review, a discussion of the cell surface of cancer cells is presented in relation to the goals of improving antigen composition of cancer cell-based vaccines. Strategies to enrich vaccines for cancer-specific antigens are also discussed.
Keywords: cellular cancer vaccine, antiangiogenic cancer vaccine, cell surface antigens
Advantages and disadvantages of cancer cell-based vaccines
The efficacy of whole-cell cancer vaccines has been investigated for more than 20 years in both preclinical models and in clinical trials in humans (1-3). The advantages of whole-cell vaccination over other types of immunotherapy that target specific antigens, is that multiple and unknown antigens may be targeted by both the innate and adaptive immune system. However, immunization with whole-cell vaccines has not resulted in significant long-term therapeutic benefits (4, 5). One possible explanation is that only a small proportion of the molecules expressed on the cell surface are specific to cancer cells, while the vast majority of cellular constituents derive from housekeeping genes, carbohydrates, nucleic acids, lipids, and other molecules that are ubiquitously expressed by normal cells (6). As a result, the immune response generated in patients administered vaccines generated from whole cancer cells is insufficient to eliminate the cancer cells present in vivo. Therefore, further improvements are necessary to improve the specificity of cancer cell-based vaccines.
Ideally, vaccines developed to treat cancer need to recognize molecules that are not found on untransformed cells (7-11). Surface antigens, proteins, and associated carbohydrates, are the targets naturally accessible to surveillance by the immune system, particularly by the humoral and cellular immune response. However, when whole cells are used for the preparation of vaccines, a greater proportion of intracellular molecules are combined with a smaller proportion of cell surface-associated molecules, thereby reducing the concentration of cancer cell-specific antigens recognized for vaccine production (Figure 1). Therefore, cancer cells used for the development of vaccines contain a high proportion of targets which are not cancer cell-specific, and an enrichment of cell surface material is needed to improve the effectiveness of cancer vaccines.
Ratios of the categories of cell surface targets which are accessible for humoral and cytotoxic immunity. Figure adapted from (118).
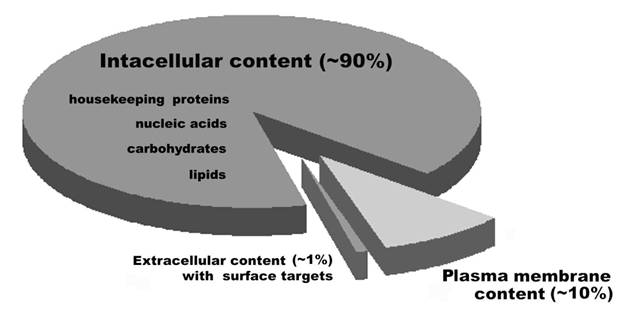
Cell surface molecules and cancer immunity
The recognition of antigens during cell-cell interactions plays an important role in immune function. For example, it is through cell-cell interactions that cancer cells are recognized and targeted for cell lysis by effector cells. Correspondingly, cancer cells have developed strategies to prevent their recognition and adhesion by immune effector cells. One way they have done this is to maintain a cell surface that is lacking immunodominant antigens (12, 13). Moreover, cancer cells present substances with immunosuppressive properties (14-19). For example, tenascin-C extracts from a glioma cell line inhibit T lymphocyte proliferation and cytokine production (20-22). Other studies have also demonstrated that glioma cells produce thick glycosaminoglycan coats to protect them from cytotoxic T-lymphocyte (CTL) responses (23-25). Galectins are another class of cell surface molecules which have been shown to have important roles in the survival, angiogenesis, and metastasis of tumors (26, 27). Furthermore, less characterized cell surface molecules have also been shown to have immunosuppressive characteristics. For example, highly malignant lymphoma cells (RAW117) have been shown to inhibit both T-cell and B-cell mitogen-induced proliferation of normal murine spleen cells. Interestingly, a similar inhibition was observed when RAW117 cells were extracted with butanol (28), and this extract was used instead of whole RAW117 cells (29). In another study where cell surface molecules from lymphoma cells were extracted with butanol, an inhibitory effect of this extract on natural killer and lymphokine activated killer cell mediated cytotoxicity was also observed (30).
In combination, these findings demonstrate that the cell surface of cancer cells lack immunodominant antigens, and predominantly consist of molecules with immunosuppressive properties.
Cell surface proteins and cancer antigenicity
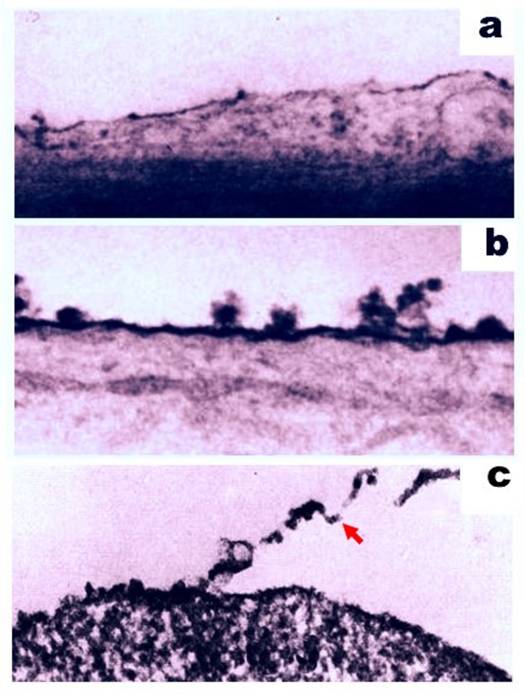
Despite the immunosuppressive activity associated with some cell surface molecules of cancer cells, antigenic motifs are also present. The ability of these two functions to co-exist on the surface of cancer cells has been investigated. For example, antibodies produced by a host organism in response to injected cells were found to selectively and specifically bind the antigenic targets of the injected cells (31-33). In another approach, cell surface molecules were removed with 0.2% trypsin, and cell antigenicity was subsequently examined (34). Trypsinization has previously been reported to release glycoproteins and sugars from the cell surface (35-38) (Figure 2), thereby leading to a loss of antigenic properties (39-42). In particular, Takeichi et al. showed that trypsin treatment of SV40-transformed 3T3 cells decreased their antigenicity in footpad assays (42). Similarly, Molinari and Platt (43) demonstrated that polyoma virus-transformed cells treated with trypsin failed to induce a delayed hypersensitivity reaction against tumor-specific antigens in footpad swelling assays. In other studies, Fakhri and Tan (44) reported that treatment of mouse plasmacytomas with 1% trypsin decreased the number of antigenic determinants present. Further experiments using mixtures of normal and sensitive cells also suggest that trypsin altered an immunologic capacity of the sensitive cells assayed (39). For example, peritoneal exudate cells from animals exhibiting delayed hypersensitivity were associated with a decrease in migration in vitro. However, this effect on migration was completely abolished when these cells were pretreated with trypsin. In combination, these data demonstrate that antigenic targets are present on the surface of cancer cells, since the removal of molecules from the cell surface results in a decrease in antigenicity.
Ruthenium staining of the cell surface of a normal rat brain cell (a) and a malignant rat neurogenic cell (b). Magnification, 40,000x. Figures adapted from (119). (c) Partial detachment of cell surface material (indicated with a red arrow) following treatment of the cell with 0.2% trypsin. Magnification, 19,000x. Figure adapted from (34).
Strategies for improving cancer cell-based vaccines
The contamination of intracellular material in the development of cancer cell-specific vaccines, and evidence regarding the presence of antigenic properties on the surface of cancer cells, have been presented. Based on these considerations, it would be advantageous to isolate the cell surface molecules of cancer cells to develop vaccines rather than using whole cell preparations. Correspondingly, the cancer cell-specificity of vaccines using cell surface antigens would be improved without a loss of native antigen diversity. Vaccination with the fraction of soluble membrane antigens of cancer cells was proposed by Hollinshead et al. many years ago (45-48). However, this vaccine development approach has not resulted in vaccines approved for human use. Therefore, here we emphasize new approach which based on vaccine development using proteolitycally-cleved cell surface antigens. Although the isolation of cell surface molecules using proteases was described many years ago (42, 49-53), as well as the use of cancer cells for vaccination, limitations in the applicability of such an approach have evolved to include considerations of protease purity and cellular damage incurred during protease treatments and antigen preparations.
Antigen pollution associated with protease impurity
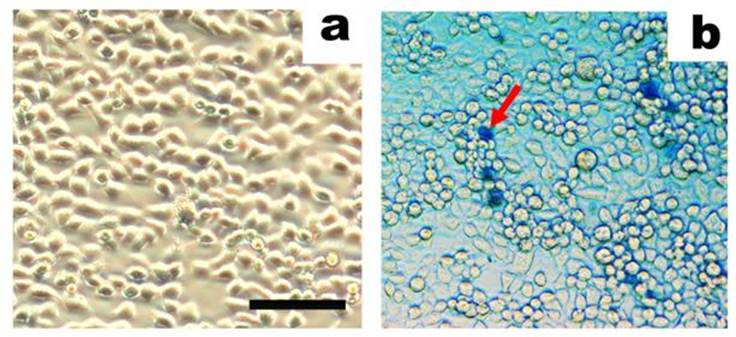
One of the earliest studies of trypsinization of tumor cells was conducted in 1958, and a significant loss of tumor mass (up to 20%) was induced without any apparent change in cell viability (54). Subsequent studies of cell-disaggregating agents and cell integrity used suspension cells (55-57) or cells from intact tissue (58, 59). In both cases, integrity of the treated cells following trypsinization was sufficient to maintain the viability of the cells (54, 56, 60, 61). However, there have been other methods described for the disaggregation of intact tissues that has been shown to cause considerable damage to the cell (58). In an investigation by Anghilery and Dermietzel, trypsinization was shown to liberate up to 10% of cell material, which contained significant amounts of lipid and nucleic acids from intracellular lipo- and nucleoproteins (50). When a mild treatment of cells with 0.1% trypsin was performed, up to 10.4% of cellular RNA and 11.4% of cellular DNA were obtained, consistent with a lysis rate of ~11% (62). However, when a higher purity trypsin was used, less than 2% of cells were lysed (63). Based on these results, trypsin impurities were identified as a cause of increased cell damage. In more recent studies where highly-purified trypsin with an activity of 15 000 U/mg was used, rates of cell lysis were less then 0.1% (Figure 3) (64). Therefore, mammalian cells can be treated with trypsin without inducing the lysis of the treated cells, if highly purified trypsin is used.
Human breast adenocarcinoma cells (MCF-7) are shown before (a) and after (b) treatment with trypsin (0.2 µg/mL, activity 15000 U/mg) for 25 min. In situ trypan blue staining was performed, and cells that are stained represent dead cells (see arrow). Images were obtained using an inverted phase contrast microscope (scale bar: 50 µm). Figure adapted from Balashova et al. (64).
Another aspect to consider regarding the use of unpurified trypsin is the possibility that the antigens collected will be contaminated with the trypsin used. Trypsin at a working concentration of ~300 µg/ml has previously been widely used in scientific experiments to cleave cell surface material (49, 65). For example, a recent study showed that the treatment of 5 ml of cancer cells with 1 ml of trypsin solution yielded only 10-20 µg of cell surface (glyco)protein fragments (64). This represents ~1% of the total protein content of a cell. Based on these results and others, the consensus of the data indicate that the concentration of trypsin used to cleave cell surface antigens significantly exceeds that of the cleaved cell surface antigens. Correspondingly, since trypsin contains numerous impurities, including other types of proteases, differently degraded forms of trypsin, and trypsin autolysis products (66), identification of the cell surface antigens treated with trypsin requires complex analytical methods, and these have been described (49, 52, 67-75). However, the need for antigen identification is more relevant to the characterization of individual antigens than for vaccine production. Thus, for vaccine production, it is important that highly purified trypsin be used in the preparation of cell surface proteins for vaccination. Correspondingly, it has recently been demonstrated that live cells treated with highly-purified trypsin resulted in a solution of cell specific peptides which were not contaminated with proteases, cytosolic proteins, or serum from the cell growth medium (76). Therefore, these results confirm that highly-purified trypsin can provide a high purity sample of cell surface antigens.
Antigen pollution associated with cell damage
The mechanism by which trypsin acts has been very well characterized. However, the cell damage that occurs as a result of treatment with proteases also includes cell damage generated by fluid shearing. It has been established that animal cells are sensitive to fluid shearing in serum-free medium (77-81). However, to obtain pure samples of cell surface antigens, cells are prepared in serum-free medium. As a result, damage of the cell membrane leads to the release of intracellular contents. As shown in Figure 1, the amount of cell surface antigens obtained from 100 cancer cells is comparable to the quantity of intracellular molecules contained within a single cell. In work by Lau and Tchao (2007) using Nara Bladder Tumour (NBT) II cells, if cells were grown on plastic surfaces vs. glass surfaces, the amount of cell damage due to fluid shear was 56% and 5%, respectively (79). Therefore, a critical aspect of preparing cell surface antigens is to minimize the destruction of cells by fluid shearing. Correspondingly, when protocol conditions were optimized and careful manipulation of the cells was maintained, an observed death rate of < 0.1% of adenocarcinoma cells was achieved following trypsinization (64) (Figure 3). For cell cultures that are more sensitive to fluid shear, an optimization of cell growth conditions and an application of cyto-protectants may be needed to prevent cell damage in serum-free medium and to decrease the rate of cell death during manipulations (77).
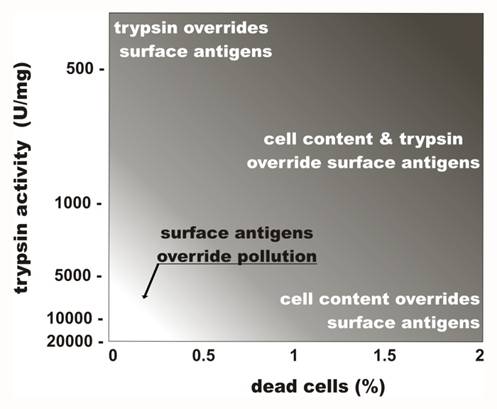
Thus, the careful treatment of live cells with highly purified proteases facilitates the collection of cell surface antigens with minimal contamination by undesired intracellular contents. Figure 4 summarizes the influence of protease impurities and cell death rates associated with fluid shearing on the purity of cell surface antigens collected.
Effects of trypsin impurity and cell death rate on the preparation of cell surface antigens. The lighter region in the lower left corner represents conditions where cell-surface antigen preparations are compatible with the preparation of vaccines. Trypsin purity is reflected in the levels of enzymatic activity. Adapted from Lokhov (118).
Proof-of-concept studies using cancer cells
To confirm that proteolytically-cleaved cell surface antigens are sufficient for developing effective vaccines, immunogenic properties of cell surface antigens and whole cancer cells were examined. In cytotoxicity assays, human CTLs were incubated with target adenocarcinoma cells, MCF-7 (64), and CTLs were stimulated with dendritic cells loaded with cell surface antigens or whole cancer cells. In these assays, cell surface antigens were 10-40% more effective at inducing cytotoxicity than the whole cell lysates (Figure 5) (64). Moreover, the total protein concentration of the whole cell lysates vs. the cell surface antigen preparations was 270 µg/mL vs. 2 µg/mL, respectively. Therefore, the higher levels of cytotoxicity that were associated with a significantly lower protein concentration demonstrates the importance of antigenic targets being free from intracellular contaminants for effective immune response stimulation. Correspondingly, the authors concluded that these results provided a proof-of-concept that the proteolytic treatment of live cancer cells can release antigenic targets that are sufficient to induce an anti-cancer immune response that exceeds that of untreated cancer cells in vitro. As a result, further studies will be needed to more completely characterize the use of cell surface antigens instead of whole cancer cell preparations in the generation of anti-cancer vaccines.
A comparison of whole cell lysates and proteolytically-cleaved cell surface antigens in mediating the lysis of breast adenocarcinoma cells in cytotoxicity assays. Target adenocarcinoma cells, MCF-7, were incubated with effector CTLs at a ratio of 4:1. On the 3rd day, target cells were carefully washed and imaged using an inverted phase contrast microscope (scale bar: 50 µm). (a) Initial cell culture of MCF-7 cells. (b) Control MCF-7 cells grown alone. (c) MCF-7 cells incubated with CTLs that had been stimulated with dendritic cells loaded with whole cell lysates. (d) MCF-7 incubated with CTLs that had been stimulated with dendritic cells loaded with cell surface antigens. Cell surface antigens show the same ability to induce cytotoxic activity in CTLs as whole cell lysates. Adapted from Balashova et al. (64).
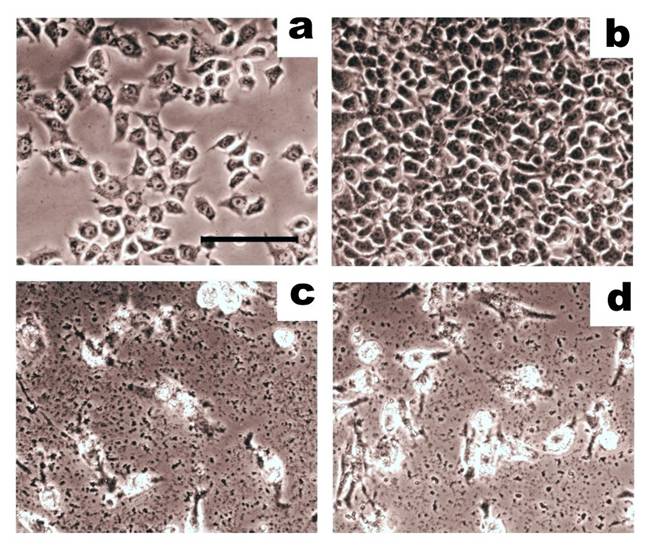
CTL-mediated lysis of human microvascular endothelial cells (HMEC) produced by whole cell lysates or cell surface antigens of HMEC in cytotoxicity assays. HMEC (40 000 cells/well) were incubated with effector cytotoxic T lymphocytes (CTL) at a ratio of 1:8 and were imaged using an inverted phase contrast microscope. (a) Initial cell culture of HMEC. (b) HMEC incubated with CTL stimulated, unloaded antigen-presenting cells. (c) HMEC incubated with CTL stimulated, antigen-presenting cells loaded with HMEC lysate. (d) HMEC incubated with CTL stimulated, antigen-presenting cells loaded with cell surface antigens prepared from HMEC. Cell surface antigens are more effective at stimulating immune cells in cytotoxicity assays than whole cell HMEC lysates. Adapted from Balashova et al. (100).
Proof-of-concept studies using endothelial cells
It is known that tumor endothelial cells actively form new blood vessels, and this additional vascular capacity is essential for tumor growth and metastasis (82-88). Tumor-associated endothelial cells proliferate in response to tumor-secreted stimulators and undergo changes in phenotype as needed. These pronounced changes distinguish them from endothelial cells of the normal vasculature (89-92). While many studies have shown that cancer cells provide antigens suitable for inducing an immune-targeted response (2, 3, 93), a similar approach has been proposed for the use of endothelial cells to develop cellular anti-angiogenic, anti-cancer vaccines. This approach was previously shown to inhibit the growth of experimental tumors in mouse models (94-99). However, endothelial cell-based vaccines share a similar limitation with cell-based vaccines in that the extracellular macromolecules of endothelial cells include both antigens and ubiquitously expressed proteins that are common to all mammalian cells, including normal endothelial cells. Therefore, the ability to isolate extracellular protein targets important for both humoral and cell-mediated immune responses by treatment of endothelial cells with proteases was a proposed strategy for the creation of anti-angiogenic vaccines, and was tested using primary cultures of human microvascular endothelial cells (HMECs) (100). With careful manipulation and use of a highly-purified protease, cell surface antigens, as well as whole cell lysates were obtained. These buffered solutions had a total protein concentration of 135 µg/mL and 2 µg/mL, respectively. However, despite this difference in concentration, cell surface antigen preparations induced more cytotoxicity than the whole cell lysates. Moreover, surface antigens obtained from tumor-activated endothelial cells were able to stimulate an immune response toward tumor-activated endothelial cells. Based on these results, the authors concluded that surface antigens of endothelial cells appear to be able to induce targeted, immune-mediated cytotoxic effects against tumor endothelial cells, and these results demonstrate the potential for endothelial surface antigens, instead of whole endothelial cells, to generate anti-angiogenic anti-cancer vaccines.
Authentication of cell surface antigens
Cultivated cell studies have indicated that cross-contamination between cell lines is widely prevalent and continues to be a major problem (101-105). Based on current estimates, up to 36% of cell lines appear to have a different origin than their initial cell lines (105, 106). Moreover, cultivated mammalian cells have a finite mitotic lifespan (107-109), which is followed by cellular degeneration and modifications of their cell surface profile (110, 111). Most existing cell lines have been passaged extensively in vitro, and therefore, considerable variation and genomic instability have been introduced (112, 113). Moreover, even cell lines that have been in culture for a relatively brief period of time, have displayed marked differences from the cultures derived from the same specimen (114). This heterogeneity is pronounced at the genomic level, and is reflected in antigen expression patterns. It is hypothesized that these changes represent a cellular response to in vitro selection pressures (114), which can subsequently lead to a divergence of cellular phenotypes over time.
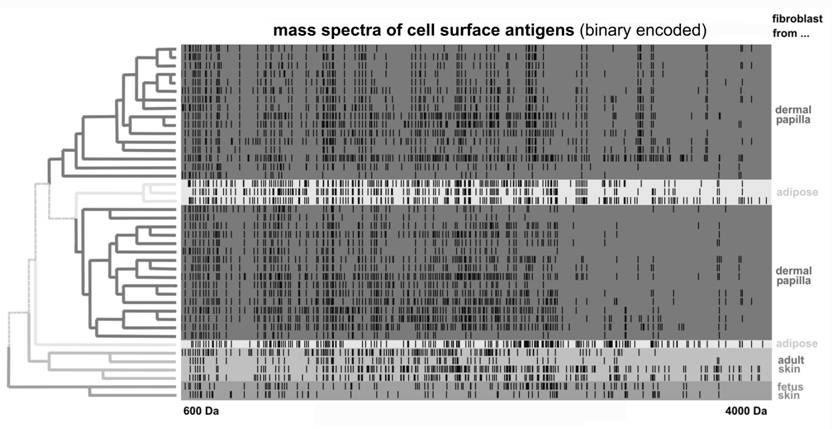
Accordingly, cultivated cancer cells intended for therapy, or for the isolation of antigens, must be authenticated and analyzed for changes in surface antigen expression. Cancer vaccines which are already in clinical trials (115) have not completed these analyses, partly because there is no routine method established for the analysis the diversity of cell surface proteins. In contrast, proteolytically-separated cell surface antigens can be easily analyzed using mass spectrometry. This technique is related to 'proteomic footprinting' that can be performed for cells, and represents a simple approach for the authentication and characterization of cells. Furthermore this method is associated with a rapid turnover time for samples and a low per sample cost (76). By comparing the composition of cell surface antigens obtained with a proteomic footprint of the reference cancer cells, the origin of the cell surface antigens obtained can be easily authenticated (Figure 7). Moreover, any divergence in this comparison can reveal changes in antigen composition that may have occurred.
Clustering of mass spectra data for cell surface antigens cleaved from fibroblasts of different origins. Fibroblast cultures were treated with trypsin under noncytolytic conditions. Mass spectra, or cell proteomic footprints, are represented in log scale and in binary format, where a dash indicates the presence of a measured peptide mass in the corresponding footprint. Footprints were clustered using the Ward method and according to the origin of the fibroblast culture analyzed. Therefore, cultures with unknown origins can be easily authenticated by identifying their relation to a footprint of a particular cluster. Adapted from Lokhov et al. (76).
Safety of cell surface antigens
A potential limitation of current cell-based vaccines is the difficulty associated with purifying cells from dangerous contaminants such as cell parasites, viruses, toxins, and prions. Improving this aspect is essential for cell-based vaccines (116, 117). Given that proteolytically-cleaved cell surface antigens include (glyco)peptides, it is possible that ultrafiltration of these preparations could remove macromolecules and supramolecular structures to render the antigen preparations safe.
Conclusions
Accumulated data regarding the isolation of cell surface antigens from cancer cells has led to the following conclusions:
- The surface of cancer cells lack immunodominant antigens yet maintain immunosuppressive properties;
- Antigenic properties of cancer cells are mediated by cell surface molecules;
- Treatment of cells with proteases remove antigens from cancer cells;
- Use of highly-purified trypsin and careful manipulation of cells allows cell surface antigens to be obtained with minimal contamination;
- Cell protectants are necessary to obtain cell surface antigens from cancer cells that are sensitive to fluid shearing;
- The use of cell surface antigens, rather than whole cell preparations, to develop vaccines may improve the detection of cancer cells by the immune system;
- By releasing cell surface antigens from the surface of cells with proteases, the antigens obtained are free from dangerous contaminants associated with whole cell preparations such as cell parasites, viruses, toxins and prions.
The efficiency of using cell surface antigens to generate anti-cancer vaccines is currently being evaluated, as well as the efficacy of using cell surface antigens for the targeting of cancer cells by the immune response in humans. Currently, there are numerous clinical trials in progress that are based on the use of cancer cell-based vaccines (115), although these vaccines are not approved for human use. However, the diversity of native cancer antigens available from the use of cancer cells, the low levels of undesirable antigens and dangerous intracellular agents that can be achieved with current methods, and the ability to authenticate antigens present, demonstrates the potential for using cell surface antigens to develop safer and more effective cellular cancer vaccines.
Acknowledgements
We would like to thank ZAO BioBohemia (Moscow, Russia) for their financial support.
Conflict of Interest
The authors declare that they have competing financial interests. Patent value may be affected by this publication.
References
1. Copier J, Dalgleish A. Overview of tumor cell-based vaccines. Int Rev Immunol. 2006;25(5-6):297-319
2. de Gruijl TD, van den Eertwegh AJ, Pinedo HM, Scheper RJ. Whole-cell cancer vaccination: from autologous to allogeneic tumor- and dendritic cell-based vaccines. Cancer Immunol Immunother. 2008;57(10):1569-77
3. Chiang CL, Benencia F, Coukos G. Whole tumor antigen vaccines. Semin Immunol. 2010;22(3):132-43
4. Copier J, Dalgleish A. Whole-cell vaccines: A failure or a success waiting to happen? Curr Opin Mol Ther. 2010;12(1):14-20
5. Bodey B, Bodey BJr, Siegel SE, Kaiser HE. Failure of cancer vaccines: the significant limitations of this approach to immunotherapy. Anticancer Res. 2000;20(4):2665-76
6. Cohen EP, Chopra A, I OS, Kim TS. Enhancing cellular cancer vaccines. Immunotherapy. 2009;1(3):495-504
7. Abercrombie M, Ambrose EJ. The surface properties of cancer cells: a review. Cancer Res. 1962;22:525-48
8. Calman F. Ultrastructural comparison of the cell coat in normal and chronic lymphocytic leukaemic blood lymphocytes by Concanavalin A labelling and cationic staining. Pathol Eur. 1975;10(3):203-14
9. Gasic GJ, Loebel F. Cytochemical identification of protein amino acids in the cell coat of mouse ascites tumor cells. Lab Invest. 1966;15(8):1310-9
10. Mallucci L, Poste GH, Wells V. Synthesis of cell coat in normal and transformed cells. Nat New Biol. 1972;235(59):222-3
11. Rittenhouse HG, Rittenhouse JW, Takemoto L. Characterization of the cell coat of Ehrlich ascites tumor cells. Biochemistry. 1978;17(5):829-37
12. Ruiz-Cabello F, Cabrera T, Lopez-Nevot MA, Garrido F. Impaired surface antigen presentation in tumors: implications for T cell-based immunotherapy. Semin Cancer Biol. 2002;12(1):15-24
13. Khong HT, Restifo NP. Natural selection of tumor variants in the generation of "tumor escape" phenotypes. Nat Immunol. 2002;3(11):999-1005
14. Bast JC. Principles of cancer biology: tumor immunology. In: (ed.) DeVita. V, Hellman SSR. Cancer Principles and Practice of Oncology. Philadelphia: J. B. Lippincott Co. 1985:125-50
15. Ebert EC, Roberts AI, O'Connell SM, Robertson FM, Nagase H. Characterization of an immunosuppressive factor derived from colon cancer cells. J Immunol. 1987;138(7):2161-8
16. Mohagheghpour N, Parhami B, Dowlatshahi K, Kadjehnouri D, Elder JH, Chisari FV. Immunoregulatory properties of human esophageal tumor extract. J Immunol. 1979;122(4):1350-8
17. Putnam JBJr, Roth JA. Identification and characterization of a tumor-derived immunosuppressive glycoprotein from murine melanoma K-1735. Cancer Immunol Immunother. 1985;19(2):90-100
18. Roth JA, Grimm EA, Gupta RK, Ames RS. Immunoregulatory factors derived from human tumors. I. Immunologic and biochemical characterization of factors that suppress lymphocyte proliferative and cytotoxic responses in vitro. J Immunol. 1982;128(5):1955-62
19. Roth JA, Osborne BA, Ames RS. Immunoregulatory factors derived from human tumors. II. Partial purification and further immunobiochemical characterization of a human sarcoma-derived immunosuppressive factor expressing HLA-DR and immunoglobulin-related determinants. J Immunol. 1983;130(1):303-8
20. Hemesath TJ, Marton LS, Stefansson K. Inhibition of T cell activation by the extracellular matrix protein tenascin. J Immunol. 1994;152(11):5199-207
21. Puente Navazo MD, Valmori D, Ruegg C. The alternatively spliced domain TnFnIII A1A2 of the extracellular matrix protein tenascin-C suppresses activation-induced T lymphocyte proliferation and cytokine production. J Immunol. 2001;167(11):6431-40
22. Parekh K, Ramachandran S, Cooper J, Bigner D, Patterson A, Mohanakumar T. Tenascin-C, over expressed in lung cancer down regulates effector functions of tumor infiltrating lymphocytes. Lung Cancer. 2005;47(1):17-29
23. Gately MK, Glaser M, McCarron RM, Dick SJ, Dick MD, Mettetal RWJr. et al. Mechanisms by which human gliomas may escape cellular immune attack. Acta Neurochir (Wien). 1982;64(3-4):175-97
24. Dick SJ, Macchi B, Papazoglou S, Oldfield EH, Kornblith PL, Smith BH. et al. Lymphoid cell-glioma cell interaction enhances cell coat production by human gliomas: novel suppressor mechanism. Science. 1983;220(4598):739-42
25. Oberc-Greenwood MA, Muul LM, Gately MK, Kornblith PL, Smith BH. Ultrastructural features of the lymphocyte-stimulated halos produced by human glioma-derived cells in vitro. J Neurooncol. 1986;3(4):387-96
26. Camby I, Le Mercier M, Lefranc F, Kiss R. Galectin-1: a small protein with major functions. Glycobiology. 2006;16(11):137R-57R
27. Liu FT, Rabinovich GA. Galectins as modulators of tumour progression. Nat Rev Cancer. 2005;5(1):29-41
28. LeGrue SJ. Noncytolytic extraction of cell surface antigens using butanol. Cancer Metastasis Rev. 1985;4(3):209-19
29. Joshi SS, O'Connor SJ, Weisenburger DD, Sharp JG, Gharpure HM, Brunson KW. Enhanced antiproliferative activity by metastatic RAW117 lymphoma cells. Clin Exp Metastasis. 1991;9(1):27-37
30. Hao W, McDonald TL, Brunson KW, Joshi SS. Enhanced immunosuppressive activity associated with metastatic lymphoma cells. Cancer Res. 1993;53(8):1921-8
31. Ben-Or S, Doljanski F. Single-cell suspensions as tissue antigens. Experimental Cell Research. 1960;20:641-4
32. Quash G, Delain E, Huppert J. Effect of antipolyamine antibodies on mammalian cells in tissue culture. Exp Cell Res. 1971;66(2):426-32
33. Nairn RC, Richmond HG, Mc EM, Fothergill JE. Immunological differences between normal and malignant cells. Br Med J. 1960;2(5209):1335-40
34. Kohnert-Stavenhagen E, Zimmermann B. Changes in the surface coat of mesenchymal cells of mouse limb buds after enzymatic cell separation. J Embryol Exp Morphol. 1980;59:145-55
35. Cook GM, Heard DH, Seaman GV. A sialomucopeptide liberated by trypsin from the human erythrocyte. Nature. 1960;188:1011-2
36. Gasic G, Gasic T. Removal and regeneration of the cell coating in tumour cells. Nature. 1962;196:170
37. Uhlenbruck G. Action of proteolytic enzymes on the human erythrocyte surface. Nature. 1961;190:181
38. Rinaldini LMJ. The isolation of living cells from animal tissues. International Review of Cytology. 1956;7:587-647
39. David JR, Lawrence HS, Thomas L. The in Vitro Desensitization of Sensitive Cells by Trypsin. J Exp Med. 1964;120:1189-200
40. Weiss L, Coombs RR. The demonstration of rupture of cell surfaces by an immunological technique. Exp Cell Res. 1963;30:331-8
41. Osunkoya BO, Mottram FC, Isoun MJ. Synthesis and fate of immunological surface receptors on cultured Burkitt lymphoma cells. Int J Cancer. 1969;4(2):159-65
42. Takeichi N, Economou GC, Boone CW. Accelerated regeneration of trypsin-treated surface antigens of simian virus 40-transformed BALB/3T3 cells induced by X-irradiation. Cancer Res. 1976;36(4):1258-62
43. Molinari JA, Platt D. Modification of surface membrane antigens by trypsin. Proc Soc Exp Biol Med. 1975;148(4):991-4
44. Fakhri O, Tan RS. Short communications. The effect of trypsin on cell surface antigens. Cell Immunol. 1975;15(2):452-6
45. Hollinshead AC, Herberman RB, Jaffurs WJ, Alpert LK, Minton JP, Harris JE. Soluble membrane antigens of human malignant melanoma cells. Cancer. 1974;34(4):1235-43
46. Hollinshead AC, Jaffurs WT, Alpert LK, Harris JE, Herberman RB. Isolation and identification of soluble skin-reactive membrane antigens of malignant and normal human breast cells. Cancer Res. 1974;34(11):2961-8
47. Hitchcock MH, Hollinshead AC, Chretien P, Rizzoli HV. Soluble membrane antigens of brain tumors. I. Controlled testing for cell-mediated immune responses in a long surviving glioblastoma multiforme patient. Cancer. 1977;40(2):660-6
48. Hollinshead A, Miller H, Tanner K, Lee O, Klausia J. Soluble cell membrane antigens associated with bladder cancer. Cancer Immunol Immunother. 1978;5:93-103
49. Glick MC, Kimhi Y, Littauer UZ. Glycopeptides from surface membranes of neuroblastoma cells. Proc Natl Acad Sci U S A. 1973;70(6):1682-7
50. Anglhileri LJ, Dermietzel P. Cell coat in tumor cells -- effects of trypsin and EDTA: a biochemical and morphological study. Oncology. 1976;33(1):17-23
51. Baumann H, Doyle D. Effect of trypsin on the cell surface proteins of hepatoma tissue culture cells. Characterization of a carbohydrate-rich glycopeptide released from a calcium binding membrane glycoprotein. J Biol Chem. 1979;254(10):3935-46
52. Baumann H, Doyle D. Turnover of plasma membrane glycoproteins and glycolipids of hepatoma tissue culture cells. J Biol Chem. 1978;253(12):4408-18
53. Warren L. The malignant cell and its membranes. Am J Pathol. 1974;77(1):69-76
54. Weiss L. The effects of trypsin on the size, viability and dry mass of sarcoma 37 cells. Exp Cell Res. 1958;14(1):80-3
55. Barnard PJ, Weiss L, Ratcliffe T. Changes in the surface properties of embryonic chick neural retina cells after dissociation. Exp Cell Res. 1969;54(3):293-301
56. Kraemer PM. Regeneration of sialic acid on the surface of Chinese hamster cells in culture. I. General characteristics of the replacement process. J Cell Physiol. 1966;68(1):85-90
57. Kraemer PM. Sialic acid of mammalian cell lines. J Cell Physiol. 1966;67(1):23-34
58. Kemp RB, Jones BM, Cunningham I, James MC. Quantitative investigation on the effect of puromycin on the aggregation of trypsin- and versene-dissociated chick fibroblast cells. J Cell Sci. 1967;2(3):323-40
59. Pitelka DR, Kerkof PR, Gagne HT, Smith S, Abraham S. Characteristics of cells dissociated from mouse mammary glands. I. Method of separation and morphology of parenchymal cells from lactating glands. Exp Cell Res. 1969;57(1):43-62
60. De Luca C. The use of trypsin for the determination of cellular viability. Exp Cell Res. 1965;40(1):186-8
61. Kraemer PM. Regeneration of sialic acid on the surface of Chinese hamster cells in culture. II. Incorporation of radioactivity from glucosamine-1-14C. J Cell Physiol. 1967;69(2):199-207
62. Allen A, Snow C. The effect of trypsin or ethylenediaminetetraacetate on the surface of cells in tissue culture. Biochem J. 1970;117(2):32P
63. Snow C, Allen A. The release of radioactive nucleic acids and mucoproteins by trypsin and ethylenediaminetetra-acetate treatment of baby-hamster cells in tissue culture. Biochem J. 1970;119(4):707-14
64. Balashova EE, Lokhov PG. Proteolytically-cleaved fragments of cell-surface proteins from live tumor cells stimulate anti-tumor immune response in vitro. J Carcinogene Mutagene. 2010;1:103
65. Buck CA, Glick MC, Warren L. A comparative study of glycoproteins from the surface of control and Rous sarcoma virus transformed hamster cells. Biochemistry. 1970;9(23):4567-76
66. Vestling MM, Murphy CM, Fenselau C. Recognition of trypsin autolysis products by high-performance liquid chromatography and mass spectrometry. Anal Chem. 1990;62(21):2391-4
67. Shin BK, Wang H, Yim AM, Le Naour F, Brichory F, Jang JH. et al. Global profiling of the cell surface proteome of cancer cells uncovers an abundance of proteins with chaperone function. J Biol Chem. 2003;278(9):7607-16
68. Jang JH, Hanash S. Profiling of the cell surface proteome. Proteomics. 2003;3(10):1947-54
69. Rodriguez-Ortega MJ, Norais N, Bensi G, Liberatori S, Capo S, Mora M. et al. Characterization and identification of vaccine candidate proteins through analysis of the group A Streptococcus surface proteome. Nat Biotechnol. 2006;24(2):191-7
70. Lund R, Leth-Larsen R, Jensen ON, Ditzel HJ. Efficient isolation and quantitative proteomic analysis of cancer cell plasma membrane proteins for identification of metastasis-associated cell surface markers. J Proteome Res. 2009;8(6):3078-90
71. Leth-Larsen R, Lund R, Hansen HV, Laenkholm AV, Tarin D, Jensen ON. et al. Metastasis-related plasma membrane proteins of human breast cancer cells identified by comparative quantitative mass spectrometry. Mol Cell Proteomics. 2009;8(6):1436-49
72. Garcia J, Faca V, Jarzembowski J, Zhang Q, Park J, Hanash S. Comprehensive profiling of the cell surface proteome of Sy5Y neuroblastoma cells yields a subset of proteins associated with tumor differentiation. J Proteome Res. 2009;8(8):3791-6
73. Leth-Larsen R, Lund RR, Ditzel HJ. Plasma membrane proteomics and its application in clinical cancer biomarker discovery. Mol Cell Proteomics. 2010;9(7):1369-82
74. Elschenbroich S, Kim Y, Medin JA, Kislinger T. Isolation of cell surface proteins for mass spectrometry-based proteomics. Expert Rev Proteomics. 2010;7(1):141-54
75. Insenser MR, Hernaez ML, Nombela C, Molina M, Molero G, Gil C. Gel and gel-free proteomics to identify Saccharomyces cerevisiae cell surface proteins. J Proteomics. 2010;73(6):1183-95
76. Lokhov P, Balashova E, Dashtiev M. Cell proteomic footprint. Rapid Commun Mass Spectrom. 2009;23(5):680-2
77. van der Pol L, Tramper J. Shear sensitivity of animal cells from a culture-medium perspective. Trends Biotechnol. 1998;16(8):323-8
78. Lau JY, Cooke P, Tchao R. Increased cell membrane sensitivity to shear force resulting from adherence on an anisotropic polymer surface. Eur Cell Mater. 2003;6(Suppl.1):106
79. Lau JY, Tchao R. Stressed polystyrene causes increased membrane sensitivity of adherent cells to fluid shear force: technical note. Eur Cell Mater. 2007;14:40-4
80. McQueen A, Bailey JE. Influence of serum level, cell line, flow type and viscosity on flow-induced lysis of suspended mammalian cells. Biotechnol Lett. 1989;11:531-6
81. Tchao R. Fluid shear force and turbulence-induced cell death in plastic tissue culture flasks. In Vitro Toxicology. 1996;9(1):93-100
82. Folkman J. Tumor angiogenesis: therapeutic implications. N Engl J Med. 1971;285(21):1182-6
83. Folkman J. What is the evidence that tumors are angiogenesis dependent? J Natl Cancer Inst. 1990;82(1):4-6
84. Ellis LM, Fidler IJ. Angiogenesis and metastasis. Eur J Cancer. 1996;32(14):2451-60
85. Pluda JM. Tumor-associated angiogenesis: mechanisms, clinical implications, and therapeutic strategies. Semin Oncol. 1997;24(2):203-18
86. Liotta LA, Steeg PS, Stetler-Stevenson WG. Cancer metastasis and angiogenesis: an imbalance of positive and negative regulation. Cell. 1991;64(2):327-36
87. Carmeliet P, Jain RK. Angiogenesis in cancer and other diseases. Nature. 2000;407(6801):249-57
88. Pralhad T, Madhusudan S, Rajendrakumar K. Concept, mechanisms and therapeutics of angiogenesis in cancer and other diseases. J Pharm Pharmacol. 2003;55(8):1045-53
89. Ruoslahti E, Rajotte D. An address system in the vasculature of normal tissues and tumors. Annu Rev Immunol. 2000;18:813-27
90. St Croix B, Rago C, Velculescu V, Traverso G, Romans KE, Montgomery E. et al. Genes expressed in human tumor endothelium. Science. 2000;289(5482):1197-202
91. Bhati R, Patterson C, Livasy CA, Fan C, Ketelsen D, Hu Z. et al. Molecular characterization of human breast tumor vascular cells. Am J Pathol. 2008;172(5):1381-90
92. Khodarev NN, Yu J, Labay E, Darga T, Brown CK, Mauceri HJ. et al. Tumour-endothelium interactions in co-culture: coordinated changes of gene expression profiles and phenotypic properties of endothelial cells. J Cell Sci. 2003;116(Pt 6):1013-22
93. Thompson PL, Dessureault S. Tumor cell Vaccines. In: (ed.) Shurin MR, Smolkin YS. Immune-Mediated Diseases: From Theory to Therapy. New York: Springer. 2007:345-55
94. Wei YQ, Wang QR, Zhao X, Yang L, Tian L, Lu Y. et al. Immunotherapy of tumors with xenogeneic endothelial cells as a vaccine. Nat Med. 2000;6(10):1160-6
95. Scappaticci FA, Nolan GP. Induction of anti-tumor immunity in mice using a syngeneic endothelial cell vaccine. Anticancer Res. 2003;23(2B):1165-72
96. Corsini E, Gelati M, Calatozzolo C, Alessandri G, Frigerio S, De Francesco M. et al. Immunotherapy with bovine aortic endothelial cells in subcutaneous and intracerebral glioma models in rats: effects on survival time, tumor growth, and tumor neovascularization. Cancer Immunol Immunother. 2004;53(11):955-62
97. Okaji Y, Tsuno NH, Kitayama J, Saito S, Takahashi T, Kawai K. et al. Vaccination with autologous endothelium inhibits angiogenesis and metastasis of colon cancer through autoimmunity. Cancer Sci. 2004;95(1):85-90
98. Chen XY, Zhang W, Wu S, Bi F, Su YJ, Tan XY. et al. Vaccination with viable human umbilical vein endothelial cells prevents metastatic tumors by attack on tumor vasculature with both cellular and humoral immunity. Clin Cancer Res. 2006;12(19):5834-40
99. Okaji Y, Tsuno NH, Saito S, Yoneyama S, Tanaka M, Nagawa H. et al. Vaccines targeting tumour angiogenesis--a novel strategy for cancer immunotherapy. Eur J Surg Oncol. 2006;32(4):363-70
100. Balashova EE, Lokhov PG. Proteolytically-cleaved fragments of cell surface proteins stimulate a cytotoxic immune response against tumor-activated endothelial cells in vitro. J Cancer Sci Ther. 2010;2(5):126-31
101. O'Brien SJ. Cell culture forensics. Proc Natl Acad Sci U S A. 2001;98(14):7656-8
102. Stacey GN. Cell contamination leads to inaccurate data: we must take action now. Nature. 2000;403(6768):356
103. MacLeod RA, Dirks WG, Matsuo Y, Kaufmann M, Milch H, Drexler HG. Widespread intraspecies cross-contamination of human tumor cell lines arising at source. Int J Cancer. 1999;83(4):555-63
104. Cabrera CM, Cobo F, Nieto A, Cortes JL, Montes RM, Catalina P. et al. Identity tests: determination of cell line cross-contamination. Cytotechnology. 2006;51(2):45-50
105. Identity crisis. Nature. 2009;457(7232):935-6
106. Findlay I, Taylor A, Quirke P, Frazier R, Urquhart A. DNA fingerprinting from single cells. Nature. 1997;389(6651):555-6
107. Hayflick L, Moorhead PS. The serial cultivation of human diploid cell strains. Exp Cell Res. 1961;25:585-621
108. Bodnar AG, Ouellette M, Frolkis M, Holt SE, Chiu CP, Morin GB. et al. Extension of life-span by introduction of telomerase into normal human cells. Science. 1998;279(5349):349-52
109. Wright WE, Shay JW. Historical claims and current interpretations of replicative aging. Nat Biotechnol. 2002;20(7):682-8
110. Huggins JW, Chesnut RW, Durham NN, Carraway K. Molecular changes in cell surface membranes resulting from trypsinization of sarcoma 180 tumor cells. Biochim Biophys Acta. 1976;426(4):630-7
111. Angus R, Collins CM, Symes MO. Expression of major histocompatibility complex (MHC) antigens and their loss on culture in renal carcinoma. Eur J Cancer. 1993;29(15):2158-60
112. Orth K, Hung J, Gazdar A, Bowcock A, Mathis JM, Sambrook J. Genetic instability in human ovarian cancer cell lines. Proc Natl Acad Sci U S A. 1994;91(20):9495-9
113. Hiorns LR, Bradshaw TD, Skelton LA, Yu Q, Kelland LR, Leyland-Jones B. Variation in RNA expression and genomic DNA content acquired during cell culture. Br J Cancer. 2004;90(2):476-82
114. Bertozzi CC, Chang CY, Jairaj S, Shan X, Huang J, Weber BL. et al. Multiple initial culture conditions enhance the establishment of cell lines from primary ovarian cancer specimens. In Vitro Cell Dev Biol Anim. 2006;42(3-4):58-62
115. Trial watch: Progress for Phase III cancer vaccines. Nat Rev Drug Discov. 2008;7(12):966-7
116. Levine MM, Sztein MB. Vaccine development strategies for improving immunization: the role of modern immunology. Nat Immunol. 2004;5(5):460-4
117. Emens LA. Roadmap to a better therapeutic tumor vaccine. Int Rev Immunol. 2006;25(5-6):415-43
118. Improving cellular cancer vaccines; Nature Precedings 2010. Lokhov PG. http://hdl.handle.net/10101/npre.2010.4356.1
119. Haugen A. Ruthenium red binding to cell coat in neoplastic neurogenic cell lines in culture. Virchows Arch B Cell Pathol. 1979;29(4):245-52
Author contact
![]() Corresponding author: Petr G. Lokhov, phone: +7 903 744 51 91, fax: +7 495 143 22 77; e-mail: lokhovpgru
Corresponding author: Petr G. Lokhov, phone: +7 903 744 51 91, fax: +7 495 143 22 77; e-mail: lokhovpgru