Impact Factor ISSN: 1837-9664

 Global reach, higher impact
Global reach, higher impactJ Cancer 2011; 2:36-51. doi:10.7150/jca.2.36 This volume Cite
Research Paper
Effects of an Indolocarbazole-Derived CDK4 Inhibitor on Breast Cancer Cells
Yuan Sun1,4, Ying-xia Li2 ![]() , Hai-jun Wu2, Si-hung Wu1, Y. Alan Wang3, Dian-zhong Luo4, D. Joshua Liao1
, Hai-jun Wu2, Si-hung Wu1, Y. Alan Wang3, Dian-zhong Luo4, D. Joshua Liao1 ![]()
1. Hormel Institute, University of Minnesota, Austin, MN 55912, USA
2. School of Pharmacy, Fudan University, Pudong, Shanghai, 201203, P.R. China
3. Discovery Center for Applied Cancer Science, Dana-Farber Cancer Institute, Harvard University, Boston, MA 02115, USA
4. Department of Pathology, Guanxi Medical University, Nanning, Guangxi, 530021, P.R.China
Received 2010-10-24; Accepted 2011-1-6; Published 2011-1-8
Abstract
Introduction: Cyclin D1 (D1) binds to cyclin-dependent kinases (CDK) 4 or 6 to form a holoenzyme that phosphorylates the Rb protein to promote cell cycle progression from G1 to S phase. Therefore, targeting CDK4/6 may be a good strategy for chemotherapy of cancer. We performed a proof-of-principle study to determine the effect of Naphtho [2, 1-α] pyrrolo [3, 4-c] carbazole-5, 7 (6H, 12H)-dione (NPCD), a novel CDK4 inhibitor, on breast cancer cell lines.
Methods: NPCD was synthesized and purified to over 99% purity verified by HPLC. MCF7, MB231, MCF15, T47D and GI101Ap human breast cancer cells were analyzed for the efficacy of NPCD with MTT and clonogenic assays, with FACS and staining for ethidium bromide and acridine orange for cell death and cell cycle profile. Western blot, reverse transcription and PCR were used for studies of gene expression, and co-immunoprecipitation for protein-complex formation.
Results: MTT assay showed that NPCD caused growth arrest and apoptosis of MCF7, MDA-MB231, T47D, MCF15 and GI101Ap cells with an IC50 ranging between 3 to 8 µM given as a single dose. The growth arrest persisted for many days after cessation of the treatment, as shown in a clonogenic assay. NPCD could induce or reduce the D1 and CDK4 protein levels, depending on the cell line, but this effect was not correlated with its efficacy. Phosphorylation of D1 at Thr286 was decreased but it unexpectedly did not correlate with the change in D1 level in the cell lines studied. Phosphorylation of the Rb protein was decreased as expected whereas the p27kip1 protein level was decreased unexpectedly. Protein levels of p21cip1, CDK2 and cyclin E were also decreased in some, but not all, of the cell lines, whereas the mRNA levels of D1, CDK4, cyclin E, CDK2, p27kip1 and p21cip1 were increased in different cell lines.
Conclusions: NPCD can cause long-lasting growth arrest and cell death of breast cancer cell lines at an IC50 of 3-8 µM. Decreased phosphorylation of Rb by D1-CDK4/6 and decreased p27kip1 protein level may be part of the underlying mechanism.
Keywords: NPCD, CDK4 Inhibitor, breast cancer cell lines
Introduction
D-type cyclins, including D1, D2 and D3, are usually induced by many growth factors and oncoproteins such as Ras, epidermal growth factor (EGF), etc [1]. D-type cyclins relay the extracellular mitogenic signals of these factors to the cell cycle machinery and drive cell cycle progression from G1 to S phase [2]. Once the cells pass the late stage of G1 phase and are about to enter S phase, the cells can complete the cell cycle without any need for further stimulus, although additional stimuli may still further accelerate completion of the ongoing cell cycle. For this reason, D-type cyclins, mainly D1, are regarded as mitogenic sensors of cells. This unique trait constitutes an important foundation for targeting D1 for cancer therapy.
D1 is amplified in about 15% of breast cancer cases in general, but in about 21% of the estrogen receptor alpha positive cases [3]. D1 amplification also occurs at high frequencies in lymphomas [4] and other malignancies, as reviewed by us [1]. It goes without saying that D1 overexpression occurs at even higher frequencies in cancers, close to 50% in breast cancer as reported in some studies [5]. D1 overexpression also distinguishes invasive and in situ breast carcinomas from non-malignant lesions [6]. Besides these alterations shared by other malignancies, D1 is exceptionally important for the mammary gland and breast cancer for two specific reasons: 1) it is the only D type cyclin expressed in the mammary gland and controls mammary gland development [7]; 2) it is a major downstream effector of estrogen receptors [8].
D1 associates with cyclin-dependent-kinase CDK4 or CDK6 to form an active holoenzyme to phosphorylate and thus inactivate Rb. Inactivated Rb releases E2F proteins to exert transcriptional activities of genes related to cell cycle progression. Although D1 also has other functions independent of CDK4 or CDK6, its activation of CDK4/6 is still considered the major mechanism for D1's oncogenic functions. Therefore, it is conceivable that inhibition of CDK4/6 kinase activity toward the Rb protein should block the D1's oncogenic functions. Based on this rationale, many investigators consider targeting D1 or its partner CDKs as a strategy for cancer treatment [9-16]. Chemical inhibitors of D1 [17-25] or CDK4 [26-36] have been developed as candidates for chemotherapeutic agents [26,37-43]. Of the CDK4 inhibitors, PD0332991 developed by Pfizer Global Research and Development as a pyridopyrimidine derivative [41,44] shows promising efficacy on several types of cancer and has already entered clinical trials [45,46]. A recent study shows that PD0332991 has good efficacy on a panel of 44 human breast cancer cell lines, with a higher sensitivity on those cells expressing higher levels of pRb or D1 proteins or lower levels of the p16 cdk4/6 inhibitor [47].
Several indolocarbazole analogs have also been developed as very potent CDK4 inhibitors [29-31,33,35,36,39,40]. For instance, naphyl[a]pyrolo[3,4-c]carbazole-5,7(6H, 12,H)-dione (NPCD) and isoquinolinyl[a]pyrrolo[34-c]carbazole (IQPC) have an IC50 on CDK4 at a range of 45-75 nM while affecting other CDKs, such as CDK2, at much higher concentrations [31,35]. However, the efficacy of these indolocarbazole analogs on cancers is unknown so far. Moreover, little is known about the molecular pathways triggered by CDK4 inhibitors in general and by indolocarbazole-derivatives in particular. The indolocarbazoles are shown to be selective G1 blockers while their bis-indolylmaleimide precursors arrest cells in the G2/M phase [31,48]. One of these compounds, 2-bromo-12,13-dihydro-indolo[2,3-a]pyrrolo[3,4-c]carbazole-5,7(6H)-dione (219476 from Calbiochem), has been shown to be able to degrade IκB and induce translocation of RelA from the cytoplasm to the nucleoplasm and then to the nucleolus, leading to suppression of NF-κB activity as a collective result [49,50].
In the present study, we determine the in vitro effect of NPCD in human breast cancer cell lines as a proof-of-principle study for targeting D1-CDK4 as a strategy for cancer therapy. We observe that NPCD arrests cells at G1 phase and causes apoptosis with an IC50 of 3-8 µM when administered as a single dose. These effects are surprisingly long-lasting as determined by a clonogenic survival assay and are associated with an unexpected decrease in p27kip1 and p21cip1 proteins. Phosphorylation of the Rb protein is decreased as expected, whereas D1 and CDK4 protein levels are reduced in some cell lines but induced in others, which has not been reported for other CDK4 inhibitory compounds such as PD0332991. To our knowledge this study is the first comprehensive one on NPCD in a panel of breast cancer cell lines that have different features of tumor biology and thus the data presented herein are novel.
Materials and Methods
Cell lines: MCF15 is a new human breast cancer cell line [51]. GI101Ap is a clone of GI101A cells kindly provided by Dr. Janet E. Price at the MD Anderson Cancer Center, the University of Texas, Houston. MCF10A cells were obtained from Karmanos Cancer Center (Detroit, Michigan), which is the former Michigan Cancer Foundation that developed the MCF series of cells, whereas MCF7, T47D and MDA-MB231 (MB231) human breast cancer cell lines were originally obtained from the American Type Culture Collection (ATCC). Among these cell lines, MCF7, MCF15 and T47D cells are estrogen receptor (ER) α negative in culture, T47D and MB231 cells have p53 mutations, whereas the ERα negative MB231 and ERα positive GI101Ap are two most commonly used metastatic cell lines, after MDA-MB435 is recently characterized to be of melanoma origin [52]. Moreover, MB231 cells are so-called triple-negative (estrogen receptor, progesterone receptor and Her-2 negative).
Reagents: Naphtho [2, 1-α] pyrrolo [3, 4-c] carbazole-5, 7 (6H, 12H)-dione (NPCD) (fig 1) was synthesized and purified by us at a purity of over 99% proved by HPLC. It was dissolved with dimethyl sulfoxide (DMSO) and kept at -20oC until use. Unless specified separately, primary antibodies used in this study were purchased from Santa Cruz Biotechnology Inc (Santa Cruz, CA), including mouse monoclonal anti-β-actin (sc-47778) as well as rabbit polyclonal anti-cyclin D1 (sc-718), anti-CDK4 (sc-260), anti-CDK6 (sc-7180), anti-CDK2 (sc-163), anti-cyclin E (sc-481), anti-Rb (sc-50), anti-pRb (Ser807/811; sc-16670), anti-p27 (sc-528), and anti-p21 (sc-397). The anti-pRb Ser780 (Cat#9307) rabbit polyclonal antibody was purchased from Cell Signaling Technology, Inc (Danvers, MA). Peroxidase-conjugated anti-mouse (NA931) and anti-rabbit (NA934) secondary antibodies were purchased from Amersham Biosciences (Piscataway, NJ).
Chemical structure of Naphtho [2, 1-α] pyrrolo [3, 4-c] carbazole-5, 7 (6H, 12H)-dione (NPCD).
3-(4, 5-Dimethylthiazol-2-yl)-2, 5-diphenyltetrazolium bromide (MTT) assay: Cells were seeded in a 96-well micro-plate at 4,000 cells per well, five wells per dose and per time point, and incubated at 37oC with 5% CO2. NPCD was added 24 hours later at indicated concentrations, with DMSO as non-treated control. The culture was continued for the indicated time period. At the end of the treatment, MTT was added into each well at a final concentration of 0.5mg/ml followed by incubation at 37oC for three hours in dark. The culture medium containing MTT was discarded and the dye crystals were dissolved in DMSO. The viable cells were detected by reading the absorbance of the metabolic MTT at wavelength 570nm using the Beckman Coulter AD340 absorbance detector (Beckman Coulter Inc, Fullerton, CA). The experiment was repeated at least three times to ensure the data reproducibility.
Clonogenic survival assay: Cells were seeded in 24-well culture plates at 1x105 cells per well and 24 hours later NPCD was added into the culture, with DMSO as non-treated control. The cells were trypsinized 72 hours after the NPCD treatment and evenly reseeded at a lower density (1x103 cells per well) in triplicate in a 6-well culture plate. Cells were maintained in culture for 10 to 15 days, with medium change every 3 days, to let the viable cells propagate to sizable colonies for quantification. The colonies were fixed with methanol-acetic acid at 3:1 ratio and then stained with 1% crystal violet for 30 minutes at room temperature. The number of colonies formed in each well was counted and photographed under the microscope. The experiment was repeated three times and the resulting data are presented after statistical processing.
Acridine orange and ethidium bromide staining: Cells were seeded in 96-well plates at 3,000 cells per well and incubated at 37oC with 5% CO2. NPCD at a final concentration of 2 or 4 µM was added 24 hours later in multiple repetitions (5 wells per dose); plates were incubated for an additional 24 or 48 hours. Prior to cell staining, the medium was discarded. The cells were washed with phosphate-buffered saline (PBS) first and then incubated with 10 µl PBS containing 10 µg/ml of Ethidium Bromide (EB) and 3 µg/ml of Acridine orange (AO). The cells stained by EB and AO were immediately visualized under the Leica DM IRB inverted fluorescence microscope (Leica Microsystems Inc. Bannockburn, IL). Multiple photos were taken at randomly-selected areas of the well to ensure that the data obtained are representative. Photographs were processed using “Image ProPlus 6.0” ultimate image analysis software (Media Cybernetics, Inc. Bethesda, MD).
Cell cycle analysis: Cells were cultured in 6-cm culture dishes until they reached 70~80% confluence. NPCD was given at an indicated concentration with DMSO as non-treated control; the cells were harvested 36 or 48 hours later. At the cessation of the treatment, both floating and adherent cells were collected, combined, washed with cold PBS, and then fixed overnight with 70% ethanol in PBS at -20oC. The cells were then washed with PBS again and incubated in the dark with in a PBS solution containing 20µg/ml propidium iodide (PI) and 200µg/ml RNase for 30 minutes at room temperature. The cells at different stages of the cell cycle were detected with a Becton Dickinson FACS Calibur flow cytometer (BD Biosciences, San Jose, CA). Intact cells were gated in the FSC/SSC plot to exclude small debris. The population of cells at different stages of the cell cycle was quantified using ModFit LT software (Verity Software House, Inc., Topsham, ME).
Cell death analysis: Annexin V and PI dual staining was used to visualize apoptotic and necrotic cells in a similar procedure as described above but the cells were not pre-fixed with 70% ethanol. Apoptotic cells were stained using the Annexin V-FITC Apoptosis Detection Kit (MBL International Corporation, Watertown, MA) following the manufacturer's instruction. All dead cells, i.e. both apoptotic and necrotic cells stained by Annexin V and PI, were analyzed by a Becton Dickinson FACS-Calibur flow cytometer (BD Biosciences, San Jose, CA), which places the Annexin V-FITC signal that detects apoptosis in FL1 and the PI-FITC signal that detects necrosis in FL2. Therefore, cells in the upper-left quadrant of the FL1/FL2 dot plot (labeled with PI-FITC only) as shown in figure 3 were necrotic, whereas cells in the bottom-right quadrant were early apoptotic. Cells in the upper-right quadrant (labeled with Annexin V-FITC and PI) were late apoptotic or necrotic.
Protein extraction and western blotting assay: Total protein samples or nuclear and cytoplasmic proteins were extracted as described by Andrew and Faller [53], followed by determination of protein concentration using a Bradford assay (Bio-Rad Laboratories Inc. Hercules, CA). An equal amount of protein from each sample was fractioned in SDS-PAGE and then transferred onto an Immobilon-P Nylon membrane (Millipore, Bedford, MA) in a tank transfer system. After being blocked with 5% milk, the membrane was incubated with specific primary antibody at an optimized concentration and then with horseradish peroxidase-conjugated secondary antibody, with three washes between each antibody. The signal was visualized with ECL chemiluminescent substrates (Pierce, Rockford, IL) on X-ray film (ISC BioExpress, Kaysville, UT). The expression levels were quantified as the band density on X-film using ImageJ software, calculated as the ratio to the β-actin expressed in the same sample, and presented as number of “+”. +++, ++ and + indicate that the ratio to β-actin is larger than 0.75, between 0.5~0.75, and less then 0.5, respectively, whereas ± and (-) indicate a very low level of expression and the absence of the signal on the X-film, respectively. The Rb protein was used as a loading control for nuclear proteins in some of the western blot assays as reported in the literature [54,55].
Co-immunoprecipitation assay: D1-CDK4 complex formation was analyzed with co-immunoprecipitation assay. Total protein lysates (500µg) from each sample were immunoprecipitated (IP) in 400µl lysate buffer containing 2µl anti-cyclin D1 (sc-718) or anti-CDK4 (sc-260) rabbit polyclonal antibody and inhibitors of various proteases, phosphotases and kinases at 4oC for 4 hours with rotation. Protein A-conjugated agarose beads (25 µl) were then added into the IP reaction with an additional 5 hours of rotation at 4oC. The antigen-antibody complexes were precipitated by a quick centrifugation, followed by four times of wash with cold PBS. Proper controls included an aliquot of rabbit serum to replace the D1 or CDK4 antibody in the IP reaction. The pellets were suspended in 20µl 2xSDS reducing western blot loading buffer and boiled for 5 minutes, followed by SDS-PAGE. The D1- or CDK4-immunoprecipitates were subjected to western blot assay to detect the D1 or CDK4 in the immunoprecipitates.
RT-PCR assay: Total RNA samples were extracted from cultured cells using TRIzol (Invitrogen, Cat. 15596-026) in a routine procedure, followed by DNaseI treatment to get rid of DNA residual. An aliquot (2.5µg) of the RNA samples was then reverse transcribed (RT) in a 25µl reaction solution to the first strand of cDNA using hexamer primer. The RT products were diluted with water to the final volume of 50 µl. For PCR amplification of each target gene, 1 µl of the diluted RT products was used as template, but the template was further diluted 10 times for PCR amplification of β-actin so as to control the PCR amplification of this highly expressed gene at the linear portion. The PCR condition (table 1) was optimized for each gene and was stopped within the linear portion of the amplification. The forward and reverse primers for each gene, listed in table 1, were designed in such a way that they are localized at two different exons of the given gene with one or several large introns in between. In this way if there still is a traceable amount of DNA residual, it either cannot be amplified due to large intron(s) or is amplified as a molecule larger than the expected size indicated in table 1. PCR products were separated and visualized in 1% agarose gel.
Primer sequences and PCR amplification conditions used in RT-PCR assay.

Statistic analyses: All MTT, FACS and clonogenic survival assays were performed in multiple repetitions in each experiment, and the experiments were repeated at least three times. The data were presented as mean ± SE. Statistical comparisons between groups were made with the student's t-test. A P value < 0.05 is considered as significant.
Results
Effect of NPCD on cell survival
MCF7, MB231, MCF15, T47D and GI101Ap cells were treated once with NPCD at escalating concentrations (1-10 μM), and the number of viable cells was determined with an MTT assay 48 or 72 hours later. The results showed that MCF7 and MCF15 cells were the most sensitive to NPCD-induced decrease in cell viability whereas GI101Ap and T47D cells were less sensitive. The sensitivity of MB231 cells to NPCD was intermediate. Nevertheless, the IC50s for all five cancer cell lines 72 hours after a single administration were close, ranging between 3-8 µM (table 2). The response of MCF10A non-transformed human breast epithelial cells was slightly less sensitive than most of these cancer cells, with IC50 between 8-10 µM (table 2).
IC50 of NPCD on five breast cancer cell lines and MCF10A non-transformed cells

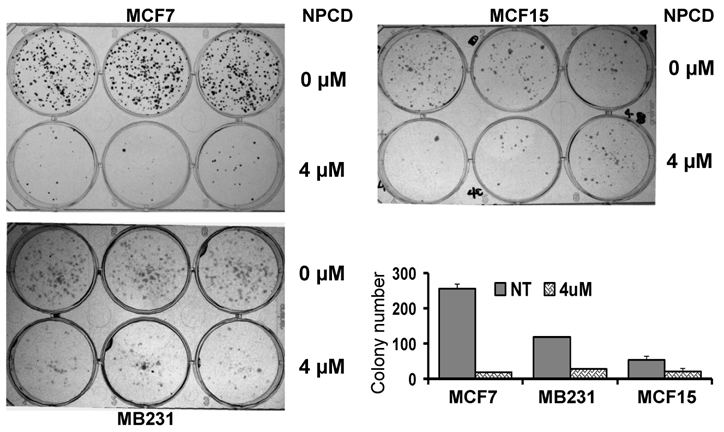
A decrease in the number of viable cells detected by MTT may be attributable to decreased cell proliferation, increased cell death, or both. To determine which of these two elements is the main mechanism, MCF7, MB231 and MCF15, the three more sensitive cell lines, were treated with 4 μM of NPCD and then subjected to a clonogenic survival assay. The number of colonies developed 10-15 days after NPCD treatment was quantified and calculated as a percentage of the corresponding non-treated control. This was used as the survival rate, which was 7.48±1% for MCF7, 23.59±0.2% for MB231, and 38.00±6% for MCF15 cells, respectively (fig 2). The photos of colonies of treated or non-treated cells from a representative experiment are also shown in figure 2. Interestingly, not only the number, but also the size, of the colonies in NPCD-treated groups was decreased, especially in MCF7 cells. Because some colonies in treated groups were too small to appear in the macroscopic photos shown in figure 2 but could be quantified under the microscope, the difference in quantitative data between the treated and non-treated groups seemed to be smaller than what was reflected by the photos, especially in MCF7 cells. Because the clonogenic assay was terminated 10-15 days after cessation of the treatment and because a smaller colony size indicates fewer cells propagated from a single survival cell in a given time period, this result indicates a persistent or long-lasting growth arrest caused by NPCD.
Results of colony survival assay. Colonies developed from MCF7, MB231 or MCF15 cells 10-15 days after being treated with 4µM NPCD, or untreated, in a representative experiment were shown, with each cell line in three wells of the 6-well culture plate. Quantitative data from three different experiments were presented in the histogram.
Effects of NPCD on cell cycle and apoptotic profiles
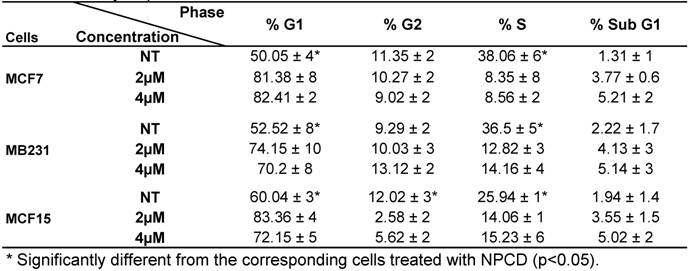
Analysis of cell cycle profile with FACS revealed that NPCD treatment of MCF7, MB231 and MCF15 cells at relatively low concentrations (2-4 μM) for 36 hours significantly arrested the cells at G1 phase, as evidenced by an increased percentage of the cells at G1 phase and a corresponding decrease in the percentage of the cells at S phase (table 3). MCF15 cells also showed a decrease in the cells at G2 phase (table 3). Apoptotic cells at the sub-G1 phase were also slightly increased by these low concentrations of NPCD at this early time point.
Cell cycle profile of breast cancer cells treated with NPCD
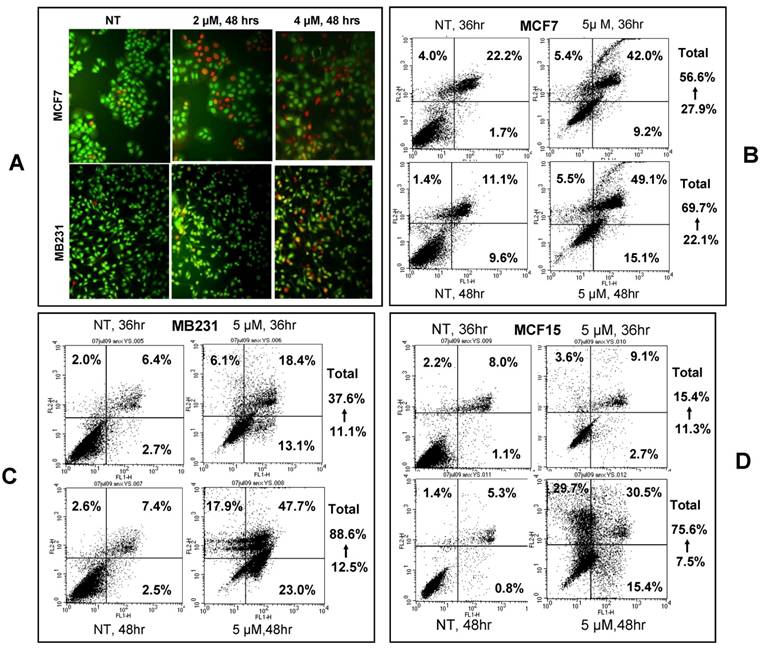
Apoptotic cells were visualized by acridine orange/ethidium bromide staining upon treatment of MCF7 and MB231 cells with 2 or 4 μM NPCD for 48 hours (fig 3A). We also used annexin V/PI staining to identify the dead cells and stratified them into categories of necrosis, early apoptosis, and both of later apoptosis and necrosis. In each graph of flow cytometry (fig 3B-D), necrosis is shown in the upper-left guardant, early apoptotic cells in the bottom-right quadrant, and later apoptotic or necrotic cells in the upper-right quadrant. The total death rate of all three fractions 36 and 48 hours after NPCD treatment showed 2- (56.6 vs 27.9%) and 3- (69.7 vs 22.1%) fold increase, respectively, for MCF7 cells, and 3- (37.6 vs 11.1%) and 7- (88.6 vs 12.5%) fold increase, respectively, for MB231 cells. Interestingly, the death rate of MCF15 cells was not changed (15.4 vs 11.3%) at 36 hours but it showed a 10-fold (75.6 vs 7.5%) increase at 48 hours. It seems that the effect occurs slightly later in MCF15 cells since MCF7 and MB231 cells had already shown an obvious increase in cell death at 36 hours. In line with these data, we observed many floating cells and cell debris in the culture dishes after NPCD treatment, which confirms the occurrence of cell death.
NPCD induced death of MCF7, MB231 and MCF15 cells. A): Acridine orange/ethidium bromide staining shows viable cells in green fluorescence and dead cells in red-orange fluorescence. B, C and D): Necrotic (upper-left quadrant), early apoptotic (bottom-right quadrant) and both late apoptotic and necrotic (upper-right quadrant) fractions of MCF7 (B), MB231 (C) and MCF15 (D) cells 36 or 48 hours after treatment with 5 µM NPCD.
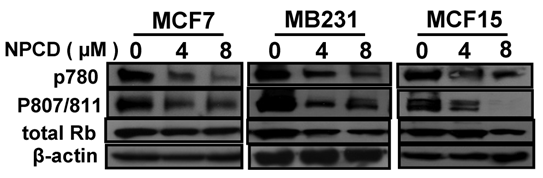
Inhibition of Rb phosphorylation by NPCD
D1-CDK4 and D1-CDK6 are known to phosphorylate the Rb protein at ser780, ser807 and ser811 [56,57]. Use of antibodies that specifically recognize these phosphorylation sites in western blot showed that the amount of Rb phosphorylated at these sites was indeed decreased upon treatment of MCF7, MB231 and MCF15 cells with NPCD at 4 or 8 μM for 48 hours (fig 4) and 72 hours (data not shown). The total amount of Rb protein was also slightly decreased in MB231 and MCF15 cells treated with the high dose of NPCD (fig 4), which may in part contribute to the decrease in the phosphorylated fraction of the Rb in these cells.
Phosphorylation of the Rb protein at ser780 or ser807/ser811 in MCF7, MB231 or MCF15 cells 48 hours after treatment with 4 or 8µM NPCD, with β-actin as loading control.
Effects of NPCD on the protein levels of G1 phase regulators
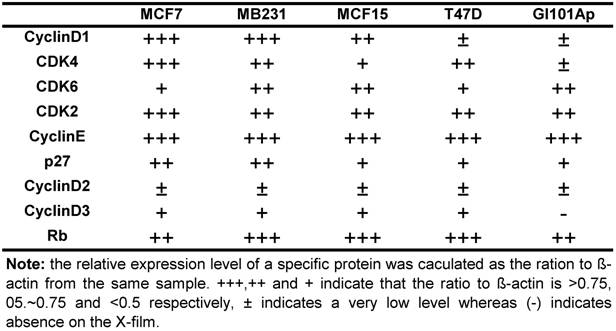
Because NPCD was developed as a CDK4 inhibitor, we wondered whether its efficacy is affected by the levels of D1 and/or CDK4. We thus conducted western blot using cell lysates of the five breast cancer cell lines to determine the protein levels of D1, D2, D3, CDK4, CDK6, CDK2, p27kip1 (p27) and p16ink4a. Western blot for each protein was repeated several times. The protein levels were quantified as the band density on X-ray-film, calculated as the ratio to β-actin expressed in the same sample, and presented as number of “+” with more “+” to indicate a higher expression level (table 4). D2 and D3 proteins were expressed at very low levels in these cells as expected, whereas p16ink4a was basically undetectable in these cell lines and thus not presented. The protein levels of D1, CDK4 and CDK6 were lower in T47D and GI101Ap cells that were relatively resistant to NPCD, suggesting a possibility that NPCD might be more effective in the cells that express a relatively higher level of these proteins, which is in line with the observation of Finn et al that breast cancer cell lines with higher D1 levels are more sensitive to PD032991 [47]. Considering that the Rb protein is the ultimate target of the G1 regulatory proteins, we also determined the total Rb protein level in the cell lines studied, and found that it varied only slightly among the cells (table 4).
Relative expression level of several G1 regulatory proteins in five human breast cancer cell lines *
Treatment with 4 or 8 μM NPCD for 72 hours significantly decreased the D1 protein levels in MCF7 and MCF15 cells (fig 5). In contrast, the D1 protein level was significantly increased by the treatment at 48 and 72 hours in MB231 cells (fig 5). NPCD treatment decreased the CDK4 protein level in MCF7 and MCF15 cells at both time points, whereas it did not obviously affect CDK4 in MB231 cells (fig 5). CDK6 protein was decreased in MCF15 cells but did not show obvious change in MCF7 and MB231 cells (fig 5). P27 protein level was dramatically decreased at 48 hours in all three MCF7, MB231 and MCF15 cells. P21 protein level was decreased in MCF7 and MCF15 cells at 72 hours, but it was not obviously changed in MB231 cells.
Western blot assay detects protein levels of cyclin D1 (D1), CDK4, CDK6, p27kip1, p21cip1, CDK2 and cyclin E in MCF7, MB231 and MCF15 cells 48 or 72 hours after treated treatment with 4 or 8 µM NPCD, compared with the untreated (0 µM) counterparts. β-actin is included as loading control.
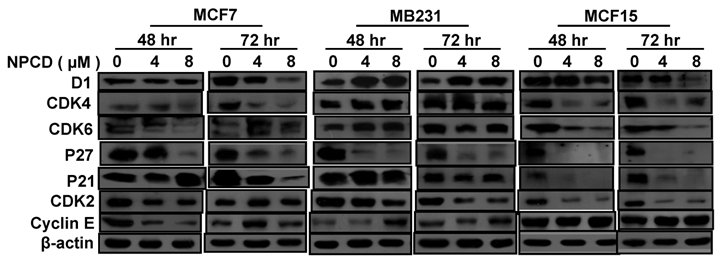
NPCD also inhibited cyclin E-CDK2 with an ID50 at 270 nM, which is much higher than the IC50 at 45 nM for the inhibition of D1-CDK4 [30,31,35]. In most occasions the levels of cyclin E and CDK2 proteins seemed to be changed in a reciprocal manner in response to NPCD treatment, i.e. an increase in cyclin E might be accompanied by a decrease in CDK2 and vice versa. In MB231 cells, CDK2 was decreased at both time points whereas cyclin E protein level was slightly increased at the high dose group. Although CDK2 levels in MCF15 cells were low and were decreased by NPCD treatment, cyclin E levels were higher in this cell line, even after NPCD treatment (fig 5). However, the reciprocal pattern was not obvious in MCF7 cells, in which NPCD did not obviously affect CDK2 while it caused a slight bi-phase change in cyclin E, i.e. an initial decrease at 48 hours followed by a recovery to normal or even a slight increase at 72 hours, especially in the low dose group (fig 5).
Cellular location of D1-CDK4 and cyclin E-CDK2 and Phosphorylation of D1 at Thr286
A major mechanism controlling D1 protein level is its phosphorylation at Thr286 by GSK-3β in the nucleus, followed by nuclear export and proteasomal degradation in the cytoplasm. Because MCF7 and MB231 cells show the opposite change in D1 levels, we further studied whether this difference is mechanistically due to cellular location and D1 phosphorylation at Thr286 after NPCD treatment for 48 hours. In MCF7 cells, NPCD significantly decreased the Thr286 phosphorylation in both nuclear and cytoplasmic fractions at high dose (8 µM) (fig 6A). This result seems to be discrepant with the observations that the level of total D1 protein in either the nucleus or the cytoplasm remains unchanged (fig 6A) or that the total D1 protein was slightly decreased at the same time point as seen in figure 5, since decreased Thr286 phosphorylation should be associated with an increase in D1 level. In the high dose group of MCF7 cells, CDK4 protein was decreased in both cytoplasmic and nuclear fractions (fig 6A). Cytoplasmic cyclin E in NPCD-treated MCF7 cells was decreased in both low- and high-dose groups while nuclear cyclin E did not show any obvious change (fig 6A). CDK2 in either the nucleus or the cytoplasm did not show any consistent change in NPCD-treated MCF7 cells.
Location of, and complex formation between, cyclin D1 (D1) and CDK4. A): Western blot assay detects protein levels of CDK4, total D1, Thr286 phosphorylated D1, CDK2 and cyclin E in the nuclear or cytoplasmic fraction of MCF7 or MB231 cells 48 hours after treatment with 4 or 8 µM NPCD, with the non-treated (0) cells as control. The Rb protein [86,87] and β-actin are included as the loading control for the nuclear and the cytoplasmic proteins, respectively. B): Co-immunoprecipitation assay detects the D1-CDK4 complex. Total cell lysates were prepared from MCF7, MB231 and MCF15 cells 48 or 72 hours after treatment with 4 or 8 µM NPCD. The protein lysates were first immunoprecipitated with D1 antibody, and the D1 immunoprecipitates were then fractioned in polyacrilamide gel and subjected to a western blot assay for detection of D1 or CDK4 as indicated. The CDK4 detected in the D1 immunoprecipitates is considered to be associated with D1.
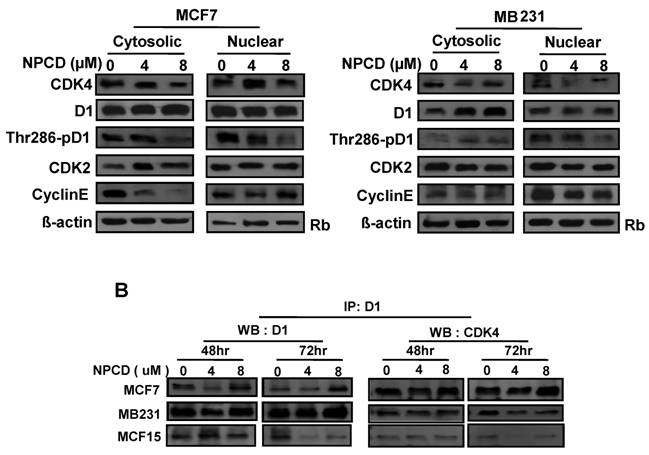
In MB231 cells, the levels of D1 and CDK4 proteins in either the nucleus or the cytoplasm did not show a consistent change. However, Thr286 phosphorylation was significantly decreased in the nuclear fraction by 8µM NPCD. Nuclear cyclin E seemed to be slightly decreased by NPCD while cytoplasmic cyclin E was not obviously affected (fig 6A). CDK2 did not show significant change in either fraction.
NPCD was designed to inhibit CDK4 activity by intervening in the ATP binding, not D1 binding [30,31]. Results from co-immunoprecipitation assay showed that NPCD did not obviously change the abundance of D1-CDK4 complex in MCF7, MB231 or MCF15 cells. A decreased D1 level was detected by the D1 immunoprecipitation and a corresponding decrease in D1-associated CDK4 protein (fig 6B) was also detected in these cells, especially in MCF15 cells, which are likely due to the decrease in total D1 protein as shown in fig 5. These results confirm that NPCD indeed does not act via intervening in the D1-CDK4 binding.
Changes in mRNA levels of G1 phase regulators upon NPCD treatment
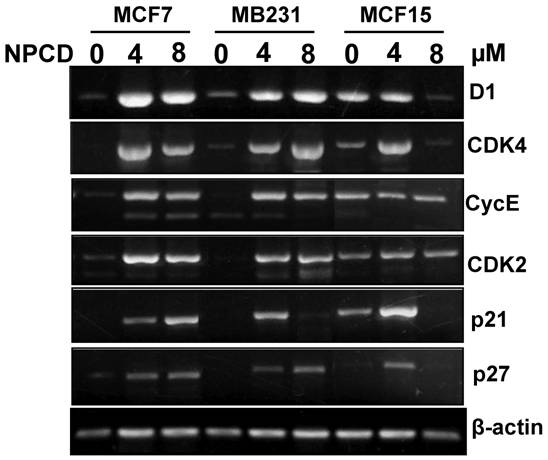
Because D1, CDK4, CDK2, cyclin E, p21cip1 and p27kip1 showed changes at the protein levels upon NPCD treatment, we performed RT-PCR to determine their mRNA levels in the cells treated with a low (4μM) or a high (8μM) concentration of NPCD for 72 hours. In the low dose group, the D1 mRNA level was induced in MCF7, MB231 and MCF15 cells, compared with the non-treated counterparts (fig 7). However, in the high dose group the increase in D1 level continued in MB231 cells but it was slightly leveled off in MCF7 and MCF15 cells (fig 7). CDK4 mRNA level was induced by NPCD in all three cell lines in a way similar to that of D1 (fig 7). The mRNA levels of cyclin E and CDK2 were induced by NPCD in MCF7 and MB231 cells but not in MCF15 cells (fig 7). P21cip1 was induced in MCF7 cells, and the induction was even higher at the high dose group. P21cip1 was also induced in MB231 and MCF15 cells, but the induction occurred only at the low dose group. P27kip1 was induced by both low and high doses of NPCD in both MCF7 and MB231 cells, but in MCF15 cells the induction occurred only in the low dose group.
RT-PCR detection of mRNA levels of different genes in MCF7, MB231 and MCF15 cells 72 hours after treatment with 4 or 8 µM NPCD or without the treatment. Note that the β-actin included as the loading control also shows a slight difference in some of the samples, which should be taken into account when a change in the expression of the desired gene is evaluated.
Discussion
Many oncoproteins and growth factors utilize D1 as a common downstream effector. In other words, D1 sensitizes the cells to various extracellular mitogenic stimuli, such as estrogen and estrogen receptors in the case of breast cancer cells. Much of the D1's role in cell cycle progression is exerted via binding to CDK4 or CDK6 to form a holoenzyme, which phosphorylates the pRb protein to drive cell proliferation. Therefore, inhibition of CDK4 or CDK6 is presumably a good strategy for cancer therapy because cancers are characterized by uncontrolled proliferation and frequently by increased D1-CDK4 activities [58,59]. We use NPCD, a novel indolocarbazole-derived inhibitor of CDK4 and CDK6, to test this therapeutic principle and explore the pathways downstream of the CDK4 inhibition. This test is needed because PD0332991 is so far the only CDK4/6 inhibitor that has entered into clinical trials for its chemotherapeutic potential. Our results show that NPCD, which has not been studied for its effect in cells, can indeed arrest breast cancer cell lines at G1 phase and cause cell death with IC50 around 3-8 µM when it is administered as a single, one-time, dose. Although a stronger potency is conceivable if multiple administrations were given to maintain the NPCD level otherwise metabolically decreased, the IC50 at 3-8 µM for cellular effects is still much higher than the reported IC50 for the in vitro inhibition of the CDK4 (45-75 nM) or CDK2 (270 nM) enzymatic activity [30,31,35]. There are several possible reasons for this discrepancy: 1) Live cells may continue producing CDK proteins once the existing CDKs are inactivated by NPCD, due to a conceivable negative feedback mechanism. 2) Live cells have other NPCD targets that also consume NPCD. At least, CDK4, CDK6 and CDK2 will simultaneously consume NPCD, thus raising the IC50 for the cellular effects. 3) A certain portion of NPCD may not be internalized into the cells while some other NPCD molecules may be metabolized. 4) The IC50 for growth inhibition and cell death should be much higher than the IC50 for enzymatic activity of CDK proteins, because the remaining activities of CDK4, CDK6 and CDK2 may be sufficient to maintain cell proliferation or viability. Besides these explanations, however, the much higher IC50 may reflect that NPCD has other cellular targets that are accountable for the observed growth inhibition and cell death, since many chemical drugs have diverse targets and effects. Probably, p27kip1 and p21cip1 are among these non-CDK targets, as discussed below. These issues deserve further explorations.
A wonderful surprise given by our results is the long-lasting growth inhibition of breast cancer cells, especially MCF7 cells, because clonogenic survival assay shows that while there are many cells that survived the NPCD treatment they cannot propagate as fast as non-treated controls to form sizable colonies even 10-15 days after the cessation of the treatment. Another reported long-lasting CDK inhibitor is R-roscovitine (CYC202), which has a relatively specific inhibitory activity towards cyclin E-CDK2 and shows a long-lasting arrest of polycystic kidney disease in animals [60]. Whether such long-lasting growth inhibition is a trait of G1 phase CDK inhibitors and what is the underlying mechanism remain to be further studied.
A basic finding of the present study is that NPCD has relatively similar efficacy on five human breast cancer cell lines, although these cells have different properties of tumor biology pertaining to such as p53, estrogen receptor, metastatic statuses, etc. Actually, even non-malignant MCF10A cells show only a small difference in the sensitivity. Use of non-transformed epithelial cell lines such as MCF10A as “normal control” in the studies of chemoresponse has disadvantages. One that is often neglected is that these cell lines require different culture conditions, such as glucocorticoid and other supplements (hEGF, insulin, cholera toxin, etc), for an optimal growth. When these cells are cultured in a routine condition used for most epithelial-derived cancer cell lines, they are more fragile than cancer cells and thus easier to die. Actually, MCF10A is not much less sensitive to some other chemotherapeutical agents either (unpublished data). If all cells are cultured in a special medium for MCF10A, the data may not be used broadly since most cancer cells are not routinely cultured with many supplements and glucocorticoid that may influence the effects of the agent to be tested. For this reason, we culture all cells in routine DMEM medium and serum without additional supplements that may affect data interpretation, although it is not optimal for MCF10A.
The effect of NPCD is associated with decreased phosphorylation of the Rb protein at Ser780, Ser807 and ser811 as expected. Of the cell lines studied, MB231 is so-called triple-negative, i.e. absence of estrogen receptor, progesterone receptor and Her-2 receptor expression. Triple-negativity is a special category of breast cancer that is notorious for its refractoriness to various chemotherapies and for its worse prognosis [61], but MB231 cells are still relatively sensitive to NPCD. However, MB231 cells express Rb protein whereas many other triple-negative cells are basal-cell like and lack Rb expression [62]. Several studies have shown that Rb-proficient cells of breast cancer [47] and other cancer types [63,64] are more sensitive to PD0332991 than the Rb-deficient counterparts. Probably, Rb-deficient breast cancer cells are relatively resistant to NPCD as well, which deserves exploration later. However, in the Rb-proficient cell lines, the protein level of Rb usually varies little and is thus used by some investigators as loading control in western blot assay [54,55]. As expected, we did not detect a significant difference in the Rb level among the cell lines studied, nor a correlation between the sensitivity to NPCD and the Rb protein level. We intend to consider that a complete lack of Rb may affect the response of cells to CDK4 inhibitors but in the Rb-proficient cells its protein level may not significantly influence the response.
Finn et al reported recently that PD0332991 is more effective on high D1 breast cancer cells [47], and Dai et al also report that D1 overexpression increases the susceptibility of human U266 myeloma cells to CDK inhibitors [65]. We found that the two relatively less sensitive cell lines, T47D and GI101Ap, express a relatively lower level of D1 and/or CDK4 compared with the other three more-sensitive cell lines, which is in line with the observations of Finn et al [47] and Dai et al [65] and dovetails with the assumption that NPCD has its effect by targeting the D1-CDK4 holoenzyme. Considering that D1 may impose resistance to other chemotherapeutic agents onto pancreatic cancer and probably also onto other cancers, as reviewed by us [1], CDK4 inhibitors may be prescribed especially to those cancers that show high D1-CDK4 activities or D1-CDK4 imposed chemotherapeutic resistance.
There are very few studies on how CDK4/6 inhibitory compounds affect the expression of D1, CDK4 and other G1 regulatory genes, and PD0332991 does not seem to have a consistent effect on D1 protein level [66]. We therefore put specific efforts on this issue. MCF7 and MCF15 seem to represent a group of breast cancer cells that show decreases in both D1 and CDK4 upon treatment with NPCD, whereas MB231 may represent another type of breast cancer cells that shows induction of D1 with unchanged CDK4 levels. CDK6 is expressed at relatively low level in these breast cancer cells and does not show a consistent pattern of changes in response to NPCD treatment. Although D1 is considered an oncogene that drives cell proliferation, it can also cause apoptosis in certain undefined situations [1]. Questions that remain to be answered include how D1 chooses to drive proliferation or cell death and whether this decision is made under influence of CDK4/6 status. Moreover, it is also unclear whether CDK4 alone has dual-functions, i.e. can cause apoptosis as well. We infer that D1, by binding to CDK4/6, may protect breast cancer cells from drug-induced cell death [1]. However, once the D1-CDK4 activity is inhibited by NPCD and conceivably also PD0332991 or other CDK4 inhibitory compounds, the D1 and CDK4 proteins become “useless” and are thus degraded via unknown mechanisms. Without D1-CDK4 sustaining their survival, the cells thus undergo growth arrest and demise. This may be the situation occurring in the cell type represented by MCF7 and MCF15 cells. In the cell type presented by MB231, in which NPCD cannot decrease or even induces D1 and CDK4, the abundant, but kinase-dead, D1-CDK4 holoenzyme may directly promote cell death since it is no longer able to phosphorylate the Rb to drive cell cycle progression. This intriguing hypothesis needs to be tested later.
Phosphorylation of D1 protein at Thr286 by GSK-3β is supposed to trigger proteasomal degradation of D1 [2]. The abundance of Thr286-phosphorylated D1 is unexpectedly decreased by NPCD in both the cytoplasmic and nuclear fractions of MCF7 cells, which opposes our original expectation that the Thr286 phosphorylation should be increased, not decreased, in MCF7 cells as a mechanism underlying the decreased level of D1 protein 72 hours post NPCD treatment as seen in figure 5. Therefore, phosphorylation at Thr286 is insufficient to degrade D1 protein in MCF7 and probably also MCF15 cells. Aspirin has been shown to rapidly degrade D1 protein by activation of p38 mitogen-activated protein kinase (MAPK), resulting in apoptosis of the cells [50]. There are some other compounds with therapeutic potential that can also cause D1 protein degradation, such as STG28 [67], silibinin [68], fucoxanthin [69] and metformin [70,71]. Of these compounds, STG28 is known to degrade D1 protein via a novel mechanism independent of Thr286 phosphorylation [67], whereas it is unclear whether degradation of D1 by the other compounds requires phosphorylation at Thr286. Therefore, the unexpected finding that a pronounced decrease in Thr286-phosphorylation is not companied with an increase in D1 protein level in MCF7 cells is of interest and deserves further studies for the underlying mechanism.
Cyclin E and CDK2 protein levels also show obvious changes in response to NPCD treatment in some cell lines. However, in most occasions the changes show a reciprocal pattern, i.e. a decrease in cyclin E is companied by an increase in CDK2 and vice versa. It is possible that an NPCD-induced decrease in cyclin E or CDK2 may trigger a negative feedback mechanism to increase the level of its partner. It is conceivable that such a reciprocal change may help stabilizing the cyclin E-CDK2 activity. Therefore, the cyclin E-CDK2 holoenzyme may not be a primary target of NPCD for its chemotherapeutic effects.
Another unexpected finding in our study is the decrease in the p27kip1 protein in all three NPCD-treated cell lines, i.e. MCF7, MB231 and MCF15 cells. P21cip1 protein level is also decreased in MCF7 and MCF15 cells, although not in MB231 cells. These results are unexpected because most chemotherapeutic agents induce, not decrease, these CDK inhibitory proteins. For instance, anticancer activities of R-roscovitine (a CDK2 inhibitor) [72], inositol hexphosphate [73], Silibinin [68] and ZD1839 [74] are associated with increased p27kip1 and/or p21cip1. On the other hand, lost expression of these CDK inhibitory genes, especially the p27kip1, is suggested as a common reason for the resistance of cancer cells to certain chemotherapeutic agents [75,76]. Therefore, NPCD may represent a new category of chemotherapeutic compounds that compensate for the weakness of those agents that require p27cip1 or p21cip1 to elicit their therapeutic effects. However, whether and how decreases of these CDK inhibitory proteins contribute to NPCD-induced cell death of cancer cells are unknown. Probably p27kip1 and p21cip1 are also cellular targets of NPCD, besides G1 phase CDKs, as a reason for a much higher IC50 for the cellular effects as discussed above. Increased abundance of the D1-CDK4 complex is known to trap more p27kip1 and/or p21cip1 in the complex, indirectly preventing cyclin E-CDK2 holoenzyme from inhibition by these CDK inhibitors. As the result, cells are enhanced to complete the G1-S phase transit and accelerate the S phase. Our results raise an intriguing question as to whether decreases in p27kip1 and/or p21cip1 per se can cause apoptosis when D1-CDK4 holoenzyme is either decreased or inactivated. This question is valid for at least two reasons: 1) p27kip1 or p21cip1 is actually required for assembling the cyclin-CDK complex and bringing the complex into the nucleus because neither D1 nor CDK4 has a canonical nuclear localization sequence whereas p27kip1 or p21cip1 has [77,78]. 2) These proteins can also function as oncogenes to promote tumor formation or to render cancer cells resistant to anticancer therapies under certain situations [79-82]. Whether PD0332991 and other CDK4/6 inhibitory compounds also decrease the protein levels of p21cip1 and p27kip1 is another interesting question waiting for an answer.
The mRNA levels of p27kip1, p21cip1, D1, CDK4, CDK2 and cyclin E are induced by NPCD at concentrations around the IC50 in different breast cancer cell lines, which is a surprise to us because we expected that NPCD might also have certain inhibitory effects on CDK7 and CDK9, just like many other CDK inhibitory compounds, although this has not really been studied. CDK7 and CDK9 are required for transcriptional elongation of mRNAs, and thus their inhibition results in decrease in the mRNA levels of most, if not all, genes [83]. Therefore, the increases in the mRNA levels of many genes observed herein indirectly suggest that NPCD may not be very potent in inhibition of CDK7 and CDK9. However, a weaker inhibition may still occur, at least in certain cells, since NPCD at 8 µM seems to decrease the levels of all the genes in MCF15 cells (fig 7). Further study on the enzymatic activity is required to clarify this issue.
In summary, we show for the first time that the CDK4 inhibitory compound NPCD can cause long-lasting growth arrest and cell death of breast cancer cell lines at an IC50 of 3-8 µM. An expected decrease in the Rb phosphorylation by D1-CDK4/6 and an unexpected decrease in p27kip1 protein level may be part of the underlying mechanism, although the D1 and CDK4 protein levels show different changes among different cell lines treated with NPCD.
Acknowledgements
We thank Dr. Fred Bogott at Austin Medical Center, Austin of Minnesota, USA, for his excellent English editing of this manuscript. This work is supported by a grant of Major scientific and technological special project for new drug creation in China (2009ZX09102-014) to Y-X Li and by a National Institute of Health grant (RO1CA100864) to D.J. Liao.
Conflict of Interest
The authors have declared that no conflict of interest exists.
References
1. Liao D, Thakur A, Wu J, Biliran H, Sarkar FH. Perspectives on c-Myc, Cyclin D1, and Their Interaction in Cancer Formation, Progression, and Response to Chemotherapy. Crit Rev Oncog. 2007;13:93-158
2. Kim JK, Diehl JA. Nuclear cyclin D1: an oncogenic driver in human cancer. J Cell Physiol. 2009;220:292-296
3. Kirkegaard T, Nielsen KV, Jensen LB, Campbell FM, Muller S, Tovey SM. et al. Genetic alterations of CCND1 and EMSY in breast cancers. Histopathology. 2008;52:698-705
4. Fernandez V, Hartmann E, Ott G, Campo E, Rosenwald A. Pathogenesis of mantle-cell lymphoma: all oncogenic roads lead to dysregulation of cell cycle and DNA damage response pathways. J Clin Oncol. 2005;23:6364-6369
5. Elsheikh S, Green AR, Aleskandarany MA, Grainge M, Paish CE, Lambros MB. et al. CCND1 amplification and cyclin D1 expression in breast cancer and their relation with proteomic subgroups and patient outcome. Breast Cancer Res Treat. 2008;109:325-335
6. Weinstat-Saslow D, Merino MJ, Manrow RE, Lawrence JA, Bluth RF, Wittenbel KD. et al. Overexpression of cyclin D mRNA distinguishes invasive and in situ breast carcinomas from non-malignant lesions. Nat Med. 1995;1:1257-1260
7. Yu Q, Geng Y, Sicinski P. Specific protection against breast cancers by cyclin D1 ablation. Nature. 2001;411:1017-1021
8. Butt AJ, McNeil CM, Musgrove EA, Sutherland RL. Downstream targets of growth factor and oestrogen signalling and endocrine resistance: the potential roles of c-Myc, cyclin D1 and cyclin E. Endocr Relat Cancer. 2005;12(Suppl 1):S47-S59
9. Aggarwal BB, Ichikawa H. Molecular targets and anticancer potential of indole-3-carbinol and its derivatives. Cell Cycle. 2005;4:1201-1215
10. Alao JP. The regulation of cyclin D1 degradation: roles in cancer development and the potential for therapeutic invention. Mol Cancer. 2007;6:24
11. Curiel DT. Gene therapy for carcinoma of the breast: Genetic ablation strategies. Breast Cancer Res. 2000;2:45-49
12. Malumbres M, Barbacid M. Is Cyclin D1-CDK4 kinase a bona fide cancer target? Cancer Cell. 2006;9:2-4
13. Mani S, Wang C, Wu K, Francis R, Pestell R. Cyclin-dependent kinase inhibitors: novel anticancer agents. Expert Opin Investig Drugs. 2000;9:1849-1870
14. Wang C, Li Z, Fu M, Bouras T, Pestell RG. Signal transduction mediated by cyclin D1: from mitogens to cell proliferation: a molecular target with therapeutic potential. Cancer Treat Res. 2004;119:217-237
15. Aubry C, Wilson AJ, Emmerson D, Murphy E, Chan YY, Dickens MP. et al. Fascaplysin-inspired diindolyls as selective inhibitors of CDK4/cyclin D1. Bioorg Med Chem. 2009;17:6073-6084
16. Corsino P, Horenstein N, Ostrov D, Rowe T, Law M, Barrett A. et al. A Novel Class of Cyclin-dependent Kinase Inhibitors Identified by Molecular Docking Act through a Unique Mechanism. J Biol Chem. 2009;284:29945-29955
17. Huang JW, Shiau CW, Yang J, Wang DS, Chiu HC, Chen CY. et al. Development of small-molecule cyclin D1-ablative agents. J Med Chem. 2006;49:4684-4689
18. Huang JW, Shiau CW, Yang YT, Kulp SK, Chen KF, Brueggemeier RW. et al. Peroxisome proliferator-activated receptor gamma-independent ablation of cyclin D1 by thiazolidinediones and their derivatives in breast cancer cells. Mol Pharmacol. 2005;67:1342-1348
19. Klasa RJ, List AF, Cheson BD. Rational approaches to design of therapeutics targeting molecular markers. Hematology Am Soc Hematol Educ Program. 2001:443-462
20. Kakeya H, Onose R, Liu PC, Onozawa C, Matsumura F, Osada H. Inhibition of cyclin D1 expression and phosphorylation of retinoblastoma protein by phosmidosine, a nucleotide antibiotic. Cancer Res. 1998;58:704-710
21. Takahashi-Yanaga F, Taba Y, Miwa Y, Kubohara Y, Watanabe Y, Hirata M. et al. Dictyostelium differentiation-inducing factor-3 activates glycogen synthase kinase-3beta and degrades cyclin D1 in mammalian cells. J Biol Chem. 2003;278:9663-9670
22. Seo BR, Lee KW, Ha J, Park HJ, Choi JW, Lee KT. Saucernetin-7 isolated from Saururus chinensis inhibits proliferation of human promyelocytic HL-60 leukemia cells via G0/G1 phase arrest and induction of differentiation. Carcinogenesis. 2004;25:1387-1394
23. Yang CS, Chin KV, Lambert JD. Cancer chemoprevention by targeting proteasomal degradation: commentary re KA Dragnev et al, Specific chemopreventive agents trigger proteasomal degradation of G1 cyclins: implications for combination therapy. Clin Cancer Res, 2004;10:2570-7. Clin Cancer Res. 2004;10:2220-2221
24. Dragnev KH, Pitha-Rowe I, Ma Y, Petty WJ, Sekula D, Murphy B. et al. Specific chemopreventive agents trigger proteasomal degradation of G1 cyclins: implications for combination therapy. Clin Cancer Res. 2004;10:2570-2577
25. Hori A, Ikeyama S, Sudo K. Suppression of cyclin D1 mRNA expression by the angiogenesis inhibitor TNP-470 (AGM-1470) in vascular endothelial cells. Biochem Biophys Res Commun. 1994;204:1067-1073
26. Senderowicz AM. Inhibitors of cyclin-dependent kinase modulators for cancer therapy. Prog Drug Res. 2005;63:183-206
27. Zhai S, Senderowicz AM, Sausville EA, Figg WD. Flavopiridol, a novel cyclin-dependent kinase inhibitor, in clinical development. Ann Pharmacother. 2002;36:905-911
28. Soni R, O'Reilly T, Furet P, Muller L, Stephan C, Zumstein-Mecker S. et al. Selective in vivo and in vitro effects of a small molecule inhibitor of cyclin-dependent kinase 4. J Natl Cancer Inst. 2001;93:436-446
29. Zhu G, Conner SE, Zhou X, Chan HK, Shih C, Engler TA. et al. Synthesis of 1,7-annulated indoles and their applications in the studies of cyclin dependent kinase inhibitors. Bioorg Med Chem Lett. 2004;14:3057-3061
30. Zhu G, Conner SE, Zhou X, Shih C, Li T, Anderson BD. et al. Synthesis, structure-activity relationship, and biological studies of indolocarbazoles as potent cyclin D1-CDK4 inhibitors. J Med Chem. 2003;46:2027-2030
31. Zhu G, Conner S, Zhou X, Shih C, Brooks HB, Considine E. et al. Synthesis of quinolinyl/isoquinolinyl[a]pyrrolo [3,4-c] carbazoles as cyclin D1/CDK4 inhibitors. Bioorg Med Chem Lett. 2003;13:1231-1235
32. Jeong HW, Kim MR, Son KH, Han MY, Ha JH, Garnier M. et al. Cinnamaldehydes inhibit cyclin dependent kinase 4/cyclin D1. Bioorg Med Chem Lett. 2000;10:1819-1822
33. Markwalder JA, Arnone MR, Benfield PA, Boisclair M, Burton CR, Chang CH. et al. Synthesis and biological evaluation of 1-aryl-4,5-dihydro-1H-pyrazolo[3,4-d]pyrimidin-4-one inhibitors of cyclin-dependent kinases. J Med Chem. 2004;47:5894-5911
34. Kubo A, Kaye FJ. Searching for selective cyclin-dependent kinase inhibitors to target the retinoblastoma/p16 cancer gene pathway. J Natl Cancer Inst. 2001;93:415-417
35. Sanchez-Martinez C, Shih C, Faul MM, Zhu G, Paal M, Somoza C. et al. Aryl[a]pyrrolo[3,4-c]carbazoles as selective cyclin D1-CDK4 inhibitors. Bioorg Med Chem Lett. 2003;13:3835-3839
36. Sanchez-Martinez C, Faul MM, Shih C, Sullivan KA, Grutsch JL, Cooper JT. et al. Synthesis of aryl- and heteroaryl[a]pyrrolo[3,4-c]carbazoles. J Org Chem. 2003;68:8008-8014
37. Senderowicz AM. Small-molecule cyclin-dependent kinase modulators. Oncogene. 2003;22:6609-6620
38. Senderowicz AM. Novel direct and indirect cyclin-dependent kinase modulators for the prevention and treatment of human neoplasms. Cancer Chemother Pharmacol. 2003;52(Suppl 1):S61-S73
39. Faul MM, Engler TA, Sullivan KA, Grutsch JL, Clayton MT, Martinelli MJ. et al. Synthetic approaches to indolo[6,7-a]pyrrolo[3,4-c]carbazoles: potent cyclin D1/CDK4 inhibitors. J Org Chem. 2004;69:2967-2975
40. Engler TA, Furness K, Malhotra S, Sanchez-Martinez C, Shih C, Xie W. et al. Novel, potent and selective cyclin D1/CDK4 inhibitors: indolo[6,7-a]pyrrolo[3,4-c]carbazoles. Bioorg Med Chem Lett. 2003;13:2261-2267
41. Toogood PL, Harvey PJ, Repine JT, Sheehan DJ, VanderWel SN, Zhou H. et al. Discovery of a potent and selective inhibitor of cyclin-dependent kinase 4/6. J Med Chem. 2005;48:2388-2406
42. VanderWel SN, Harvey PJ, McNamara DJ, Repine JT, Keller PR, Quin JIII. et al. Pyrido[2,3-d]pyrimidin-7-ones as specific inhibitors of cyclin-dependent kinase 4. J Med Chem. 2005;48:2371-2387
43. Fry DW, Harvey PJ, Keller PR, Elliott WL, Meade M, Trachet E. et al. Specific inhibition of cyclin-dependent kinase 4/6 by PD 0332991 and associated antitumor activity in human tumor xenografts. Mol Cancer Ther. 2004;3:1427-1438
44. Zhang C, Yan Z, Arango ME, Painter CL, Anderes K. Advancing bioluminescence imaging technology for the evaluation of anticancer agents in the MDA-MB-435-HAL-Luc mammary fat pad and subrenal capsule tumor models. Clin Cancer Res. 2009;15:238-246
45. Menu E, Garcia J, Huang X, Di LM, Toogood PL, Chen I. et al. A novel therapeutic combination using PD 0332991 and bortezomib: study in the 5T33MM myeloma model. Cancer Res. 2008;68:5519-5523
46. Wang L, Wang J, Blaser BW, Duchemin AM, Kusewitt DF, Liu T. et al. Pharmacologic inhibition of CDK4/6: mechanistic evidence for selective activity or acquired resistance in acute myeloid leukemia. Blood. 2007;110:2075-2083
47. Finn RS, Dering J, Conklin D, Kalous O, Cohen DJ, Desai AJ. et al. PD 0332991, a selective cyclin D kinase 4/6 inhibitor, preferentially inhibits proliferation of luminal estrogen receptor-positive human breast cancer cell lines in vitro. Breast Cancer Res. 2009;11:R77
48. Sanchez-Martinez C, Shih C, Zhu G, Li T, Brooks HB, Patel BK. et al. Studies on cyclin-dependent kinase inhibitors: indolo-[2,3-a]pyrrolo[3,4-c]carbazoles versus bis-indolylmaleimides. Bioorg Med Chem Lett. 2003;13:3841-3846
49. Thoms HC, Dunlop MG, Stark LA. CDK4 inhibitors and apoptosis: a novel mechanism requiring nucleolar targeting of RelA. Cell Cycle. 2007;6:1293-1297
50. Thoms HC, Dunlop MG, Stark LA. p38-mediated inactivation of cyclin D1/cyclin-dependent kinase 4 stimulates nucleolar translocation of RelA and apoptosis in colorectal cancer cells. Cancer Res. 2007;67:1660-1669
51. Shen KC, Miller F, Tait L, Santner SJ, Pauley R, Raz A. et al. Isolation and characterization of a breast progenitor epithelial cell line with robust DNA damage responses. Breast Cancer Res Treat. 2006;98:357-364
52. Rae JM, Creighton CJ, Meck JM, Haddad BR, Johnson MD. MDA-MB-435 cells are derived from M14 Melanoma cells--a loss for breast cancer, but a boon for melanoma research. Breast Cancer Res Treat. 2007;104:13-19
53. Andrews NC, Faller DV. A rapid micropreparation technique for extraction of DNA-binding proteins from limiting numbers of mammalian cells. Nucleic Acids Res. 1991;19:2499
54. Banerjee S, Zhang Y, Ali S, Bhuiyan M, Wang Z, Chiao PJ. et al. Molecular evidence for increased antitumor activity of gemcitabine by genistein in vitro and in vivo using an orthotopic model of pancreatic cancer. Cancer Res. 2005;65:9064-9072
55. Banerjee S, Zhang Y, Wang Z, Che M, Chiao PJ, Abbruzzese JL. et al. In vitro and in vivo molecular evidence of genistein action in augmenting the efficacy of cisplatin in pancreatic cancer. Int J Cancer. 2007;120:906-917
56. Grafstrom RH, Pan W, Hoess RH. Defining the substrate specificity of cdk4 kinase-cyclin D1 complex. Carcinogenesis. 1999;20:193-198
57. Pan W, Cox S, Hoess RH, Grafstrom RH. A cyclin D1/cyclin-dependent kinase 4 binding site within the C domain of the retinoblastoma protein. Cancer Res. 2001;61:2885-2891
58. Dai Y, Grant S. Small molecule inhibitors targeting cyclin-dependent kinases as anticancer agents. Curr Oncol Rep. 2004;6:123-130
59. Graf F, Koehler L, Kniess T, Wuest F, Mosch B, Pietzsch J. Cell Cycle Regulating Kinase Cdk4 as a Potential Target for Tumor Cell Treatment and Tumor Imaging. J Oncol. 2009:106378
60. Bukanov NO, Smith LA, Klinger KW, Ledbetter SR, Ibraghimov-Beskrovnaya O. Long-lasting arrest of murine polycystic kidney disease with CDK inhibitor roscovitine. Nature. 2006;444:949-952
61. Dawson SJ, Provenzano E, Caldas C. Triple negative breast cancers: clinical and prognostic implications. Eur J Cancer. 2009;45(Suppl 1):27-40
62. Gauthier ML, Berman HK, Miller C, Kozakeiwicz K, Chew K, Moore D. et al. Abrogated response to cellular stress identifies DCIS associated with subsequent tumor events and defines basal-like breast tumors. Cancer Cell. 2007;12:479-491
63. Michaud K, Solomon DA, Oermann E, Kim JS, Zhong WZ, Prados MD. et al. Pharmacologic inhibition of cyclin-dependent kinases 4 and 6 arrests the growth of glioblastoma multiforme intracranial xenografts. Cancer Res. 2010;70:3228-3238
64. Rivadeneira DB, Mayhew CN, Thangavel C, Sotillo E, Reed CA, Grana X. et al. Proliferative suppression by CDK4/6 inhibition: complex function of the retinoblastoma pathway in liver tissue and hepatoma cells. Gastroenterology. 2010;138:1920-1930
65. Dai Y, Hamm TE, Dent P, Grant S. Cyclin D1 overexpression increases the susceptibility of human U266 myeloma cells to CDK inhibitors through a process involving p130-, p107- and E2F-dependent S phase entry. Cell Cycle. 2006;5:437-446
66. Baughn LB, Di LM, Wu K, Toogood PL, Louie T, Gottschalk R. et al. A novel orally active small molecule potently induces G1 arrest in primary myeloma cells and prevents tumor growth by specific inhibition of cyclin-dependent kinase 4/6. Cancer Res. 2006;66:7661-7667
67. Wei S, Yang HC, Chuang HC, Yang J, Kulp SK, Lu PJ. et al. A novel mechanism by which thiazolidinediones facilitate the proteasomal degradation of cyclin D1 in cancer cells. J Biol Chem. 2008;283:26759-26770
68. Kaur M, Velmurugan B, Tyagi A, Deep G, Katiyar S, Agarwal C. et al. Silibinin suppresses growth and induces apoptotic death of human colorectal carcinoma LoVo cells in culture and tumor xenograft. Mol Cancer Ther. 2009;8:2366-2374
69. Das SK, Hashimoto T, Kanazawa K. Growth inhibition of human hepatic carcinoma HepG2 cells by fucoxanthin is associated with down-regulation of cyclin D. Biochim Biophys Acta. 2008;1780:743-749
70. Liu B, Fan Z, Edgerton SM, Deng XS, Alimova IN, Lind SE. et al. Metformin induces unique biological and molecular responses in triple negative breast cancer cells. Cell Cycle. 2009;8:2031-2040
71. Alimova IN, Liu B, Fan Z, Edgerton SM, Dillon T, Lind SE. et al. Metformin inhibits breast cancer cell growth, colony formation and induces cell cycle arrest in vitro. Cell Cycle. 2009;8:909-915
72. Appleyard MV, O'Neill MA, Murray KE, Paulin FE, Bray SE, Kernohan NM. et al. Seliciclib (CYC202, R-roscovitine) enhances the antitumor effect of doxorubicin in vivo in a breast cancer xenograft model. Int J Cancer. 2009;124:465-472
73. Roy S, Gu M, Ramasamy K, Singh RP, Agarwal C, Siriwardana S. et al. p21/Cip1 and p27/Kip1 Are essential molecular targets of inositol hexaphosphate for its antitumor efficacy against prostate cancer. Cancer Res. 2009;69:1166-1173
74. Yabuuchi S, Katayose Y, Oda A, Mizuma M, Shirasou S, Sasaki T. et al. ZD1839 (IRESSA) stabilizes p27Kip1 and enhances radiosensitivity in cholangiocarcinoma cell lines. Anticancer Res. 2009;29:1169-1180
75. Bertagnolli MM, Warren RS, Niedzwiecki D, Mueller E, Compton CC, Redston M. et al. p27Kip1 in stage III colon cancer: implications for outcome following adjuvant chemotherapy in cancer and leukemia group B protocol 89803. Clin Cancer Res. 2009;15:2116-2122
76. Bedard PL, de AE, Cardoso F. Beyond trastuzumab: overcoming resistance to targeted HER-2 therapy in breast cancer. Curr Cancer Drug Targets. 2009;9:148-162
77. Cheng M, Olivier P, Diehl JA, Fero M, Roussel MF, Roberts JM. et al. The p21(Cip1) and p27(Kip1) CDK 'inhibitors' are essential activators of cyclin D-dependent kinases in murine fibroblasts. EMBO J. 1999;18:1571-1583
78. Murakami H, Horihata M, Andojo S, Yoneda-Kato N, Kato JY. Isolation and characterization of cytoplasmic cyclin D1 mutants. FEBS Lett. 2009;583:1575-1580
79. Gartel AL. p21(WAF1/CIP1) and cancer: a shifting paradigm? Biofactors. 2009;35:161-164
80. Abukhdeir AM, Park BH. P21 and p27: roles in carcinogenesis and drug resistance. Expert Rev Mol Med. 2008;10:e19
81. Blain SW. Switching cyclin D-Cdk4 kinase activity on and off. Cell Cycle. 2008;7:892-898
82. Chu IM, Hengst L, Slingerland JM. The Cdk inhibitor p27 in human cancer: prognostic potential and relevance to anticancer therapy. Nat Rev Cancer. 2008;8:253-267
83. Wesierska-Gadek J, Krystof V. Selective cyclin-dependent kinase inhibitors discriminating between cell cycle and transcriptional kinases: future reality or utopia? Ann N Y Acad Sci. 2009;1171:228-241
84. Banerjee S, Zhang Y, Ali S, Bhuiyan M, Wang Z, Chiao PJ. et al. Molecular evidence for increased antitumor activity of gemcitabine by genistein in vitro and in vivo using an orthotopic model of pancreatic cancer. Cancer Res. 2005;65:9064-9072
85. Banerjee S, Zhang Y, Wang Z, Che M, Chiao PJ, Abbruzzese JL. et al. In vitro and in vivo molecular evidence of genistein action in augmenting the efficacy of cisplatin in pancreatic cancer. Int J Cancer. 2007;120:906-917
86. Banerjee S, Zhang Y, Ali S, Bhuiyan M, Wang Z, Chiao PJ. et al. Molecular evidence for increased antitumor activity of gemcitabine by genistein in vitro and in vivo using an orthotopic model of pancreatic cancer. Cancer Res. 2005;65:9064-9072
87. Banerjee S, Zhang Y, Wang Z, Che M, Chiao PJ, Abbruzzese JL. et al. In vitro and in vivo molecular evidence of genistein action in augmenting the efficacy of cisplatin in pancreatic cancer. Int J Cancer. 2007;120:906-917
Author contact
![]() Corresponding author: Ying-xia Li, Ph.D, School of Pharmacy, Fudan University, Pudong, Shanghai, 201203, P.R. China. Phone: 86-21-51980127; Email: liyx417com. D. Joshua Liao, Ph.D., Hormel Institute, University of Minnesota, 801 16th Ave, NE, Austin, MN 55912, USA. Tel: 1-507-437-9665; Fax: 1-507-437-9606; Email: djliaoumn.edu
Corresponding author: Ying-xia Li, Ph.D, School of Pharmacy, Fudan University, Pudong, Shanghai, 201203, P.R. China. Phone: 86-21-51980127; Email: liyx417com. D. Joshua Liao, Ph.D., Hormel Institute, University of Minnesota, 801 16th Ave, NE, Austin, MN 55912, USA. Tel: 1-507-437-9665; Fax: 1-507-437-9606; Email: djliaoumn.edu