Impact Factor ISSN: 1837-9664

 Global reach, higher impact
Global reach, higher impactJ Cancer 2011; 2:107-115. doi:10.7150/jca.2.107 This volume Cite
Review
Human Cancer Classification: A Systems Biology- Based Model Integrating Morphology, Cancer Stem Cells, Proteomics, and Genomics
Halliday A Idikio ![]()
Department of Pathology and Laboratory Medicine, University of Alberta, Edmonton, ALBERTA T6G 2B7, CANADA
Received 2011-1-19; Accepted 2011-2-18; Published 2011-2-22
Abstract
Human cancer classification is currently based on the idea of cell of origin, light and electron microscopic attributes of the cancer. What is not yet integrated into cancer classification are the functional attributes of these cancer cells. Recent innovative techniques in biology have provided a wealth of information on the genomic, transcriptomic and proteomic changes in cancer cells. The emergence of the concept of cancer stem cells needs to be included in a classification model to capture the known attributes of cancer stem cells and their potential contribution to treatment response, and metastases. The integrated model of cancer classification presented here incorporates all morphology, cancer stem cell contributions, genetic, and functional attributes of cancer. Integrated cancer classification models could eliminate the unclassifiable cancers as used in current classifications. Future cancer treatment may be advanced by using an integrated model of cancer classification.
Keywords: Human Cancer, Cancer Stem cells, Multiple Omics, Integrated Classification
Introduction
Human cancers occur worldwide. In 2008, 12.7 million new cancers and 7.6 million cancers were recorded; incidence and mortality rates varied with regions and levels of income around the world (1). These results require refocusing on all attributes of cancer.
Differentiating features of malignant and benign lesion are well established; these include rapid growth, increased cell turn-over, invasive growth, metastases, vascular or lymphatic channel invasion for malignant lesions. There are many exceptions to these attributes of cancer. There are overlaps between benign and malignant lesions. Benign (non-malignant) tumors do show chromosome aberrations; uveal melanomas and blue nevi share mutations in G protein (2). A good example is the recent attempt among dermatopathologists to segregate some melanocytic lesions as atypical melanocytic proliferations with low malignant potential (MELTUMP) (3). Sometimes a cancer at given site/organ is classified as containing two cell types; for example, pancreatic cancer with neuroendocrine and acinar/ductal components (4). What model can accommodate unclassifiable cancers in a specific location? Cancer classification schemes always reserve a group as unclassifiable. How can this group be eliminated?
The last two decades have witnessed the surge in molecular profiling (5, 6) and has already expanded into predictive and diagnostic molecular classification of cancers (7, 8). As in the diagnosis of cancers, current molecular classification schemes are still dependent on morphologic variables. These classifications schemes use cell of origin as seen by light and electron microscopy. Inherently, all organs can generate multiple cancer types as multiple cell types exist in these organs- “the holy-grail of all subspecialties”. Furthermore, cancer subtypes are generated under the banner of a single, specific cell type of origin concept. Take the example of the common Basal cell carcinoma- it has variants and subtypes such as nodular, superficial, adenoid, morpheaform, infiltrative, keratotic, pigmented, basosquamous, clear cell, granular, eccrine, apocrine, fibroepitheliomatous, adamantoid, and basosebaceous (9). Do these entities have unique biological features or simply morphological variants of interest only to the diagnostic pathologists? Will the “cancer stem cell origin concept” cure this malady?
The current move to genomics {(gene and transcripts, kinomes, microRNAs, single nucleotide polymorphisms (SNPs), gene copy number variation (CNVs) and proteomics (antibody microarray and mass spectrometry)} brings change to the diagnostic information needed for treatment. Along with the genomic profiling, are efforts at targeted and gene therapy. Because of accumulated experience, in diagnosis, classifications and treatment of cancer that depends on morphology, the shift to genomic methods should be comprehensive and adequate for day-to-day clinical use. While future cancer classification schemes or models may not require morphological attributes, current dependencies on morphological phenotype requires its inclusion (10-12). Morphologic cancer phenotyping does not need to hide the compendium of genetic alterations, interactions with environment and alterations in transcriptional and protein interaction networks that are present in all cancers (11, 13).
Hallmarks of Cancer Cell
Hanahan and Weinberg (2000) (14, 15) listed the seven attributes of cancer; 1) Self sufficiency in growth signals, 2) Insensitivity to anti-growth signals, 3) Evading apoptosis, 4) Limitless replicative potential, telomerase and telomeres 5) Sustained angiogenesis, 6) Tissue invasion and metastasis, and 7) Genome instability. All seven attributes have received great attention in the past decade. Growth and anti-growth signaling are really complex (13). Protein-protein interaction and signaling networks, growth signaling pathways, the role of ubiquitination and protein degradation, and dysfunctional protein networks (16-18) and interactions are complex, described as hubs, modules and motifs (13). Information on cancer cell death and provocation by drugs and irradiation now requires all cell death types to be considered- apoptosis, necrosis, autophagy (19, 20). We now must include the pivotal role of microRNAs (21, 22), and methylation patterns (23). For example, microRNA-185 suppress cancer growth by interfering with Six1; when absent in cancers leads to increase growth and progression (24). Recent efforts have uncovered the role of transposons in the induction of cancer in mouse models; the studies are generating previously unknown cancer related genes (25). Class II (DNA transposons) and class I retrotransposons contribute to DNA instability (26). Cancer cells use aerobic glycolysis to meet energy needs (Warburg effect) and presumed to be a response to hypoxia and tumor micro-environment; changes in metabolic needs of cancer cells such as need for glutamine and activation of hypoxia-inducible-factor (HIF) are interconnected to oncogene activation (27-29). These interacting functionalities of cancer cells impact prognostic and predictive models based on one or two functional attributes of cancer (30).
Origin of Cancer Cells -The Cancer Stem Cell Model
The traditional model of cancers envisaged a “normal cell” transformed to “atypical or dysplastic“ cell with progression into invasive of malignant cell. This is the model that only assumes stochastic generation of cells capable of the behavior of metastasis and progression and cellular heterogeneity of cancers. The stochastic model is used to explain heterogeneity in cancers such as in prostate cancer (Fig 1). The stochastic model will have to assume that all genetic aberrations conferring advantages to the cancer cells “must be maintained in all subsequent cells as growth and proliferation continues and some maturation occurs”. As cancers can also undergo senescence, apoptosis, autophagy and necrosis, the stochastic model must account for these changes (31, 32). Cancer senescence occurs via telomere shortening, oxidative stress, and oncogene activation, that can impair cancer progression (33-35).The stochastic model has to account for local recurrence and metastasis after long post-treatment intervals. Cell of origin models cannot exclude all interactions between cancer cells and any influence from the stroma (36, 37). Recent computational stochastic models, in part based on the hallmarks of cancer, suggest that onset of cancers depend on the first two (2) mutations and early-onset and late-onset cancer initiate these mutations at different times (38). Cancer initiation via DNA damage response and repair, induction of senescence and p53 mutation (39), the generation of driver mutations can be accommodated in stochastic and cancer stem cell models. Driver mutations in cancer initiation are suggested to accumulate over time. Recent detailed studies of human cancers and cancer cell lines show extensive and highly localized mutations following DNA damage and repair response; 2-3 % of human cancers develop these single chromosome mutations labeled as chrothripsis and are suggested as the genesis of driver mutations generated (40). There are no unique driver mutations for the stochastic model.
Patterns and heterogeneity in Cancerous Prostate Tissue (Hematoxylin and eosin stain). A. Normal prostate showing luminal and basal cells. B. Prostate cancer with definable glandular pattern, usually part of Gleason pattern 3. C. Prostate cancer with aggregation, clustering or individual infiltrating cells as described for Gleason 4.
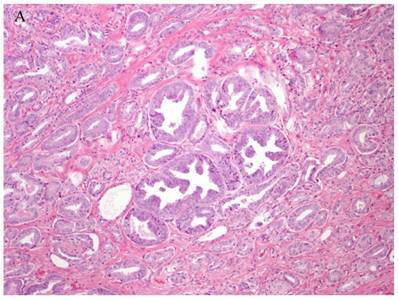
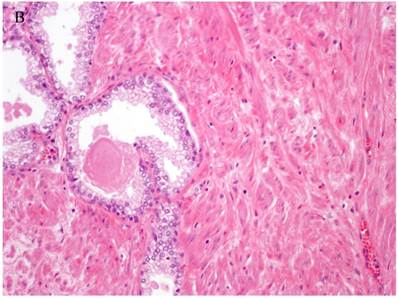
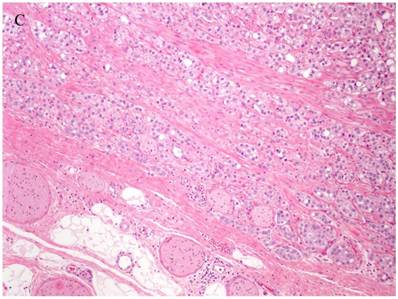
Cancers of certain presumed cell types, synovial cells, occur in locations without defined synovium or other related cell type. Clear cell sarcoma is seen in deep soft tissues and is a copy of all features of melanoma except for genetic aberrations. How do we explain these cancers on the cell of origin concept? Prostate cancer cell of origin thought to be luminal cells, are now shown to be derived from basal cells with the attributes of androgen-independence (41).
The cancer stem cell model has been used to explain cancer cell origin, initiation, progression and metastasis (42-44) (Fig 2). Cancer stem cells as origin of cancers has attributes of hierarchical organization, may be under-estimated and assumed to be a minor population (45, 46). As in their resident or embryonic stem cell counterparts, there are known regulators, such as p53 and WNT signaling pathways. Cancer stem cells show c-Myc transcription profile, similar to embryonic stem cells (47). These cancer stem cells, initially described in breast cancers (48), are now described in liver, ovarian, prostate, head and neck, colon and brain cancers, melanomas (49-54). The cancer stem cells have their specific microenvironments to allow for their specific functions; epigenetic modifications may make cancer stem cells not reliant on its specific niche (42). Cancer stem cells are usually projected as a minor population of all cancer cells (46, 55). The number of identified cancer initiating stem cells may be affected by the background of animals used in xenotranspnatation; in a mouse xenograft model of melanoma, 25% of cancer initiating cells could be found (50).
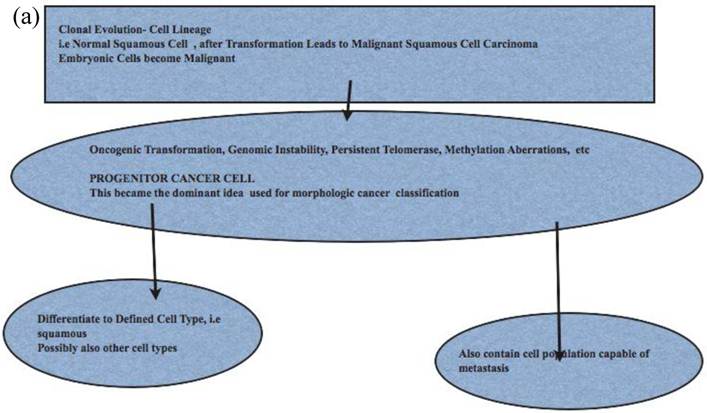
Comparison of Stem Cell and Lineage/Clonal Evolution Models of Cancer Cell Origin. (a)The lineage/clonal evolution model is used in morphological classification of cancer. One cell type gives rise to one cancer type. Squamous epithelium gives rise to squamous cell carcinoma. Pathologists do encounter squamous cell carcinoma in the urothelium of the urinary bladder. How we explain this is by “metaplasia of urothelium to squamous epithelium” and perhaps then to squamous cell carcinoma. An easier explanation will be cancer stem cell model as these have the capacity to become any cell type. (b) Cancer stem cells, with their inherent functional capacities including reduced cell death, are of interest when cancers are treated by irradiation or chemotherapy. Human cancers, examined in detail and extensively, do contain heterogeneous cell types; lung cancers are a good example. This leads to difficulties in some classification schemes depending on lineage.
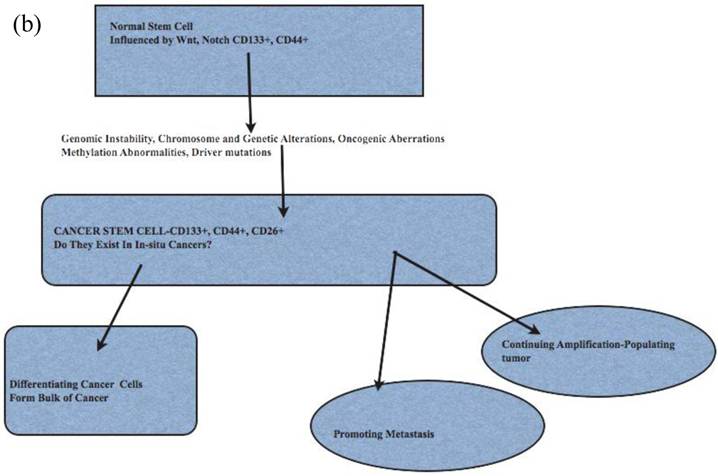
The functional attributes of cancer stem cells such as (i) evasion of cell death, i.e apoptosis, (ii) telomere activation, (iii) colony formation, tumor initiation and differentiation are suited to their role in human cancer (45). The contribution to recurrence, metastasis and treatment, especially radiotherapy-resistance, is now better appreciated (56, 57). Cancer stem cell markers for several human cancers are listed in Table 1. Cancer stem cells markers have functional attributes such as adhesion, cell invasion (CD44) and interactions with GLI1 and focal adhesion kinase (FAK) (CD24) (58).
Biomarkers for Human Cancer Stem Cells
| Cancer Type | Markers |
|---|---|
| Breast | CD44+CD24_/low |
| Ovary | CD133+/CD44+, CD117, Oct4, STELLAR, Nanog and ABCG2/BCRP1 |
| Lung | Cd133+(ABCG2, Oct4, ESA) |
| Brain | CD133+ |
| Colon | CD133+ CD44+ EpCam+, CD166+ (CD29+, CD24+) |
| Pancreas | CD44+EpCam+ CD24+ |
Cancer Metastases and Cancer Stem Cell Model
Cancer metastasis occurs either through progressive acquisition of metastatic potential or the traits are acquired early during cancer initiation; this implies that metastases occurs early or late in cancer (59-61). The acquired traits include survival and evasion of cell death, dormancy, migration, immune escape, and ability to seed, home on targets (62). Support for late evolution of metastasis was described in pancreatic cancer using genome sequencing ; this study indicated clones with metastatic capacity evolve late ( 5years), and are present in the primary tumor (63, 64). Breast cancer cells reaching the bone marrow share stem cell features and markers CD24 and CD44 as determined by double immunohistochemistry in bone marrow; these stem-like cells ranged from 33-100% in metastases (65). CD26+CD24+ positive cancer cells found in colon cancers, not initiating CD133+CD26+CD44 positive cells, define occurrence of metastases (66). Some have separated cancer stem cells from pancreatic cancers capable of initiating metastases (67).
Personalized Genomic Medicine and Cancer Classification
How will the push for personalized medicine affect the present morphology based and the future trend of molecular cancer classification? Personalized medicine, as envisioned will require individual cancer genomics and proteomics for maximum benefit of targeted treatment for the individual; the implications of genomic cancer medicine should encourage use of integrated cancer classification models (68-73).
Integrated Model of Cancer Classification
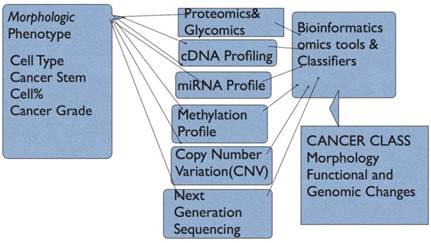
The model envisaged (Fig 3) takes into account all elements of a cancer. We now can provide morphologic classes and subtypes, extract proteomic and gene profiles and gene copy number variations including cytogenetic and array comparative genomics. An added feature is that the functions of proteins and signaling pathways can be derived from gene expression and proteomics. This means that altered or dysfunctional pathways can be supported by adding cytogenetic or CNV data. There are emerging integrated models that use both genomics, exon resequencing, and proteomics in cancer analysis (74-76). Recent prostate cancer survival and post-surgical recurrence used modeling based on some aspects of the integrated classification model including mutation patterns, CNVs, targeted signaling pathway deregulation, miRNA and cDNA profiling; new oncogenes, and CNV-based disease risk profiles over above morphology grades were found (77).
Figure Model of Cancer Classification. In this model, the Phenotype is represented by Morphological Characteristics/and subtypes; Proteomic profile can be derived from high-throughput tissue microarrays and immunohistochemistry plus automated computer-assisted quantitation with normalized intensities, protein microarrays and mass spectrometry; array comparative genomics for Copy Number Variation(CNV) and chromosomal aberrations, Genomic profiling using cDNA microarrays, and finally microRNA profiling. This provides protein profile and cDNA profiles for Gene Ontology and functional annotation. Signally pathways active or repressed can be derived. The CNV and microRNA data provide information to explain active oncogene induced pathways and miRNA targeted pathways that impact proliferation, cell survival, metastasis etc.
- Proteomics: Normal and cancer tissues can be used for comparative proteomics. The methods available are mass spectrometry, protein and antibody microarray and tissue microarray. In mass spectrometry, tissue and cell protein content is quantitatively determined by analyzing peptide content and via bioinformatics the protein content, protein types and classes, protein interaction networks and via gene ontology functional protein mapping (78-80). Tissue microarray uses tissue cores for immunohistochemistry and indirect analysis of protein levels (81-83). In antibody and protein-antigen arrays, protein or antibody spotted on an array is probed with fluorophore-labelled protein or antibody and analyzed like cDNA microarrays (84-87). Flow cytometry can be used to catalogue both surface membrane, cytoplasmic and nuclear proteins in cells. Mass spectrometry, tissue microarray can be used to validate other profiling methods. Mass spectrometry is quantitative and can estimate both modified and unmodified proteins; 2-dimensional gel electrophoresis and protein isolation can be followed by mass spectrometry (78, 80, 88, 89). Glycomics and glycan profiling can add to the protein profile using both mass spectrometry and lectin microarrays to generate aberrant cell migration (90, 91).
- cDNA Profiling and Transcriptomics; cDNA microarray profiling is the most common highthroughput method for determining expression levels of mRNA in cells and tissues (5). Isolated mRNA is transformed to cDNA, which is used to probe optimally designed oligonucleotide array that helped to generate new classes of breast cancer, leukemias and lymphomas. Using transcriptomics for clinical cancer treatment is an area of intense research ( translational research) ( 12, 68, 72). Direct RNA sequencing overcomes drawbacks of cDNA based methods, detect low quantities of mRNA, detect chimeric transcripts, can use RNA from fixed tissues, and uses sequencing-by-synthesis (92).
- Copy Number variatiopn (CNV): Normal and cancer cells show variations in genes. The most common variation is single nucleotide polymorphisms (SNPs). Variations greater than 1kilobase of DNA is called CNV. Other variations include microsattelite instability (MSH), variable tandem repeats, transposable elements, deletions, inversions, and duplications. Furthermore, transcripts of gene fusions provide a wealth of information on the functional input of fused genes(64, 93-97)
- Methylation status: Gene promoter methylation at the cytosine-guanine sites (CpG islands) leads to gene transcription suppression. Demethylation leads to transcript activation. Methylation status of cancer genes adds complexity to interpretation of gene profiling. Finding methylation status involves use of(i) methylation-sensitive or methylation-specific endonucleases, (ii) Sodium bisulphate treatment and DNA sequencing (iii) target amplification by capture and ligation (23, 98, 99).
- miRNA Profiling: microRNAs (18-24 nucleotides) are present in plants and animals. miRNAs play significant roles in normal development, cellular responses and in human cancer. miRNAs represses mRNA trascription via partial complimentary alignment with targer mRNAs. miRNA profiling adds to complexity of cancer cell and tissue profiling. Alignment with their targets tells us why certain transcripts may be down-regulated (22, 100-105).
- Gene Sequencing: Human genome sequencing ushered in the grand promise of genomics medicine in 2000. Now individual genes and chromosomes, and gene fusions are sequenced; these efforts will have impact on the information sets necessary for diagnosis, treatment and predicting outcome in cancer (38). This enables direct analysis of mutations within the gene components of interest. Gene annotation enables linking of sequences to active transcripts (96). Next generation sequencing can overcome some drawbacks of qPCR and cDNA microarray and can enable assessment of cancer gene mutations, copy number variations (CNV), SNPs, miRNA and transcription profiling (106). Emerging sequencing methods, labeled third generation sequencing, include single-molecule-real-time sequencing (SMRT), direct sequencing using direct imaging and sequencing using nanopores (107).
- Cancer Grading: Cancer grading to reflect the extent of differentiation is done for many cancers. Grading systems vary and depend on cancer type. Morphologic attributes are used to grade (score) prostate cancer (108, 109). Cancer grades will form an integral part of the model. Cancer stem cell compartment within the tumor can be estimated by proteomics- immunohistochemical methods using antibodies to CD24, CD44, CD133 and CD26.
The integrated model can be generated using comprehensive bioinformatics and data mining tools. OmicsAnalyzer is one such tool, which is a plug-in for Cytoscape (cytoscape.org), a web based platform for protein/gene network modeling, functional annotation and analysis. (110). Bioinformatics tools for classification, data mining and modeling are available ( R-project.org). The integrated model of cancer classification can help stratify individuals for target treatment and disease outcome based on the treatment (Table 2).
Anticipated data sets from a biopsy for decision making in targeted cancer treatment. Gene profiling signatures (growth, oncogene and stemness), and proteomics can be correlated with CNVs, mutations and gene fusions. Protein interaction maps can suggest treatment targets, possible pathways of resistance.
| Cancer morphology | Adenocarcinoma, Grade 2 |
| 10% Mucinous component | |
| 10% Cancer stem cells | |
| Proteomics/Transcriptome/Methylome | Up-regulated cell death pathway |
| Up-regulated growth factor signaling | |
| Hyper-methylation of kinases | |
| Decreased expression of autophagy pathway | |
| miRNA clusters in some autophagy genes | |
| De-regulated metabolic genes | |
| Mutations (Sequencing/CNV) | Mutations in 24 genes |
| Gene fusions | |
| Copy number changes in 10 genes | |
| Clustered mutations in 8 genes | |
| BRAF, KRAS, EGFR, and p53 mutations |
Conclusion
Cancer cells are endowed with capacities for uninhibited proliferation, invasion and metastasis. Cancer initiation, driver mutations, progression and metastases are common attributes for both stochastic and cancer stem cell models; cancer stem cells possess attributes that enhance survival in all environments hence more suitable as model of cancer. Integrated models that capture every essence of a cancer could enhance the ability to target different components of cancer for maximum therapeutic effect.
Conflict of Interest
The author has declared that no conflict of interest exists.
References
1. Ferlay J, Shin H-R, Bray F, Forman D, Mathers C, Parkin D. Estimates of worldwide burden of cancer in 2008: GLOBOCAN2008. Int J Cancer. 2010;127:2893-2917
2. Van Raamsdonk C, Bezrookove V, Green G. et al. Frequent somatic mutations of GNAQ in uveal melanoma and blue nevi. Nature. 2009;457:599-602
3. Cerroni L, Barnhill R, Elder D. et al. Melanocytic Tumors of Uncertain Malignant Potential. American J Surgical Pathology. 2008;34(3):314-326
4. Stelow E, Shaco-Levy R, Bao F, Garcia J, Klimstra D. Pancreatic Acinar Cell Carcinomas With Prominent Ductal Differentiation: Mixed Acinar Ductal Carcinoma and Mixed Acinar Endocrine Carcinoma. American J Surgical Pathology. 2010;34(4):510-518
5. Golub T, Slonim D, Tamayo P. et al. Molecular Classification of Cancer: Class Discovery and Class Prediction by Gene Expression Monitoring. Science. 1999;286(5439):531-537
6. Segal Ep, Friedman N, Kaminski N, Regev A, Koller D. From signature to models: understanding cancer using microarrays. Nature Genetics. 2005;37:S38-45
7. Bair E, Tibshirani R. Semi-Supervised Methods to Predict Patient Survival from Gne Expression Data. PLoS Biology. 2004;2:0511
8. van de Vijver M, He Y, van't Veer LJ. et al. A Gene Expression Signature As A Predictor Of Survival In Breast Cancer. NEJM. 2002;347(25):1999-2009
9. Crowson A. Basal cell carcinoma: biology, morphology and clinical implications. Modern Pathology. 2006;19:S127-S147
10. Berman J. Modern classification of neoplasms: reconciling differences between morphologic and molecular approaches. BMC Cancer. 2005;5:100
11. Loscalzo J, Kohane I, Barabasi A-L. Human disease classification in the postgenomic ea: A complex systems approach to human pathobiology. Molecular Systems Biology. 2007;3:124
12. Webb C, Pass H. Translation research: from accurate diagnosis to apprpriate treatment. Journal of Translational Medicine. 2004;2:35
13. Barabasi A-L, Oltvali Z. Network Biology: Understanding The Cells Functional Organization. Nature Reviews Genetics. 2004;5:101-113
14. Hanahan D, Weinberg R. The Hallmarks of Cancer. Cell. 2000;100:57-70
15. Hahn W, Weinberg R. Modeling the Molecular Circuitary of Cancer. Nature Reviews Cancer. 2002;2:331-341
16. Chen Z, Sun L. Nonproteolytic Functions of Ubiquitin in Cell Signaling. Molecular Cell. 2009;33:275-286
17. Goodarzi H, Elemento O, Tavazoie S. Revealing Global Regulatory Perturbations across Human Cancers. Molecular Cell. 2009;36:900-911
18. Lemmon M, Schessinger J. Cell Signaling by Receptor Tyrosine Kinases. Cell. 2010;141:1117-1134
19. Amaravadi R, Thompson C. The Roles of Therapy-Induced Autophagy and Necrosis in Cancer Treatment. Clin Cancer Res. 2007;13(24):7271-7279
20. Baehrecke E. Autophagy: dual roles in life and death? Nature Reviews Molecular Cell Biology. 2005;6:505-510
21. Carthew R, Sontheimer E. Origins and Mechanisms of miRNAs and siRNAs. Cell. 2009;136:642-655
22. Esquela-Kerscher A, Slack F. Oncomirs- microRNAs with a role in cancer. Nature Reviews Cancer. 2006;6:259-269
23. Muntean A, Hess J. Epigenetic Dysregulation in Cancer. American Journal of Pathology. 2009;175(4):1353-1361
24. Imam J, Buddavarapu K, Lee-Chang J. et al. MicroRNA-185 suppresses tumor growth and progression by targeting the Six1 oncogene in human cancers. Oncogene. 2010;29:4971-4979
25. Copeland N, Jenkins N. Harnessing the transposons for cancer gene discovery. Nature Reviews Cancer. 2010;10:696-706
26. O'Donnell K, Burns K. Mobilizing diversity: transposable element insertions in genetic variation and disease. Mobile DNA. 2010;1:21
27. Gatenby R, Gillies R. Why do cancers have high aerobic glycolysis. Nature Reviews Cancer. 2004;4(11):891-899
28. Kroemer G, Pouyssegur J. Tumor Cell Metabolism: Cancer's Archilles' Heel. Cancer Cell. 2008;13:472-482
29. Levine A, Puzio-Kuter A. The Control of the Metabolic Switch in Cancers by Oncogenes and Tumor Suppressor Genes. Science. 2010;330:1340-1344
30. Bild A, Potti A, Nevins J. Linking oncogenic pathways with therapeutic opportunities. Nature Reviews Cancer. 2006;6:735-740
31. Collado M, Blasco M, Serrano M. Cellular Senescence in Cancer and Aging. Cell. 2007;130:223-233
32. Levine B, Kroemer G. Autophagy in the Pathogenesis of Disease. Cell. 2008;132:27-42
33. Sharpless N, DePinho R. Telomeres, stem cells, senescence, and cancer. J Clinical Investigation. 2004;113(2):160-168
34. Feldser D, Greider C. Short Telomeres Limit Tumor Progression in Vivo by Inducing Senescence. Cancer Cell. 2007;11(5):461-470
35. Di Micco R, Fumagalli M, Cicalese A. et al. Oncogene-induced senescence is a DNA damage response triggered by DNA hyper-replication. Nature. 2006;444:638-642
36. Kassenbrock K, Plaks V, Werb Z. Matrix Mettaloproteinases: Regulators of the Tumor Microenvironment. Cell. 2010;141(1):52-57
37. Qian B-Z, Pollard J. Macrophage Diversity Enhances Tumor Progression and Metastases. Cell. 2010;141(1):39-51
38. Bozic I, Antal T, Ohtsuki H. et al. Accumulation of driver and passenger mutations during tumor progression. PNAS. 2010;107(43):18545-18550
39. Halazonetis T, Gorgoulis V, Bartek J. An Oncogene-Induced DNA Damage Model for Cancer Development. Science. 2008;319:1352-1355
40. Stephens P, Greenman C, Fu B. et al. Massive Genomic Rearrangement Acquired in a Single Catastrophic Event during Cancer Development. Cell. 2011;144(1):27-40
41. Goldstein A, Huang J, Guo C, Garraway I, Witte O. Identification of a Cell of Origin for Human Prostate Cancer. Science. 2010;329:568-571
42. Clarke M, Fuller M. Stem Cells and Cancer: Two Faces of Eve. Cell. 2006;124:1111-1115
43. Jordan C, Guzman M, Noble M. Cancer Stem Cells. New Engl J Med. 2006;355:1253-1256
44. Fodde R. The Stem of Cancer. Cancer Cell. 2009;15(2):87-90
45. Shackelton M, Quintana E, Fearon E, Morrison S. Heterogeneity in Cancer: Cancer Stem Cells versus Clonal Evolution. Cell. 2009;138:822-829
46. Adams J, Strasser A. Is Tumor Growth Sustained by Rare Cancer Stem Cells or Dominant Clones? Cancer Research. 2008;68(11):4018-4021
47. Kim J, Woo A, Chu J. et al. A Myc Network Accounts for Similarities between Embryonic Stem and Cancer Cell Transcription Programs. Cell. 2010;143(2):313-324
48. Al-Hajji M, Wicha M, Benito-Hernandez A, Morrison S, Clarke M. Prospective identification of breast cancer stem cells. PNAS. 2003;100:3983-3988
49. Vescovi A, Galli R, Reynolds B. Brain tumor stem cells. Nature Reviews Cancer. 2006;6:425-436
50. Quintana E, Shackelton M, Sabel M, Fullen D, Johnson T, Morrison S. Efficient tumor formation by single human melanoma cells. Nature. 2008;456:593-598
51. Hu L, McArthur C, Jaffe R. Ovarian cancer stem-like side-population cells are tumorigenic and chemortesistant. British J Cancer. 2010;102:1276-1283
52. Lawson D, Witte O. Stem cells in prostate cancer initiation and progression. The Journal of Clinical Investigation. 2007;117(8):2044-2050
53. Sell S, Leffert H. Liver Cancer Stem Cells. J Clin Oncol. 2008;26(17):2800-2805
54. Ponnusamy M, Batra S. Ovarian cancer: emerging concept on cancer stem cells. J ournal of Ovarian Research. 2008;1:4
55. Kern S, Shibata D. The Fuzzy Math of Solid Tumor Stem Cells: A Perspective. Cancer Research. 2007;67:89850-8988
56. Rich J. Cancer Stem Cells in Radiation Resistance. Cancer Research. 2007;67:8980-8984
57. Eyler C, Rich J. Survival of the Fittest: Cancer Stem Cells in Therapeutic Resistance and Angiogenesis. J Clin Oncol. 2008;26(17):2839-2845
58. Keysar S, Jimeno A. More than Markers: Biological Significance of Cancer Stem Cell-Defining Molecules. Mol Cancer Ther. 2010;9(9):2450-7
59. Talmadge J. Clonal Selection of Metastasis within the Life History of a tumor. Cancer Research. 2007;67(24):11471-11475
60. Klein C. The Metastasis Cascade. Science. 2008;321:1785-1787
61. Weinberg R. Leaving Home Early:Reexamination of the Canonical Models of Tumor Progression. Cancer Cell. 2008;14:283-284
62. Bogenrieder T, Herlyn M. Axis of evil: molecular mechanisms of cancer metastasis. Oncogene. 2003;22:6524-6536
63. Yachida S, Jones S, Bozic I. et al. Distant metastasis occurs late during genetic evolution of pancreatic cancer. Nature. 2010;467:1114-1117
64. Campbell P, Yachida S, Mudie L. et al. The patterns and dynamics of genomic instability in metastatic pancreatic cancer. Nature. 2010;467:1109-1113
65. Balic M, Lin H, Young L. et al. Most Early Disseminated Cancer Cells Detected in Bone Marrow of Breast Cancer Patients Have a Putative Breast Cancer Stem Cell Phenotype. Clinical Cancer Research. 2006;12(19):5615-5621
66. Pang R, Law W, Chu A. et al. A Subpopulation of CD26+ Cancer Stem Cells with Metastatic Capacity in Human Colorectal Cancer. Cell Stem Cell. 2010;6:603-615
67. Herman P, Huber S, Herrier T. et al. Distinct Populations of Cancer Stem Cells Determine Tumor Growth and Metastatic Activity in Human Pancreatic Cancer. Cell Stem Cell. 2007;1:313-323
68. de Bono J, Ashworth A. Translating cancer research into targeted therapeutics. Nature. 2010;467:543-549
69. Varmus H. Ten Years On- The Human Genome and Medicine. New England J ournal of Medicine. 2010;362:2028-2029
70. Bentley D. Genomes for medicine. Nature. 2004;429:440-445
71. Evans W, Relling M. Moving towards individualized medicine with pharmacogenomics. Nature. 2004;429:464-468
72. Strausberg R, Simpson A, Old L, Riggins G. Oncogenomics and the development of new cancer therapies. Nature. 2004;429:469-474
73. Feero W, Guttmacher A, Collins F. Genomic Medicine- An Updated Primer. N Engl J Med. 2010;362:2001-2011
74. Kim S, Hahn W. Cancer genomics: integrating form and function. Carcinogenesis. 2007;28(7):1387-1392
75. Nevins J, Potti A. Mining gene expression profiles: expression signatures as cancer phenotypes. Nature Reviews Genetics. 2007;8:601-609
76. Rhodes D, Chinnaiyan A. Integrative analysis of cancer transcriptome. Nature Genetics. 2005;17:S31-S37
77. Taylor B, Schultz N, Hieronymus H. et al. Integrative Genomic Profiling of Human Prostate Cancer. Cancer Cell. 2010;18:11-22
78. Nomura D, Dix M, Cravatt B. Activity-based protein profiling for biochemical pathway discovery in cancer. Nature Reviews Cancer. 2010;10:630-638
79. Colinge J, Bennett K. Introduction to Computational Proteomics. PLoS Computational Biology. 2007;3(7):e114
80. Domon B, Aebersold R. Options and considerations when selecting a quantitative proteomics strategy. Nature Biotechnology. 2010;28(7):710-721
81. Kononen J, Bubendorf L, Kallioniemi A. et al. Tissue microarrays for high-throughput molecular profiling of tumor specimens. Nature Medicine. 1998;4(7):844-847
82. Celis J, Gromov P. Proteomics in translational cancer research: Toward an integrated approach. Cancer Cell. 2003;3(1):9-16
83. Celis J, Gromov P, Gromova I, Moreira J, Cabezon T, Ambartsumian Nea. Integrating Proteomic and Functional Genomic Technologies in Discovery-driven Translational Breast Cancer Research. Mol and Cellular Proteomics. 2003;2:369-377
84. Brennan D, O'Connor D, Rexhepaj E, Ponten F, Gallagher W. Antibody-based proteomics: fast-tracking molecular diagnostics in oncology. Nature Reviews Cancer. 2010;10:605-617
85. Iadevaia S, Lu Y, Morales F, Mills G, Ram P. Identification of Optimal Drug Combinations Targeting Cellular Networks: Integrating Phospho-Proteomics and Computational Network Analysis. Cancer Research. 2010;70(17):6704-6714
86. Xu Q, Lam K. Protein and Chemical Microarrays- Powerful Tools for Proteomics. J Biomed and Biotechnology. 2003;5:257-266
87. MacBeath G. Protein microarrays and proteomics. Nature genetics Supplement. 2002;32:526-532
88. Domon B, Aebersold R. Mass Spectrometry and Protein Analysis. Science. 2006;312:212-217
89. Nesvizhskii A, Vitek O, Aebersold R. Analysis and validation of proteomic data generated by tandem mass spectrometry. Nature Methods. 2007;4(10):787-797
90. Hu S, Wong D. Lectin microarray. Proteomics Clin Appl. 2009;3(2):148-154
91. North S, Hitchen P, Haslam S, Dell A. Mass spectrometry in the analysis of N-and-O -linked glycans. Curr Opin Struct Biol. 2009;19(5):498-506
92. Ozsolak F, Platt A, Jones D. et al. Direct RNA sequencing. Nature. 2009;461:814-818
93. Yeang C-H, McCormick F, Levine A. Combinatorial patterns of somatic gene mutations in cancer. The FASEB J. 2008;22:2605-2622
94. Beroukhim R, CH M, Porter D, Wei G, Raychaudhuri Sea. The landscape of somatic copy-number alteration across human cancers. Nature. 2010;463:899-905
95. Freeman J, Perry G, Feuk L. et al. Copy number variation: New insights in genome diversity. Genome Research. 2006;16:949-961
96. Maher C, Kumar-Sinha C, Cao X. et al. Transcriptome sequencing to detect gene fusions in cancer. Nature. 2009;458:97-101
97. Network TCGAR. Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature. 2008;455(23 Oct):1061-1068
98. Nautiyal S, Carlton V, Lu Y. et al. High-throughput method for analyzing methylation of CpGs in targeted genomic regions. PNAS. 2010;107(28):12587-12592
99. Lister R, Pelizzola M, Dowen R. et al. Human DNA methylomes at base resolution show widespread epigenomic differences. Nature. 2009;462:315-322
100. Zhang H, Li Y, Lai M. The microRNA network and tumor metastasis. Oncogene. 2010;29:937-948
101. Moazed D. Small RNAs in transcritional gene silencing and genome defence. Nature. 2009;457:413-420
102. Lee Y, Dutta A. MicroRNAs in cancer. Annu Rev Pathol. 2009;4:199-227
103. Garofalo M, Condorelli G, Croce C, Condorelli G. MicroRNAs as regulators of death receptors signaling. Cell Death and Differentiation. 2009;17:200-208
104. Bandyopadhyay S, Mitra R, Maulik U, Zhang M. Development of the human cancer microRNA network. Silence. 2010;1:6
105. Sotiropoulou G, Pampalakis G, Lianidou E, Mourelatos Z. Emerging roles of microRNAs as molecular switches in the integrated circuit of thecancercell. RNA. 2009;15:1443-1461
106. Dahl F, Stenberg J, Fredrikson S. et al. Multigene amplification and massively parallel sequencing for cancer mutation discovery. PNAS. 2007;104(22):9387-9392
107. Schadt E, Turner S, Kasarskis A. A window into third-generation sequencing. Human Molecular Genetics. 2010;19(2):R227-R240
108. Gleason D, Mellinger G. Prediction of prognosis for prostatic adenocarcinoma by combined histologic grading and clinical staging. J Urology. 1974;111:58-64
109. Gleason D. Histologic grading of prostate cancer: a perspective. Human Pathology. 1992;23(3):273-279
110. Xia T, Hemert J, Dickerson J. OmicsAnalyzer: a Cytoscape plug-in suite for modeling omics data. Bioinformatics. 2010;26(23):2995-2996
Author contact
![]() Corresponding author: Halliday Idikio MD, FRCPC, University of Alberta, Department of Pathology and Laboratory Medicine, 5B4.11 Walter MacKenzie Health Sciences Center, 8440-112 th Street, Edmonton, ALBERTA T6G 2B7, CANADA. Telephone 1-780-407-7271; Fax 1-780-407-3009; E-mail hidikioca
Corresponding author: Halliday Idikio MD, FRCPC, University of Alberta, Department of Pathology and Laboratory Medicine, 5B4.11 Walter MacKenzie Health Sciences Center, 8440-112 th Street, Edmonton, ALBERTA T6G 2B7, CANADA. Telephone 1-780-407-7271; Fax 1-780-407-3009; E-mail hidikioca