Impact Factor ISSN: 1837-9664

 Global reach, higher impact
Global reach, higher impactJ Cancer 2011; 2:507-514. doi:10.7150/jca.2.507 This volume Cite
Technical Report
Rapid Generation of In Vitro Multicellular Spheroids for the Study of Monoclonal Antibody Therapy
Yen T. Phung1, Dario Barbone2, V. Courtney Broaddus2, Mitchell Ho1 ![]()
1. Laboratory of Molecular Biology, Center for Cancer Research, National Cancer Institute, National Institutes of Health, Bethesda, Maryland, United States of America;
2. Lung Biology Center, University of California San Francisco, San Francisco, California, United States of America
Received 2011-9-27; Accepted 2011-10-12; Published 2011-10-13
Abstract
Tumor microenvironments present significant barriers to penetration by antibodies and immunoconjugates and are difficult to study in vitro. Cells cultured as monolayers typically exhibit less resistance to therapy than those grown in vivo. Therefore, it is important to develop an alternative research model that better represents in vivo tumors. We have developed a protocol to produce multicellular spheroids, a simple and more relevant model of in vivo tumors that allows for further investigations of the microenvironmental effects on drug penetration and tumor cell killing. The protocol is used to produce in vitro three-dimensional tumor spheroids from established human cancer cell lines and primary cancer cells isolated from patients without the use of any extracellular components. To study the ability of tumor-targeting immunoconjugates to penetrate these tumor spheroids in vitro, we have used an immunotoxin targeting mesothelin, a surface protein expressed in malignant mesotheliomas. This method for producing consistent, reproducible 3D spheroids may allow for improved testing of novel monoclonal antibodies and other agents for their ability to penetrate solid tumors for cancer therapy.
Keywords: multicellular spheroids, protocol, monoclonal antibody therapy
INTRODUCTION
Treatment of solid tumors remains a major challenge despite the availability of a vast number of anti-cancer agents. Antibody-based therapies represent a promising new approach for treating solid tumors. However, a major challenge involves delivering sufficient amounts of antibodies and immunoconjugates within tumor masses. For an anti-cancer antibody agent to be successful, it must (a) be effective in the tumor microenvironment, and (b) reach the tumor cells in optimal quantities to exert a therapeutic effect5. Tumors are more resistant to therapy than cancer cells cultured as monolayers. This can be explained by “multicellular resistance,” a mechanism for drug resistance attributed to cell-cell contacts, cell-matrix contacts, and the three-dimensional (3D) shape found in tissue5. The majority of the ~1013 cells in the human body are only within a few cell diameters of a blood vessel. This feature facilitates the delivery of oxygen and nutrients to the cells that form the tissues of the body (Fig. 1)5 and also enables the efficient delivery of most medicines.
Schematic overview of the in vivo tumor and the in vitro tumor spheroid. Comparison of the components between a tumor and spheroid model.
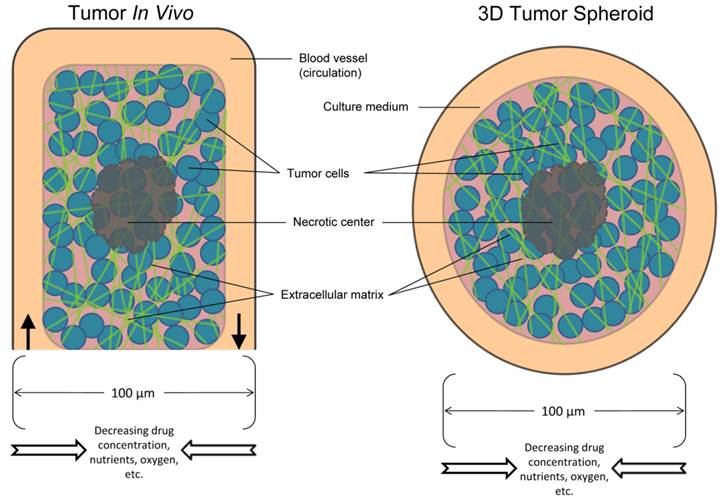
In light of this, the solid tumor microenvironment has several major characteristics, including hypoxia, large distances between blood vessels, high interstitial fluid pressure, structure and composition of the extracellular matrix, and cell-cell adhesion. Overall, these features of the tumor microenvironment limit the delivery of anticancer drugs to cancer cells that are situated far from blood vessels. Multicellular resistance acquired by tumor cells may contribute to difficulties in translating promising findings from in vitro studies into clinical therapy. In vitro multicellular cancer spheroids have begun to bridge the complexity gap between monolayer cell culture and in vivo tumors and have become valuable models in the study of drug resistance. Spheroids exhibit many features of the tumor microenvironment and model the avascular region of tumors that is dependent on diffusion (Fig. 1)4. A simple, reliable, high-throughput and less expensive in vitro tumor model would be useful for characterizing and screening antibodies and immunoconjugates for cancer therapy.
Here, we describe a detailed protocol to establish an in vitro 3D tumor spheroid model. This model can be used to identify potential new therapeutic targets that are highly expressed in mesothelioma cells in 3D spheroids, but not in monolayers, and therefore be relevant in the 3D tumor. Furthermore, this protocol may be easily applied to in vitro studies of other tumor-targeting antibodies and immunoconjugates in vitro. It may also be a useful system for screening tumor penetrating monoclonal antibodies by antibody engineering technologies such as phage display 7.
Development of the protocol
Most studies of anticancer drugs consider only cellular and/or genetic mechanisms at the level of the single cell. However, drug penetration is a highly important additional mechanism and requires a more complex cellular environment to study. Indeed, in vitro spheroid models have become the most commonly used tools to assess drug penetration. Although animal studies, when feasible, hold the advantage of mimicking the clinical environment most closely, in vitro spheroids offer the benefit of being able to examine the distribution of drugs in the absence of complicating factors such as pharmacokinetics, which often differ between mice and humans. Not only are tumor spheroids an excellent model to evaluate drug penetration, they play an increasingly meaningful role in drug discovery and development.
In 2006, Ivascu and Kubbies at Roche Pharmaceutical Research Oncology in Germany first reported a simple method to generate tumor spheroids for potential high-throughput functionality and toxicity analysis.4 Briefly, a defined number of cancer cells ranging from 1,000 to 20,000 were seeded into wells of poly(2-hydroxyethylmethacrylate)-coated, 96-well, round- or conical-bottom plates in standard growth medium and centrifuged for 10 minutes at 1000 x g. Within 24 hours of culturing, this procedure generated individual spheroids in each well with homogeneous sizes, morphologies, and stratification of proliferating cells found in the rim that also include dying cells in the core region.4 In addition, by adding basement membrane extract Matrigel to some cell lines, they were able to improve the structure from an aggregate to spheroid morphology. In 2008, after evaluating several techniques, V. Courtney Broaddus' group at the University of California San Francisco (USA) first established mesothelioma spheroids for the study of apoptotic resistance using multicellular spheroids1, modifying the method originally reported by Ivascu and Kubbies.1 Interestingly, although Broaddus' study did not use any basement membrane extract, they found the formation of spheroids to be stably intact.
Our laboratory at the National Cancer Institute (NCI) focuses on producing human monoclonal antibodies (mAbs) for the development of cancer therapy. Although leukemia treatments involving mAbs have been in clinical use for years, this approach has not been as successful for solid tumors. The proliferation of tumor cells forces blood vessels apart, reducing vascular density and creating a population of cells distant (>100μm) from vessels.5 Drugs generally do not penetrate further than three to five cell diameters from blood vessels, thereby depriving more distantly located tumor cells of any drugs. Penetrating antibody technology is increasingly seen by many to be the holy grail of antibody therapy. A limitation in our ability to identify and evaluate effective penetrating antibody reagents has been the lack of an in vitro model that recapitulates the features of a solid tumor. In the last two years, in collaboration with our group, the Takayama laboratory at the University of Michigan has produced two publications related to 3D spheroid cultures 10, 11. One study established co-cultures by introducing different types of cells on the membrane or in the bottom chamber of commercially available Transwells. They found that cancer cells formed 3D spheroids while maintaining their viability and used this procedure to evaluate mouse embryonic stem (mES) cell differentiation based on heterogeneous cell-cell interactions11. Co-culture of mES cells and HepG2 human hepatocellular cancinoma (HCC) cells decreased SOX17 expression of mES cells, and direct cell-cell contact further decreased SOX17 expression, indicating that co-culture with HepG2 cells inhibits endoderm differentiation. Another study described high-throughput 3D spheroid culture and drug testing using a 384-well format hanging drop culture plate10.
In collaboration with V. Courtney Broaddus, we made a 3D spheroid model culture using a human mesothelioma cell line and primary cell lines isolated from the ascites of malignant mesothelioma patients. Using this model, we treated spheroids with SS1P, a recombinant immunotoxin targeting mesothelin6, and evaluated how the microenvironment affected the ability of the drug to penetrate a 3D cancer cell mass and kill cancer cells.8 Mesothelin is highly expressed in mesothelioma and several other human cancers, including pancreatic adenocarcinomas, ovarian cancers, lung adenocarcinomas and cholangiocarcinoma.2, 3, 9 Although mesothelioma cells grown as monolayers or as spheroids express comparable levels of mesothelin, we observed that spheroids were at least 100 times less sensitive to SS1P and that penetration within spheroids was limited after four hours. The core area of spheroids revealed a substantial number of tight junctions, along with a significant increase in expression of E-cadherin, a protein involved in the assembly and sealing of tight junctions and highly expressed in malignant mesothelioma. Moreover, siRNA silencing and antibody inhibition targeting E-cadherin enhanced SS1P immunotoxin therapy in vitro.8
Advantages and limitations of the present protocol
3D spheroid culture is often complicated to follow and varies frequently according to individual practices. It can be cumbersome for labs with limited resources and expensive to manage. However, it is worthwhile to mention that although not all features of solid cancers, such as the influence of stroma and immune cells, are modeled by multicellular spheroids, spheroids do reflect many important properties of solid tumors, including the development of an extracellular matrix, tight junctions between epithelial cells, and gradients of nutrient concentration and cell proliferation from the exterior to the center. Spheroids can develop central necrosis and regions of hypoxia. By incubating spheroids with an anticancer drug, it then becomes possible to examine the kinetics of drug penetration. Unlike a previous method12, the protocol described here is relatively simple and straightforward to learn, rapid in producing samples, and economically feasible.
Experimental Design
NCI-H226 (American Type Culture Collection or ATCC, Rockville, MD) is a human lung squamous carcinoma cell line. HepG2 and Hep3B (ATCC) are two human liver cancer (HCC) cell lines. NCI-M-03 and NCI-M-13 (Raffit Hassan, NCI, Bethesda, MD) are two human mesothelioma primary cell lines isolated from the ascites of malignant mesothelioma patients treated at the NCI.8
MATERIALS
REAGENTS FOR FORMATION OF SPHEROIDS
- Poly-HEMA (Sigma; Cat. #P3932)
- 95% ethanol (in milliQ water)
- U-bottomed 96-well plates (Greiner; Cat. #650185)
- SS1P (Ira Pastan, Laboratory of Molecular Biology, NCI, Bethesda, MD)
- RPMI 1640 Media (Invitrogen; Cat. #21870-076) containing 10% fetal bovine serum (FBS), 1% L-glutamine (Invitrogen, Carlsbad, CA) and 1% penicillin-streptomycin (Invitrogen, Carlsbad, CA)
- FreeStyle 293 Media (Invitrogen; Cat. #12338-018)
- Accutase (BD Biosciences; Cat. #561527)
- T-75cm2 tissue culture flasks (Sarstedt; Cat. #83.1813.002)
ADDITIONAL REAGENTS FOR CHARCTERIZATION OF SPHEROIDS
- Coomassie Plus Protein Assay Reagent (Thermo Scientific; Cat. #1856210)
- RNeasy Mini Kit (Qiagen; Cat. #74104)
- Cell Counting Kit-8 (Dojindo Laboratories; Cat. #CK04)
- Costar white opaque-walled 96-well plates adequate for cell culture (Corning; Cat. #3922)
- CellTiter-Glo Luminescent Cell Viability Assay (Promega; Cat. #G7572)
REAGENT SETUP
Lysis buffer
To make a stock solution of lysis buffer, combine one “Complete Mini-EDTA free” protease inhibitor tablet (Roche; Cat. #1873580), 10 mL of RIPA buffer, and 1 mL of 20% SDS.
Poly-HEMA
Prepare a 120 mg/mL stock solution of poly-HEMA in 95% ethanol. Incubate while stirring with a sterile magnetic bar at room temperature (15-20°C) overnight. To make a working solution of Poly-HEMA, pipette 1 mL of Poly-HEMA stock solution into 23 mL of 95% ethanol to obtain a final concentration of 5 mg/mL (this volume is enough to coat four plates). Vortex solution and leave at room temperature (15-20°C) until ready to coat. Prepare fresh working solution every time new plates are made.
EQUIPMENT
Device (plate shaker) for mixing multiwell plates
Luminometer or charge-coupled device (CCD) camera imaging device capable of reading multiwell plates
PROCEDURE
Preparation of non-adsorbent poly-HEMA plates
1 Pipette 60 μL of poly-HEMA stock solution into each well of a 96-well U-bottomed plate.
TROUBLESHOOTING
2 Evaporate with lids on at room temperature (15-20°C) inside of a sterile hood for 72 hours.
TROUBLESHOOTING
3 After plates are completely dried, seal with parafilm, and store at 4ºC.
4 Before use, bring to room temperature (15-20ºC) and sterilize in the hood with lids off for 30 minutes.
Preparation of Spheroids
5 Maintain cell line as adherent monolayer cultures in RPMI 1640 medium supplemented with 10% FBS, L-glutamine, and penicillin-streptomycin.
6 Incubate at 5% CO2 with balance of air at 37°C.
7 Seed at 2x105cells/mL in T-75cm2 tissue culture flasks.
8 Change cell media twice a week.
9 Rinse the cells with PBS and add 5 mL of Accutase to detach cells and incubate at 37°C for 3-5 minutes.
10 Neutralize with 5 mL of growth media and perform a cell count.
11 Centrifuge at 216 x g at room temperature (15-20°C) for 5 minutes.
12 Dilute cell concentration to 5x104 cells/mL with growth media in order to form 10,000-cells per well spheroids (approximately 1 million cells are needed per plate).
13 Pipette 200 µL of the cell suspension into each well of the 96-well plate pre-coated with poly-HEMA.
14 Centrifuge the plate at 216 x g at room temperature (15-20°C) for 10 minutes.
15 Incubate for 24-48 hours at 37°C, 5% CO2.
TROUBLESHOOTING
Preparation of Cell Lysate from Spheroids and Monolayers
Spheroids
16 Pipette growth media and spheroid completely from each well and collect in a 50 mL tube.
17 Centrifuge at 216 x g at room temperature (15-20°C) for 5 minutes, aspirate the supernatant, and resuspend the cells with 1.5 mL of PBS.
18 Centrifuge at 216 x g at room temperature (15-20°C) for 5 minutes, aspirate the supernatant, and resuspend the cells in 50 µL of lysis buffer (for approximately 1 million cells).
19 Vortex thoroughly.
20 Repeat four cycles of freezing at 80ºC for approximately 10 minutes and thawing at 37ºC for approximately 20 minutes.
21 Centrifuge at 216 x g at room temperature (15-20°C) for 1 minute, and collect all of the supernatant.
22 Measure concentration by Coomassie stain, a reliable standard for protein quantification. Load approximately 50-100 µg of cell protein lysate per lane for western blot analysis.
Monolayers
23 Aspirate growth media of monolayer cells and rinse three times with PBS.
Δ CRITICAL STEP Allow monolayer cells to grow for two to three days after seeding until 50-60% confluent.
24 Add 2 mL of PBS and scrape monolayer cells on ice, and collect in a 15 mL tube.
25 Add another 2 mL of PBS to the plate, scrape, and collect the remaining cells.
26 Centrifuge at 216 x g at room temperature (15-20°C) for 5 minutes, aspirate the supernatant, and resuspend the cells in 50 µL of lysis buffer (for approximately 1 million cells)
27 Vortex thoroughly.
28 Repeat four cycles of freezing and thawing (-80º/37ºC).
29 Centrifuge at 216 x g at room temperature (15-20°C) for 5 minutes, and collect all supernatant.
30 Measure concentration by Coomassie stain. Load approximately 50-100 µg of cell protein lysate per lane for western blot analysis.
TROUBLESHOOTING
Purification of RNA for Microarray Analysis
31 Collect monolayer and spheroid cell lysate on three separate days.
Δ CRITICAL STEP Save a sufficient amount of cell lysate for RNA quality validation.
32 Follow manufacturer's kit instructions to purify RNA.
TROUBLESHOOTING
WST Assay
33 Form 3D spheroids on a 96-well poly-HEMA coated plate.
34 Allow monolayers and spheroids to grow for 24 hours.
35 Add 20 µL of SS1P/BL22 (negative control)/Cycloheximide (CMH) (positive control) to 200 µL of RPMI growth media per well for a 1:10 dilution.
TROUBLESHOOTING
36 Allow monolayers and spheroids to incubate for 72 hours with immunotoxin.
37 Prepare a sufficient volume of WST reagent in serum-free FreeStyle media (e.g. 1 mL of reagent in 9 mL of media).
38 Remove growth media and replace with 100 µL/well of a working solution of WST reagent- FreeStyle media mentioned in the previous step.
39 Transfer all 100 µL containing each spheroid from each well to a flat-bottom 96-well plate.
40 Allow to incubate for 2-4 hours at 37ºC until brown-yellow color develops.
41 Measure absorbance at 450 nm with reference wavelength of 650 nm.
ATP Assay
42 Form 3D spheroids on a 96-well poly-HEMA coated plate.
43 Allow monolayers and spheroids to grow for 24 hours.
44 Seed 10,000 cells/well for monolayer cells and one 10,000-cell spheroid/well containing 200 µL of RPMI growth media in a 96-well flat bottom plate.
45 Add 20 µL of SS1P and BL22 (negative control) from 1000 ng/mL to 0.0001 ng/mL to each well.
TROUBLESHOOTING
46 Incubate for 72 hours at 37ºC.
47 Transfer monolayer cells and spheroids via a pipette to opaque-walled multiwell plates in culture medium (100 µL/well for 96-well plates).
48 Prepare control wells containing medium without cells to obtain background luminescence value.
49 Equilibrate the plate and its contents at room temperature (15-20ºC) for approximately 30 minutes.
50 Add a volume of CellTiter-Glo Reagent equal to the volume of cell culture medium present in each well (e.g., add 100 µL of reagent to 100 µL of growth media containing cells for a 96-well plate).
51 Mix contents for 2 minutes on an orbital shaker to induce cell lysis.
52 Allow plate to incubate at room temperature (15-20ºC) for 10 minutes to stabilize luminescent signal.
53 Measure luminescence.
TROUBLESHOOTING
TROUBLESHOOTING
- For proper storage, the stock solution of poly-HEMA can be stored at room temperature (15-20ºC) indefinitely.
- It is optional to evaporate with the lids on at room temperature (15-20°C) on the bench for 1 to 2 weeks for slower drying, or to evaporate at 37°C inside of a humidity-free bacterial incubator for 48 hours. Depending on the type of cells, some may form tighter spheroids when incubated in this manner using pre-coated poly-HEMA plates.
- Within six hours, mesothelioma cells will begin to form as an aggregate and should be left alone as they may be easily disturbed by pipetting. However, by 24 hours, mesothelioma cells form into a disk-like shape called a multicellular spheroid and can be transferred gently by a pipette without dissociating.
- For proteins that may be more difficult to visualize or detect (e.g. E-Cadherin), use at least 100 µg of cell protein lysate per lane for western blot analysis.
- Selected GeneChip Human Genome U133 Plus 2.0 Array was performed by NCI Affymetrix Group (Frederick, MD) for reliability. For optimal results, the recommended final concentration of RNA for array analysis is 100 ng/mL in a 6 µL volume per sample.
- To properly prepare SS1P at the original concentration (0.25 mg/mL): For 100 ng/mL final concentration, add 20 µL of 1 µg/mL SS1P into 100 µL growth media per well. For 1000 ng/mL final concentration, add 20µl of 10µg/ml SS1P into 200µl growth media per well. Original concentration of BL22 (1 µg/mL): For 100 ng/mL final concentration, add 20 µL of 1 µg/mL BL22 into 200 µL growth media per well. Original concentration of CHM (100 µg/mL): For working solution, dilute 1 µL in 99 µL of growth media. For 1:1000 final concentration, add 20 µL of CHM working solution into 200 µL growth media per well.
- To properly prepare SS1P at the original concentration (0.25 mg/mL): Dilute to 10,000 ng/mL working solution (adding 20 µL of this in 200 µL of cell suspension results in a final concentration of 1000 ng/mL). For a 1000 ng/mL working solution, add 100 µL of 10,000 ng/mL SS1P in 900 µL of growth media (adding 20 µL of this in 200 µL of cell suspension results in a final concentration of 100 ng/mL). Continue to serially dilute 100 µL of SS1P in 900 µL of growth media. Original concentration of BL22 (684 µg/mL): Dilute to 10,000 ng/mL working solution (adding 20 µL of this in 200 µL of cell suspension results in a final concentration of 1000 ng/mL). For a 1000 ng/mL working solution, add 100 µL of 10,000 ng/mL BL22 in 900 µL of growth media (adding 20 µL of this in 200 µL of cell suspension results in a final concentration of 100 ng/mL). Continue to serially dilute 100 µL of BL22 in 900 µL of growth media.
- It is highly recommended to do more than one type of cell proliferation or viability assay (e.g. WST and ATP).
TIMING
Step 1-4, Preparation of non-adsorbent Poly-HEMA plates: 3 days
Step 5-15, Preparation of Spheroids: 2 days
Step 16-30, Preparation of Cell Lysate: 3 hours
Step 31-32, Purification of RNA for Microarray Analysis: 1 day
Step 33-41, WST Assay for SS1P-Treated Monolayers and Spheroids: 4 days
Step 42-53, ATP Assay for SS1P-Treated Monolayers and Spheroids: 4 days
ANTICIPATED RESULTS
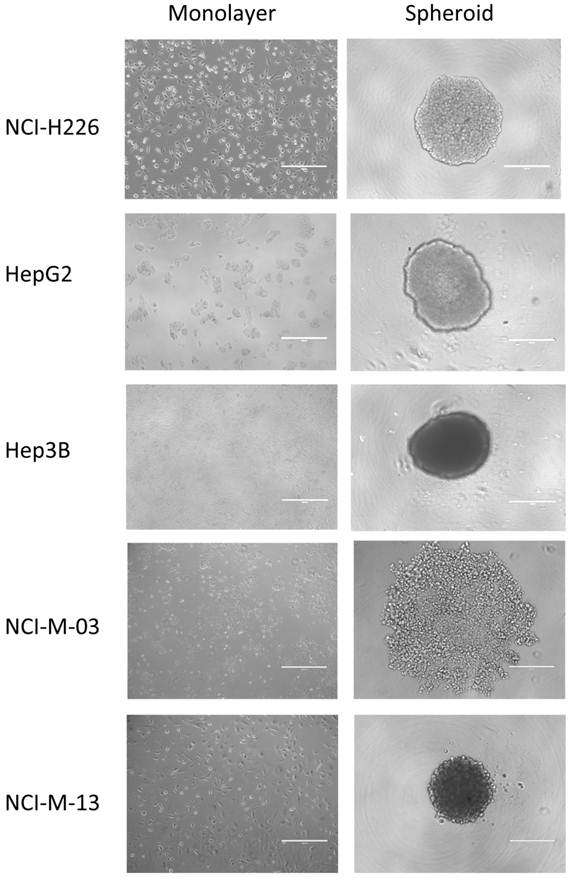
Using the experimental conditions described in this protocol, almost all of the established cancer cell lines and primary cells isolated from patients are able to form 3D spheroids in vitro (Fig. 2). We have used mesothelioma and HCC cells and found that the formation of spheroids is not tumor type-dependent.
Establishment of in vitro tumor spheroids. Microscopic images of monolayers and spheroids of human cancer cell lines, NCI-H226 (mesothelioma), HepG2 (hepatocellular carcinoma or HCC), Hep3B (HCC), and primary mesothelioma lines, NCI-M-03 and NCI-M-13, isolated from patients taken after 24 hours. Scale bars, 400 µm.
Within only 2 days after seeding cells, spheroids are ready for tumor penetration studies of antibodies or immuunoconjugates, RNA extraction for microarray analysis, protein lysis for proteomics analysis or discovery of tumor penetration antibodies by phage display and other antibody technologies.
To investigate how tumor microenvironments affect the killing activity and penetration of an antibody agent, monolayers and spheroids were treated with SS1P and a negative control. Cell growth inhibition (WST) and cell viability (ATP) assays showed that the IC50 of SS1P for spheroids was >1000 ng/mL, at least 100 times the IC50 for monolayers, ~10 ng/mL, after 72 hours. Both assays revealed that greater than 50% of the cancer cells from spheroids could not be killed by SS1P concentrations as high as 1,000 ng/mL. Finally, we tested SS1P on primary lines isolated from malignant mesothelioma patients and confirmed that SS1P was far less effective on spheroids.
We explored two different experimental approaches to examine the roles of protein regulation in regard to immunotoxin penetration. The first involved silencing E-Cadherin using siRNA. A reduction in protein expression (greater than 80%) was observed followed by an increased sensitization to SS1P therapy. Finally, an inhibitory antibody against E-Cadherin also demonstrated enhanced anti-tumor activity only if added before, but not after, spheroid formation.
Acknowledgements
This work was supported in part by the Intramural Research Program of the National Institutes of Health (NIH), National Cancer Institute (NCI), Center for Cancer Research (CCR), and in part by the Department of Defense PR080717. Dr. Mitchell Ho is supported by the 2011 NCI Director's Intramural Innovation Award (Principal Investigator Award). Dr. Ho is a co-inventor on patents assigned to the United States of America, as represented by the Department of Health and Human Services, for the investigational products. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript. The content of this publication does not necessarily reflect the views or policies of the Department of Health and Human Services, nor does mention of trade names, commercial products, or organizations imply endorsement by the U.S. Government. We thank the NIH Fellows Editorial Board for editorial assistance.
Author Contributions
MH conceived and designed the experiments. YP performed the experiments. MH, VCB and YP analyzed the data. YP and MH wrote the manuscript. YP, MH, VCB and DB revised the manuscript.
Conflict of Interest
There is no conflict of interest and the financial disclosure has been described in the Acknowledgements.
References
1. Barbone D, Yang T.M, Morgan J.R, Gaudino G, Broaddus V.C. Mammalian target of rapamycin contributes to the acquired apoptotic resistance of human mesothelioma multicellular spheroids. J. Biol. Chem. 2008;283:13021-13030
2. Hassan R, Ho M. Mesothelin targeted cancer immunotherapy. Eur. J. Cancer. 2008;44:46-53
3. Ho M. et al. Mesothelin expression in human lung cancer. Clin. Cancer Res. 2007;13:1571-1575
4. Ivascu A, Kubbies M. Rapid generation of single-tumor spheroids for high-throughput cell function and toxicity analysis. J Biomol Screen. 2006Dec;11(8):922-32
5. Minchinton A.I, Tannock I.F. Drug penetration in solid tumours. Nat. Rev. Cancer. 2006;6:583-92
6. Pastan I, Hassan R, Fitzgerald D.J, Kreitman R.J. Immunotoxin therapy of cancer. Nat. Rev. Cancer. 2006;6:559-65
7. Winter G, Griffiths A.D, Hawkins R.E, Hoogenboom H.R. Making antibodies by phage display technology. Annu. Rev. Immunol. 1994;12:433-55
8. Xiang X. et al. The development and characterization of a human mesothelioma in vitro 3D model to investigate immunotoxin therapy. PLoS One. 2011;6:e14640
9. Yu L. et al. Mesothelin as a potential therapeutic target in human cholangiocarcinoma. J Cancer. 2010;1:141-9
10. Tung YC, Hsiao AY, Allen SG, Torisawa YS, Ho M, Takayama S. High-throughput 3D spheroid culture and drug testing using a 384 hanging drop array. Analyst. 2011;136:473-8
11. Torisawa YS, Mosadegh B, Cavnar SP, Ho M, Takayama S. Transwells with Microstamped Membranes Produce Micropatterned Two-Dimensional and Three-Dimensional Co-Cultures. Tissue Eng Part C Methods. 2011;17:61-7
12. Hjelstuen MH, Rasch-Halvorsen K, Brekken C, Bruland O, de L Davies C. Penetration and binding of monoclonal antibody in human osteosarcoma multicell spheroids. Comparison of confocal laser scanning microscopy and autoradiography. Acta Oncol. 1996;35:273-9
Author contact
![]() Corresponding author: Dr. Mitchell Ho, Ph.D., Antibody Therapy Section, Laboratory of Molecular Biology, Center for Cancer Research, National Cancer Institute, National Institutes of Health, 37 Convent Drive, Room 5002C, Bethesda, MD 20892-4264, USA. Tel: (301) 451-8727; Fax: (301) 480-4667.
Corresponding author: Dr. Mitchell Ho, Ph.D., Antibody Therapy Section, Laboratory of Molecular Biology, Center for Cancer Research, National Cancer Institute, National Institutes of Health, 37 Convent Drive, Room 5002C, Bethesda, MD 20892-4264, USA. Tel: (301) 451-8727; Fax: (301) 480-4667.