Impact Factor ISSN: 1837-9664

 Global reach, higher impact
Global reach, higher impactJ Cancer 2012; 3:158-165. doi:10.7150/jca.4211 This volume Cite
Mini-Review/Case Report
Combination of the Deacetylase Inhibitor Panobinostat and the Multi-Kinase Inhibitor Sorafenib for the Treatment of Metastatic Hepatocellular Carcinoma - Review of the Underlying Molecular Mechanisms and First Case Report
Susanne Gahr1,2, Till Wissniowski1,3, Steffen Zopf1, Deike Strobel1, Anette Pustowka4, Matthias Ocker1,5 ![]()
1. Department of Medicine 1, University Hospital Erlangen, Erlangen, Germany
2. Klinikum Nuremberg Nord, Department of Pneumology, Nuremberg, Germany
3. Division of Gastroenterology, University Hospital Marburg, Marburg, Germany
4. Novartis Pharma GmbH, Nuremberg, Germany
5. Institute for Surgical Research, Philipps University Marburg, Marburg, Germany
Received 2012-2-8; Accepted 2012-3-29; Published 2012-4-9
Abstract
Advanced hepatocellular carcinoma still represents an unmet medical need that has only a limited overall survival despite the introduction of the multi-kinase inhibitor sorafenib. Recently, inhibitors of histone and other protein deacetylases have been established as novel therapeutic approaches to cancer diseases. We here review the molecular rationale for combining these two novel targeted therapies and report a patient with metastasized hepatocellular carcinoma who showed a partial remission of primary and metastatic lesions for five months after a combination therapy with sorafenib and the orally available pan-deacetylase inhibitor panobinostat.
Keywords: hepatocellular carcinoma, sorafenib, panobinostat, deacetylase inhibitor
Introduction
Hepatocellular carcinoma (HCC) is the most common primary tumor of the liver and represents the 2nd and 5th most common cause of cancer related death in men and women, respectively, worldwide[1-2]. HCC also shows rising incidence and mortality rates in Western countries due to the high prevalence of chronic viral hepatitis, alcoholic liver diseases and steatoheaptitis[3-5].
Although several molecular alterations have been identified in HCC[2, 6], so far only the multi-kinase inhibitor sorafenib proved a significant treatment advantage in liver cancer[7], while other targeted therapies like small molecule receptor tyrosine kinase inhibitors or monoclonal anti-growth factor (receptor) antibodies failed[8]. As sorafenib therapy is associated with high treatment costs[9] and still unsatisfying overall response rates[8], novel further treatment approaches are urgently needed.
Inhibitors of histone deacetylases (HDAC) have been established as potent novel anticancer therapies in hematologic and solid tumors[10-12]. We have shown previously that expression of HDAC isoenzymes is associated with overall survival in HCC patients[13] and that various HDAC inhibitors can induce cell death and synergize with chemotherapeutics in HCC models[14-16]. Panobinostat (LBH589) is a novel oral pan-deacetylase inhibitor that has shown strong pro-apoptotic and anti-proliferative effects in HCC cell lines and a xenograft model[17] and is currently under investigation for various tumor entities[18-19].
Classically, deacetylase inhibitors (DACi) are considered to modulate the transcriptional control (Figure 1) of various genes by interfering with HDAC isoforms in the nucleus and thus control chromatin packing and conformation, leading to either an “opened” and transcriptionally active or “closed” and transcriptionally suppressed DNA structure[10]. Here, a strong role for the p53-dependent regulation of the endogenous cell cycle inhibitor p21cip1/waf1 has been described for various DACi like panobinostat, vorinostat and others[20]. Recently, experimental data suggests that also regulatory miRNAs are strongly influenced by DACi and that this contributes significantly to the observed changes in gene expression[21-23]. Our own preclinical data also indicate that pan-DACi like panobinostat also influence the acetylation status of a variety of cytosolic and non-histone proteins[24], although the acetylome is still not well defined[25]. This could affect the protein folding and function of several so far unidentified proteins leading to the activation of the unfolded protein response, ER stress-mediated apoptosis and autophagy as alternative mechanisms of cell death execution[17, 26-27]. The influence of pan-DACi on cellular protein stability and function has also been demonstrated to be involved in the known anti-angiogenic effects of panobinostat, e.g. by destabilizing the hypoxia sensor Hif-1α[28] and we have shown previously in a xenograft mouse model that panobinostat leads to a significan reduction in tumor vascularization[17]. DACi can further influence signal transduction pathways related to angiogenesis by modulating the expression of growth factors like VEGF[29] or of downstream kinases like mitogen activated protein kinases (MAPK) [17, 30-31].
Molecular pathways affected by panobinostat and sorafenib. Besides the classical effects of HDACi on transcriptional regulators (e.g. p53, miRNAs, HDAC) and the regulation of p21cip1/waf1 expression, panobinostat is also capable to induce canonical (death-receptor and mitochondria related) and alternative cell death pathways (e.g. ER stress, unfolded protein response and autophagy). Although the effects of deacetylase inhibitors on protein folding and function are not completely understood, the inhibition of non-nuclear HDAC enzymes influences protein stability and enzymatic activity of various cellular proteins, including the destabilization of the hypoxia sensor Hif-1α and the function of different protein kinases. In addition, the expression of these proteins can also be affected via the shown influence on transcriptional control processes. The multi-kinase inhibitor sorafenib is predominantly inhibiting the RAS-RAF-MAPK pathway and thus inhibits processes related to angiogenesis, proliferation and survival. Together, both compounds synergize at the level of protein kinases and their downstream effectors leading to inhibition of tumor growth.
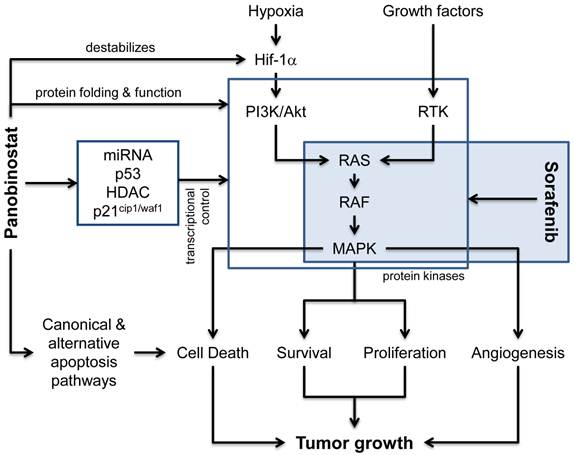
Signaling via protein kinase cascades is a hallmark of tumor cell proliferation, survival and cancer related angiogenesis. Monoclonal antibodies against pro-angiogenic growth factor receptors (e.g. bevacizumab) as well as small molecule kinase inhibitors (e.g. everolimus, sorafenib) targeting downstream signal transduction molecules have recently been introduced into treatment regimes for various advanced solid tumors. Sorafenib has been approved for the treatment of advanced stages of HCC and inhibits the RAF-Erk-MAPK signaling in tumor cells as well as in endothelial cells, thus exerting a dual effect on cell viability, proliferation and angiogenesis[32-33]. Inhibition of survival pathways by sorafenib has now been shown to lead to cell death via autophagy induction, too, and several studies also showed a synergism when combined with DACi[34-37]. Recently, the combination of panobinostat and sorafenib has been shown to exert additive anti-proliferative and pro-apoptotic effects in HCC cell culture and xenograft models[38]. The following model of action for this combination is suggested (Figure 1): DACi and kinase inhibitors synergize at the level of protein kinases, modulating these central tumorigenic pathways. Inhibition of DAC modulates protein kinase expression and activity and shifts the cellular microenvironment towards cell death induction which is then additionally hit by the kinase inhibition.
We therefore initiated a trial with the standard dose of sorafenib (800 mg daily) and a dose-escalation of panobinostat starting at 20 mg on day 1 and day 4 for 2 weeks and sorafenib alone in week 3. This cycle was repeated until disease progression or withdrawal of informed consent. The trial was approved by the local ethics committee of the University Hospital Erlangen, Germany, and is registered at clinicaltrials.gov (NCT00823290).
We here report a case of advanced metastatic HCC treated with the standard dose of sorafenib and the oral pan-deacetylase inhibitor panobinostat, which could provide a novel treatment option for advanced HCC.
Case Report
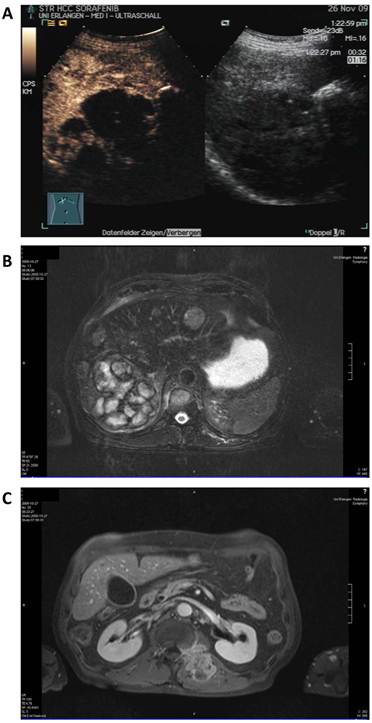
A 68-year old man was diagnosed with advanced multilocular HCC on the basis of ethyltoxic liver cirrhosis (Child-Pugh A) in September 2009. At the time of diagnosis, a moderately differentiated metastasis to the thyroid gland and several metastases to the autochthonic spine musculature and the vertebral bodies with stenoses of the spinal canal (L I) and the neuroforamina (Th XII to L I) were present (Figure 2).
Baseline radiologic assessment of the patient in September 2009 before initiation of sorafenib therapy. A: sonography showing a 10 cm lesion in the liver. B: Magnetic resonance imaging (MRI) of the liver revealing a large lesion in the right liver lobe and several diffuse smaller nodules in the total liver. C: MRI scan of the soft tissue metastasis in the autochthonic spine musculature.

Therapy with sorafenib at 800 mg per day was started in September 2009 and showed a mixed response in MRI scan 6 weeks after treatment start with partly necrotic or decreased lesions but also increasing and novel lesions in the liver and stable soft tissue and bone metastases. Sonography revealed a large lesion (11 cm) in the right liver lobe which was more than 75% necrotic but also 8 additional lesions up to 5 cm (Figure 3).
Staging after sorafenib monotherapy and before initiation of additional panobinostat treatment. A: Contrast enhanced (left) and native sonography (right) showing necrosis (> 75%) in the largest lesion. B: MRI scan of the liver showing a mixed radiologic response compared to baseline. C: MRI scan of the soft tissue metastasis.
After obtaining informed consent, therapy with panobinostat was started in November 2009: sorafenib was continued at 800 mg daily and panobinostat at 20 mg was administered orally on days 1 and 4 for two consecutive weeks followed by one week of sorafenib therapy alone, representing a three week therapy cycle which was repeated 5 times.
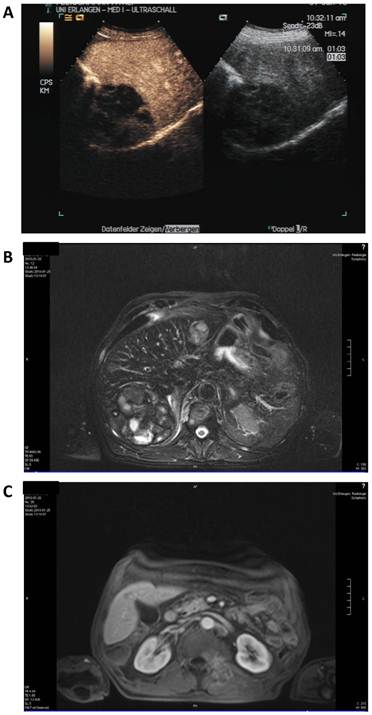
Staging was performed after three cycles in January 2010 (Figure 4). Here, contrast enhanced sonography showed constant lesions in liver segments V, VI and VII (94*80*75 mm) as well as in segment II (29*32 mm) with signs of cirrhosis but no ascites. MRI scanning showed slightly decreasing hemorrhagic lesions with signs of liquid transformation in both liver lobes and of the soft tissue metastasis in the autochthonic spine musculature. No new lesions were discovered. The patient reported diarrhea, exsiccosis, hypocalcaemia and nausea during the last cycle that responded to supportive treatment.
Staging after 8 weeks of sorafenib and panobinostat combination therapy. A: Contrast enhanced (left) and native sonography (right). B: MRI scan of the liver. C: MRI scan of the soft tissue metastasis.
After eight cycles of sorafenib and panobinostat, MRI staging in April 2010 showed a further regression of the lesions in the liver and the spine musculature. Contrast sonography showed a necrotic lesion in segment VI and a hypervascularized but still constant lesion on segment VIII.
By request of the patient the therapy with panobinostat was stopped in May 2010 as the patient experienced lack of appetite and slight weight loss (3 kg since February 2010), which was perceived as increasingly wearing by the patient and lead to withdrawal of consent. Sorafenib was further continued at the standard dose.
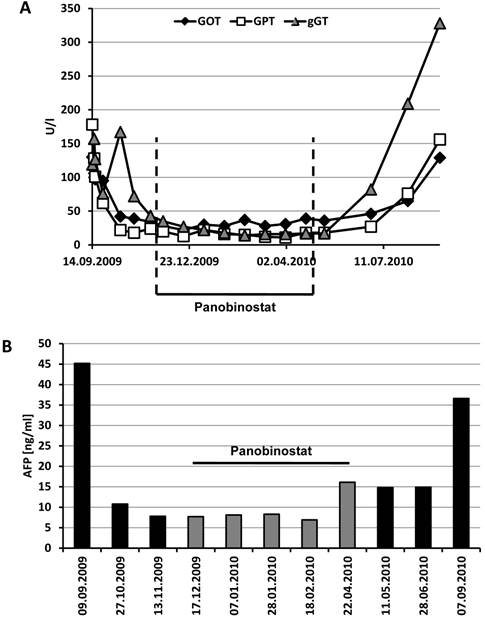
Although transaminases and γ-GT were initially elevated (maximum GOT 130 U/l, GPT 178 U/l, γ-GT 167 U/l), these parameters rapidly normalized under sorafenib therapy and stayed in the normal range until the end of treatment with panobinostat (Figure 5A). Albumin and prothrombin time as markers of liver synthesis capacity were slightly decreased throughout the treatment period without signs of clinical symptoms. Interestingly, the initially elevated tumor marker α-fetoprotein (AFP) returned to normal values already under sorafenib therapy despite a lack of radiologic response at this stage (Figure 5B). AFP levels remained below 10 ng/ml until the end of the panobinostat treatment period, too, but raised to 36.6 ng/ml at the end of the observation period. This increase in AFP was also paralleled by an increase in transaminases and γ-GT at this stage indicating a deterioration of liver function and progress of the tumor disease under sorafenib monotherapy. Hematologic assessment revealed leucopenia, low hemoglobin, erythrocytes and hematocrit under initial sorafenib therapy. These values remained stable also under additional panobinostat therapy with even an amelioration of leucopenia to normal range after three cycles. Differential blood count showed a stably lymphopenia and monocytosis during the whole treatment period. All other laboratory parameters remained in the normal range throughout the study period.
Time course of transaminases and AFP under sorafenib and panobinostat treatment. (A) Serum levels of GOT, GPT and γ-GT remained in the normal range under panobinostat (< 50 U/l for GOT and GPT, < 60 U/l for γ-GT) but rapidly increased again after cessation of panobinostat intake. (B) The tumor marker α-fetoprotein (AFP) decreased under the combination therapy with sorafenib and panobinostat but rised again under sorafenib monotherapy (normal level < 10 ng/ml). Panobinostat treatment period is indicated in the figure.
Discussion
We here report the first case of a combination therapy of the multikinase inhibitor sorafenib with the oral pan-deacetylase inhibitor panobinostat in a patient with advanced metastasized hepatocellular carcinoma.
Sorafenib has been established as the first-line therapy for advanced HCC[7]. Yet, the overall response rate still remains unsatisfying with only around 3%[8]. Typically, sorafenib induces a stable disease in the majority of patients. Our patient initially showed a mixed response to sorafenib as a single agent with some lesions showing a radiologic response while other lesions increased in size or even appeared anew. The add-on therapy with 20 mg of the pan-deacetylase panobinostat further ameliorated the clinical situation. Besides beneficial effects on tumor burden, no additional hematologic or serologic toxicities were observed, indicating a good tolerability of the combination therapy in patients with advanced HCC but early stage cirrhosis (Child-Pugh A). The serologic assessment of transaminases, γ-GT and AFP showed a quick and significant response of the patient to both, the initial sorafenib treatment and the addition of panobinostat, and thus confirmed the current concept of AFP as a predictive biomarker for HCC[39]. Our findings also confirm the pivotal role of selecting adequate imaging techniques for assessing treatment response[40].
Several preclinical studies report additive effects between multikinase inhibitors and inhibitors of histone and other protein deacetylases. Our own data with panobinostat in HCC cell lines and a xenograft model show an inhibition of proliferation pathways via upregulation of the endogenous cell cycle inhibitor p21cip1/waf1, a classical target of HDAC inhibitors[20], but independent of growth factor related kinases[17]. Yet, panobinostat was also capable of inhibiting MAPK signaling under these experimental conditions. Interestingly, MAPK is also the final downstream target of receptor tyrosine kinases and the Ras-Raf signaling pathway, which is the main target of sorafenib[41]. The dual blockade of cell cycle progression could thus represent a molecular basis for the observed potent effects of the combination therapy.
The synergism between sorafenib and histone deacetylase inhibitors has been proposed to be also mediated by the activation of CD95-mediated extrinsic apoptotic pathways[35, 42-43]. Additionally, panobinostat is also capable of inducing cell death in liver cancer models by activating alternative pathways of apoptosis induction, e.g. via activation of ER stress and autophagy mechanisms[17, 24]. Interestingly, also sorafenib has recently been demonstrated to activate autophagy and ER stress mechanisms in various settings[36-37, 44]. Several papers now reported a synergism in inducing these alternative cell death mechanisms by a combination of sorafenib and histone deacetylase inhibitors[34-35, 43].
These preclinical findings and experimental models provide a clear rationale for the good clinical tolerability and efficacy of the combination of sorafenib and panobinostat: both compounds lead to cell cycle arrest and proliferation inhibition via two independent pathways and both compounds can induce cell death by interacting with classical apoptosis signaling as well as alternative forms of cell death like autophagy. Additional effects of deacetylase inhibitors like inhibition of angiogenesis[17, 31] can further contribute to these additive effects here. Yet, these models need to be further investigated, esp. in clinical settings. Recently, the oral HDAC inhibitor resminostat also showed a favourable pharmacokinetic and safety profile in a phase I/II trial in patients with advanced and sorafenib-resistant HCC[45].
In summary, this case report demonstrates a good tolerability and efficacy of a combination therapy of sorafenib and panobinostat in a patient with HCC that needs to be evaluated further in additional clinical trials, also to identify the discussed molecular pathways of this treatment option.
Acknowledgements
We thank Petra Meier and Annette Remberg for their technical assistance.
Competing Interests
The trial was supported by Novartis Pharma GmbH, Nuremberg, Germany, which is the manufacturer of panobinostat. M.O. received honoraria from Novartis Pharma GmbH, Nuremberg, Germany and A.P. is an employee of Novartis Pharma GmbH, Nuremberg.
References
1. Jemal A, Bray F, Center MM, Ferlay J, Ward E, Forman D. Global cancer statistics. CA Cancer J Clin. 2011;61:69-90
2. Okamoto K, Neureiter D, Ocker M. Biomarkers for novel targeted therapies of hepatocellular carcinoma. Histol Histopathol. 2009;24:493-502
3. Jemal A, Siegel R, Xu J, Ward E. Cancer statistics, 2010. CA Cancer J Clin. 2010;60:277-300
4. Bosetti C, Levi F, Boffetta P, Lucchini F, Negri E, La Vecchia C. Trends in mortality from hepatocellular carcinoma in Europe, 1980-2004. Hepatology. 2008;48:137-45
5. El-Serag HB, Rudolph KL. Hepatocellular carcinoma: epidemiology and molecular carcinogenesis. Gastroenterology. 2007;132:2557-76
6. Llovet JM, Bruix J. Molecular targeted therapies in hepatocellular carcinoma. Hepatology. 2008;48:1312-27
7. Llovet JM, Ricci S, Mazzaferro V, Hilgard P, Gane E, Blanc JF. et al. Sorafenib in advanced hepatocellular carcinoma. N Engl J Med. 2008;359:378-90
8. Greten TF, Korangy F, Manns MP, Malek NP. Molecular therapy for the treatment of hepatocellular carcinoma. Br J Cancer. 2009;100:19-23
9. Carr BI, Carroll S, Muszbek N, Gondek K. Economic evaluation of sorafenib in unresectable hepatocellular carcinoma. J Gastroenterol Hepatol. 2010;25:1739-46
10. Schneider-Stock R, Ocker M. Epigenetic therapy in cancer: molecular background and clinical development of histone deacetylase and DNA methyltransferase inhibitors. IDrugs. 2007;10:557-61
11. Atadja PW. HDAC inhibitors and cancer therapy. Prog Drug Res. 2011;67:175-95
12. Lane AA, Chabner BA. Histone deacetylase inhibitors in cancer therapy. J Clin Oncol. 2009;27:5459-68
13. Quint K, Agaimy A, Di Fazio P, Montalbano R, Steindorf C, Jung R. et al. Clinical significance of histone deacetylases 1, 2, 3, and 7: HDAC2 is an independent predictor of survival in HCC. Virchows Arch. 2011Aug;459(2):129-39
14. Gahr S, Peter G, Wissniowski TT, Hahn EG, Herold C, Ocker M. The histone-deacetylase inhibitor MS-275 and the CDK-inhibitor CYC-202 promote anti-tumor effects in hepatoma cell lines. Oncol Rep. 2008;20:1249-56
15. Ocker M, Alajati A, Ganslmayer M, Zopf S, Luders M, Neureiter D. et al. The histone-deacetylase inhibitor SAHA potentiates proapoptotic effects of 5-fluorouracil and irinotecan in hepatoma cells. J Cancer Res Clin Oncol. 2005;131:385-94
16. Herold C, Ganslmayer M, Ocker M, Hermann M, Geerts A, Hahn EG. et al. The histone-deacetylase inhibitor Trichostatin A blocks proliferation and triggers apoptotic programs in hepatoma cells. J Hepatol. 2002;36:233-40
17. Di Fazio P, Schneider-Stock R, Neureiter D, Okamoto K, Wissniowski T, Gahr S. et al. The pan-deacetylase inhibitor panobinostat inhibits growth of hepatocellular carcinoma models by alternative pathways of apoptosis. Cell Oncol. 2010;32:285-300
18. Prince HM, Bishton MJ, Johnstone RW. Panobinostat (LBH589): a potent pan-deacetylase inhibitor with promising activity against hematologic and solid tumors. Future Oncol. 2009;5:601-12
19. Atadja P. Development of the pan-DAC inhibitor panobinostat (LBH589): successes and challenges. Cancer Lett. 2009;280:233-41
20. Ocker M, Schneider-Stock R. Histone deacetylase inhibitors: signalling towards p21cip1/waf1. Int J Biochem Cell Biol. 2007;39:1367-74
21. Humphreys KJ, Cobiac L, Le Leu RK, Van der Hoek MB, Michael MZ. Histone deacetylase inhibition in colorectal cancer cells reveals competing roles for members of the oncogenic miR-17-92 cluster. Mol Carcinog. 2012
22. Shin S, Lee EM, Cha HJ, Bae S, Jung JH, Lee SM. et al. MicroRNAs that respond to histone deacetylase inhibitor SAHA and p53 in HCT116 human colon carcinoma cells. Int J Oncol. 2009;35:1343-52
23. Lee EM, Shin S, Cha HJ, Yoon Y, Bae S, Jung JH. et al. Suberoylanilide hydroxamic acid (SAHA) changes microRNA expression profiles in A549 human non-small cell lung cancer cells. Int J Mol Med. 2009;24:45-50
24. Ocker M. Deacetylase inhibitors - focus on non-histone targets and effects. World J Biol Chem. 2010;1:55-61
25. Spange S, Wagner T, Heinzel T, Kramer OH. Acetylation of non-histone proteins modulates cellular signalling at multiple levels. Int J Biochem Cell Biol. 2009;41:185-98
26. Di Fazio P, Ocker M, Montalbano R. New Drugs, Old Fashioned Ways: Er Stress Induced Cell Death. Curr Pharm Biotechnol. 2011
27. Ocker M, Höpfner M. Apoptosis-modulating drugs for improved cancer therapy. Eur Surg Res. 2012 doi:10.1159/00036875
28. Qian DZ, Kato Y, Shabbeer S, Wei Y, Verheul HM, Salumbides B. et al. Targeting tumor angiogenesis with histone deacetylase inhibitors: the hydroxamic acid derivative LBH589. Clin Cancer Res. 2006;12:634-42
29. Lemoine M, Derenzini E, Buglio D, Medeiros LJ, Davis RE, Zhang J. et al. The pan-deacetylase inhibitor panobinostat induces cell death and synergizes with everolimus in Hodgkin lymphoma cell lines. Blood. 2012
30. LaBonte MJ, Wilson PM, Fazzone W, Russell J, Louie SG, El-Khoueiry A. et al. The dual EGFR/HER2 inhibitor lapatinib synergistically enhances the antitumor activity of the histone deacetylase inhibitor panobinostat in colorectal cancer models. Cancer Res. 2011;71:3635-48
31. Ellis L, Hammers H, Pili R. Targeting tumor angiogenesis with histone deacetylase inhibitors. Cancer Lett. 2009;280:145-53
32. Liu L, Cao Y, Chen C, Zhang X, McNabola A, Wilkie D. et al. Sorafenib blocks the RAF/MEK/ERK pathway, inhibits tumor angiogenesis, and induces tumor cell apoptosis in hepatocellular carcinoma model PLC/PRF/5. Cancer Res. 2006;66:11851-8
33. Adnane L, Trail PA, Taylor I, Wilhelm SM. Sorafenib (BAY 43-9006, Nexavar), a dual-action inhibitor that targets RAF/MEK/ERK pathway in tumor cells and tyrosine kinases VEGFR/PDGFR in tumor vasculature. Methods Enzymol. 2006;407:597-612
34. Park MA, Reinehr R, Haussinger D, Voelkel-Johnson C, Ogretmen B, Yacoub A. et al. Sorafenib activates CD95 and promotes autophagy and cell death via Src family kinases in gastrointestinal tumor cells. Mol Cancer Ther. 2010;9:2220-31
35. Park MA, Mitchell C, Zhang G, Yacoub A, Allegood J, Haussinger D. et al. Vorinostat and sorafenib increase CD95 activation in gastrointestinal tumor cells through a Ca(2+)-de novo ceramide-PP2A-reactive oxygen species-dependent signaling pathway. Cancer Res. 2010;70:6313-24
36. Ullen A, Farnebo M, Thyrell L, Mahmoudi S, Kharaziha P, Lennartsson L. et al. Sorafenib induces apoptosis and autophagy in prostate cancer cells in vitro. Int J Oncol. 2010;37:15-20
37. Shi YH, Ding ZB, Zhou J, Hui B, Shi GM, Ke AW. et al. Targeting autophagy enhances sorafenib lethality for hepatocellular carcinoma via ER stress-related apoptosis. Autophagy. 2011
38. Lachenmayer A, Toffanin S, Cabellos L, Alsinet C, Hoshida Y, Villanueva A. et al. Combination therapy for hepatocellular carcinoma: Additive preclinical efficacy of the HDAC inhibitor panobinostat with sorafenib. J Hepatol. 2012
39. Vora SR, Zheng H, Stadler ZK, Fuchs CS, Zhu AX. Serum alpha-fetoprotein response as a surrogate for clinical outcome in patients receiving systemic therapy for advanced hepatocellular carcinoma. Oncologist. 2009;14:717-25
40. Llovet JM, Di Bisceglie AM, Bruix J, Kramer BS, Lencioni R, Zhu AX. et al. Design and endpoints of clinical trials in hepatocellular carcinoma. J Natl Cancer Inst. 2008;100:698-711
41. Wilhelm SM, Carter C, Tang L, Wilkie D, McNabola A, Rong H. et al. BAY 43-9006 exhibits broad spectrum oral antitumor activity and targets the RAF/MEK/ERK pathway and receptor tyrosine kinases involved in tumor progression and angiogenesis. Cancer Res. 2004;64:7099-109
42. Zhang G, Park MA, Mitchell C, Hamed H, Rahmani M, Martin AP. et al. Vorinostat and sorafenib synergistically kill tumor cells via FLIP suppression and CD95 activation. Clin Cancer Res. 2008;14:5385-99
43. Walker T, Mitchell C, Park MA, Yacoub A, Graf M, Rahmani M. et al. Sorafenib and vorinostat kill colon cancer cells by CD95-dependent and -independent mechanisms. Mol Pharmacol. 2009;76:342-55
44. Bareford MD, Park MA, Yacoub A, Hamed HA, Tang Y, Cruickshanks N. et al. Sorafenib Enhances Pemetrexed Cytotoxicity through an Autophagy-Dependent Mechanism in Cancer Cells. Cancer Res. 2011;71:4955-67
45. Bitzer M, Horger M, Ganten T, Ebert MP, Siveke J, Woerns MA. et al. Clinical Update of the SHELTER Study: A Phase I/II Trial of HDAC Inhibitor Resminostat in Patients with Sorafenib-Resistant Hepatocellular Carcinoma (HCC). Ann Oncol. 2011;22:v76-v
Author contact
![]() Corresponding author: Prof. Dr. med. Matthias Ocker, Institute for Surgical Research, Philipps University Marburg, Baldingerstrasse, 35033 Marburg, Germany. ockeruni-marburg.de; Phone: +49-6421-5868930; Fax: +49-6421-5868926
Corresponding author: Prof. Dr. med. Matthias Ocker, Institute for Surgical Research, Philipps University Marburg, Baldingerstrasse, 35033 Marburg, Germany. ockeruni-marburg.de; Phone: +49-6421-5868930; Fax: +49-6421-5868926