Impact Factor ISSN: 1837-9664

 Global reach, higher impact
Global reach, higher impactJ Cancer 2012; 3:196-206. doi:10.7150/jca.4484 This volume Cite
Research Paper
Establishment of Human Ultra-Low Passage Colorectal Cancer Cell Lines Using Spheroids from Fresh Surgical Specimens Suitable for In Vitro and In Vivo Studies
Satyajit Ray1, Russell C. Langan1, John E. Mullinax1, Tomotake Koizumi1, Hong-Wu Xin1, Gordon W. Wiegand1, Andrew J. Anderson1, Alexander Stojadinovic2,3, Snorri Thorgeirsson4 ![]() , Udo Rudloff1, Itzhak Avital1,5
, Udo Rudloff1, Itzhak Avital1,5 ![]()
1. Surgery Branch, Center for Cancer Research, National Cancer Institute, NIH, Bethesda, MD;
2. Department of Surgery, Division of Surgical Oncology, Walter Reed National Military Medical Center, Bethesda, MD;
3. Department of Surgery, Uniformed Services University of the Health Sciences, Bethesda, MD;
4. Laboratory of Experimental Carcinogenesis, Center for Cancer Research, National Cancer Institute, NIH, Bethesda, MD;
5. Bon Secours Cancer Institute, Richmond VA, USA
Received 2012-4-17; Accepted 2012-5-6; Published 2012-5-7
Abstract
Colorectal cancer (CRC) holds the third highest incidence and cancer related mortality rate among men and women in the United States. Unfortunately, there has been little progression made in the treatment of this deadly disease once it has spread beyond the colon. It has been hypothesize that colon cancer stem cells are implicated in CRC carcinogenesis, metastasis, and therapeutic resistance. One of the difficulties in testing these hypotheses is the current use of established high-passage cancer cell lines. Long term, high-passage established cell lines have cells with stem like properties as they propagate almost indefinitely. These cells are thought to be different than the original cancer stem cells in fresh tumors. In order to investigate cancer stem cells, and molecularly profiling tumors with high fidelity to the original primary tumor, one needs to establish suitable primary ultra-low passage cell lines from fresh surgical specimens. Here we report the establishment of tumor initiating colon cancer ultra-low passage cell lines by a combination of gentle mechanical, enzymatic dissociation, spheroid formation, and followed by two generation xenografts from fresh tumors obtained at time of operation. Tumors generated were characterized by morphology, flow cytometry, immunofluorescence, and by gene expression. In the future, such a technology can be used to produce expeditiously enough material to test for mutations, genetic signatures and molecular subtyping readily available for clinical therapeutic decision making.
Keywords: Colon cancer, Fresh Tumor, Cell lines, Spheroids, Cancer Stem Cells.
Introduction
Colorectal cancer (CRC) is one of the most aggressive solid organ cancers and holds the third highest incidence and mortality rate amongst the US population [1]. Among patients with CRC, approximately 50% will eventually develop metastases and once metastases ensue prognosis is poor. Recent work suggests the existence of cancer stem cells in solid organ cancers including neoplasms of the colon and rectum [2-4]. Literature suggests the existence of cancer stem cells in neoplasms of the colon and rectum [2-4]. It has been hypothesize that colon cancer stem cells are implicated in CRC carcinogenesis, metastasis, and therapeutic resistance. Isolation of live cancer stem cells has been a limiting step for further investigation of these hypotheses. An in vitro model of CRC better reflecting the original tumor must be developed through the establishment of ultra-low passage primary cell lines from clinically available fresh surgical specimens.
Though difficult, the establishment of clinically relevant ultra-low passage cell lines from fresh tumors is potentially more conducive for the study of cancer because they are likely to represent more accurately the primary tumor. Cells derived from fresh tumors may not directly grow in vitro, from each patient every time leading to waste of precious samples and data. Previously, several studies reporting on isolation and culture of fresh human colon epithelial tumor cells were critiqued for inefficiency [6-9]. Attempts were made to establish cell culture from fresh human colon tumors on a short term basis [10]. Importantly, in the future, parallel testing of mutations, genetic signatures, chemotherapy and targeted therapy sensitivities will likely be done routinely for every patient. Therefore, there is a tremendous need for a technology that will generate expeditiously ultra-low passage cancer cell lines producing large quantities of cells, for multiple clinical testing. The importance of generating ultra-low passage cancer cell lines cannot be over emphasized. In this study, we report a successful method for the establishment of ultra-low passage colon cancer primary cell lines from fresh surgical specimens using a combination of gentle mechanical, enzymatic dissociation, spheroid formation, and followed by two generation xenografts formation.
Materials and Methods
Patients/tissue specimens
Fresh tumors were obtained from patients with radiographic, biochemical, and histologically proven CRC who were treated at the Clinical Research Center, National Cancer Institute (NCI), National Institutes of Health (NIH), USA. All patients were enrolled on a tissue procurement protocol. Fresh tumors were obtained during surgical resection of CRC primary and metastatic sites (Table 1). For purposes of this study, we selected one patient with early stage cancer and another with advanced metastatic cancer. The tissue procurement protocol was approved by the NCI Institutional Review Board prior to enrollment and treatment of all patients (IRB Protocol: 09-C-0079).
Characteristics of patient's and tumors used to establish ultra-low passage colorectal cancer cell lines.
| CSCL-01-WE | CSCL-03-BA | |
|---|---|---|
| Age at Diagnosis | 41 | 44 |
| Gender | Female | Female |
| Primary Disease Site | Rectum | Colon |
| Stage | IV | I |
| Primary Pathology | T4N3M1 | T1N0M0 |
| Grade | Moderately Differentiated Adenocarcinoma | Well Differentiated Adenocarcinoma |
| Classification of specimen used for culture | Fresh Tumor | Fresh Tumor |
Tumor cell isolation and primary culture
Immediately following resection, tumors were placed into 40C normal saline with antibiotics and antifungal, and transported to the laboratory. Once in the laboratory, using sterile technique, excess fat and normal tissue were removed and the tumors were washed with 1X HBSS. Using sharp dissection, tumors were minced into 1-3 mm3 chunks and approximately 3g of minced tumor was transferred into a gentleMACS C tube (Miltenyi Biotec, Germany) which contained 5 ml of dissociation medium. Dissociation medium contained DMEM/F12 (1:1) growth medium (Mediatech, USA) supplemented with 1 mg/ml of collagenase, type IV (Sigma, USA) and 10U/ml of pulmozyme (DNase, Dornase alfa inhalation solution, Genentech, USA). Minced tumors were dissociated gently by gentleMACS dissociator (Miltenyi Biotec) to yield cell suspensions with a high viability rate. Cell suspensions were created by the gentleMACS dissociator (Miltenyi Biotec, Germany) through a series of programmable gentle dissociations and incubations in humidified atmosphere containing 5% CO2 at 370C. Cell suspensions were then filtered through sterile 70 µm nylon cell strainers (BD Falcon) washed with 50 ml 1X HBSS and centrifuged at 1400 rpm (40C) for 10 min. Cells were re-suspended using standard growth medium. Standard growth medium contained DMEM/F12 (1:1) supplemented with 10% fetal bovine serum (Gibco, USA), 10% glutamine (Gibco, USA), 2% penicillin/streptomycin solution (Gibco, USA), and 1.25 µg/ml of Fungizone (Amphotencin B, Gibco, USA). Cell count and viability were determined by the trypan blue dye exclusion method using a T4 Cellometer (Nexcelom Bioscience LLC, USA).
Primary organoid cultures
Cell suspensions were plated into ultra-low attachment tissue culture flask (Type and producer) at a concentration of 1x106/ml and cultured in humidified atmosphere containing 5% CO2 at 370C. At this stage, cells were grown in suspension for 7 days until formation of organoid bodies. Growing cells in suspended spheroid-bodies is thought to increase the relative proportion of stem-like cells, and to rid of the fibroblast thus, better potential to result in a stable cell line. Suitability for cell propagation and morphology were determined in vitro by a phase-contrast microscope (Olympus). Spheroid bodies were passaged by fluid aspiration and filtration, and used for xenografting immediately or cryopreserved for future studies, at a concentration of 5x106/ml with freezing medium in liquid nitrogen.
Establishment of ultra-low passage stable human colorectal cancer cell lines
Spheroid-bodies were harvested from primary fresh tumor suspension cultures, and injected subcutaneously into Nude SCID mice, strain: Crl:SHO-PrkdcscidHrhr (Charles River, USA). 3x106 cells were resuspended in 100 µl of 25% matrigel (BD Biosciences, USA) diluted with 1XHBSS, and injected subcutaneously into mice. After the tumor grew approximately to 2 cm2, xenografts were harvested and redigested and cultured as described above. Cell suspensions were placed into ultra-low attachment tissue culture flask for spheroid formation at a concentration of 1x106/ml with the same growth medium and conditions. Spheroids were harvested after 6 days, resuspended with 25% matrigel as described above and were re-injected into the same strain of mice. After the tumor grew approximately to 2cm2, xenografts were harvested, redigested, and cultured as described above. However, this time (passage 3, surgery-to-cell line) into normal adherent tissue culture flasks generating ultra-low passage stable cell lines. Subsequently, cells were grown for several passages with DMEM/F12 (1:1) growth medium supplemented with 10% FBS (Gibco, USA), 10% glutamine (Gibco, USA), and 1% penicillin streptomycin (Gibco, USA) to test for growth stability.
Histological and morphological evaluation of tumors
At each passage of tumor cells, mouse xenografts were fixed in 4% paraformaldehyde and embedded into paraffin blocks. Embedded tissues were then sliced into 6 microns sections. Sections were fixed onto glass microscope slides and H&E staining was performed. Cellular morphology and histology was determined by a phase-contrast microscope (Olympus) by a board-certified Pathologist.
Fluorescence confocal microscopy
To validate the cell of origins, we used fluorescence confocal microscopy to test cell cultures established from mice xenografts for carcinoembryonic antigen (CEA) expression. To retrieve the antigen, 50,000 cells were centrifuged onto a charged glass slide and fixed with 4% paraformaldehyde for 15 minutes at room temperature. Slides were subsequently immersed in 10 mM Sodium Citrate Buffer, pH 6.0 and heated for 3 minutes at 1200C in a pressure boiler. Cells were permeabilized in PBS containing 0.25 % Triton-X-100 for 10 minutes at room temperature. To assess for CEA, goat polyclonal pan CEA (D-19) primary antibody (10 µg/ml, Santa Cruz Biotechnology, USA) and Alexa Fluor® 546 donkey anti-goat IgG (H+L) (1:200 dilution, Invitrogen, USA) secondary antibody were applied for 1 hour at room temperature. To assess CD45 expression, mouse monoclonal anti-human CD45 primary antibody (10 µg/ml, Invitrogen, USA), and Alexa Fluor® 633 goat anti-mouse IgG (H+L) (1:200 dilution, Invitrogen, USA) secondary antibody were applied for 1 hour at room temperature. Slides were mounted with Vectashield/DAPI stain (Vector Laboratories H-1200) and stored at 4°C in darkness prior to confocal microscopy. Confocal images were captured with Zeiss AIM software on a Zeiss LSM 510 Confocal System (Carl Zeiss Inc.) with a Zeiss Axiovert 100M inverted microscope and 50 mW argon UV laser tuned to 364 nm and a 1 mW HeNe laser tuned to 546 nm, or a 5 mW HeNe laser tuned to 633 nm. A 20x objective was used at digital zoom settings of 1 or 2. Emission signals after sequential excitation of DAPI (blue) and Alexa-Fluor 555 (red) or Alexa Fluor 568 (pseudo- red) by 364 nm and 543 nm or 633 nm laser lines were acquired with a BP 385-470 filter and LP 560 filter or LP 650 filter, respectively, using individual photomultipliers. Bone marrow cells of NOD/SCID gamma mouse and CD45 positive fraction of human whole blood cells were used as controls.
Analysis of CEA and CD45 expression using flow cytometry
CEA and CD45 expression was assessed in the newly established cell lines by fluorescence-activated cell sorting (FACS). In order to analyze the cells, 100,000 cells were stained separately with PE-conjugated mouse monoclonal antibody (mAb) against Human CEA (Abcam, USA; CAT# ab42796) and PE-conjugated mouse monoclonal antibody (mAb) against Human CD45 (BD Biosciences, USA; CAT# 555483). Cells were incubated with the antibodies for 30 min at 4°C in total darkness. Cells were washed three times with PBS containing 10 % FCS (Crystalgen, USA; CAT# FCS-50) and analyzed on a BDFacsAriaII flow cytometer (BD Biosciences, San Jose, USA). Bone marrow cells of NOD/SCID gamma mouse and CD45 positive fraction of human whole blood cells were used as controls.
Validation of CEA and CD45 by real-time quantitative reverse-transcriptase polymerase chain reaction (qRT-PCR)
Total RNA was isolated from the various cell lines tested using a miRNeasy Mini kit with RNase-Free DNase Set (Qiagen, USA) according to the manufacturer's protocol. The concentration and the purity of the total RNA was determined using ultraviolet spectrophotometry (NanoDrop Spectrophotometer ND-1000, USA). Sample quality was deemed acceptable if the OD 260/280 nm ratio was >1.9. Samples were stored at -80°C until downstream applications were undertaken. Reagents for real-time qRT-PCR, genomic DNA elimination, reverse-transcription, pre-amplification, and primer mix were purchased from SABiosciences (Frederick, MD, USA). GAPDH housekeeping gene was used as internal control. The reverse-transcription cocktail was prepared according to the manufacturer's instruction. After eliminating genomic DNA, equal volume of reverse-transcription cocktail was added to the RNA sample and incubated at 42°C for 30 minutes. The reaction was stopped by heating at 95°C for 5 minutes. Real-Time qRT-PCR was performed using SABioscience RT2 master mix in an ABI 7900 HT system (Applied Biosystems, USA) following the supplier's protocol. The thermal conditions for real-time qRT-PCR were set at 95°C for 10 minutes for denaturation, followed by 40 cycles of polymerase chain reaction with denaturation at 95°C for 15 seconds and annealing/extension at 60°C for 2 minutes. Ct values were analyzed using the SABioscience Company's online software.
Results
Establishment of ultra-low passage human colorectal cancer cell lines
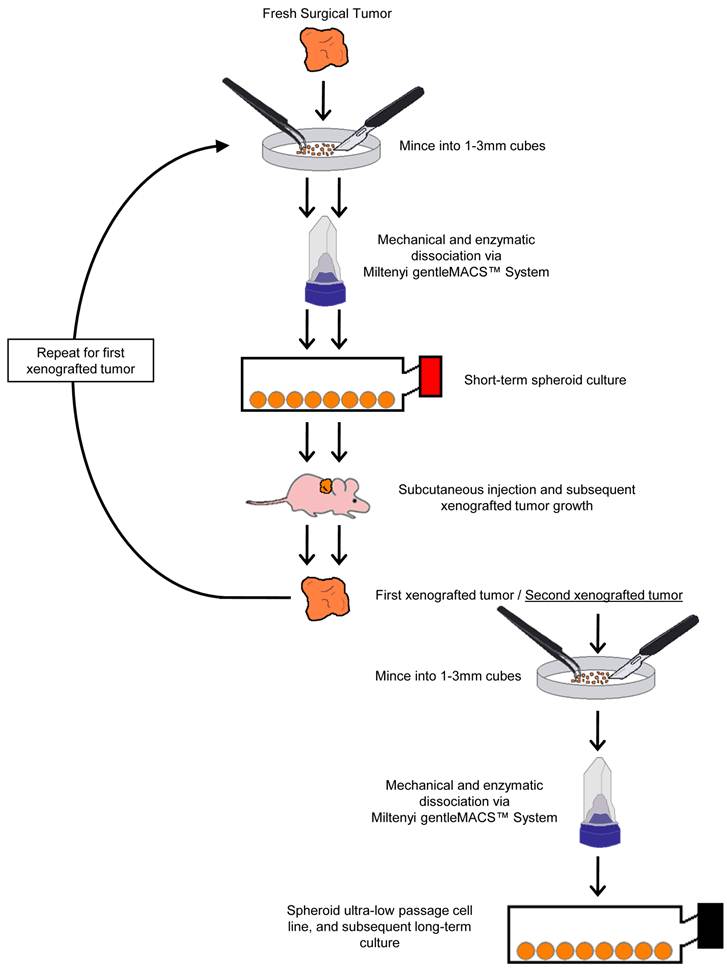
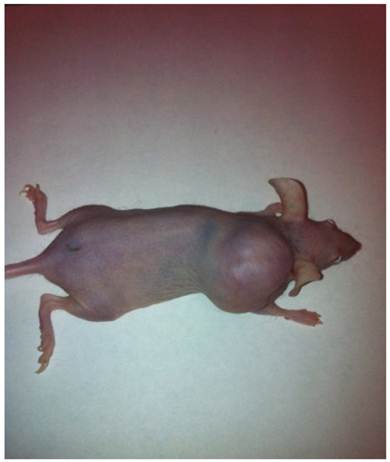
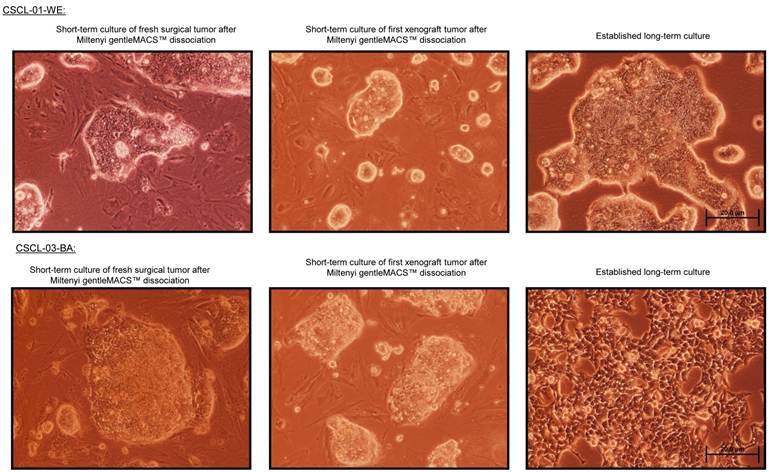
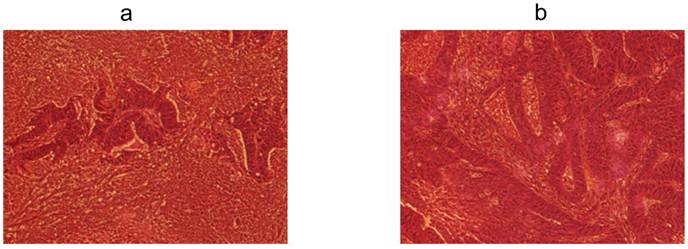
Two colorectal cancer cell lines (CSCL-01-WE and CSCL-03-BA) were established from primary tumors of two colorectal cancer patients (Table 1). Figure 1 summarizes the methodological approaches in details. Figure 2 shows a tumor formation (approximately 2cm2) by CSCL-01-WE cells after only 4 weeks of subcutaneous injection of 3 million cells. Combination of gentle mechanical and enzymatic dissociation of resected tumor provided us adequate number of cells for culture with relatively high efficiency 55-60% viability. These cell lines grew both on tissue culture treated adherent flaks as a colony and formed spheroids in ultra-low attachment flask. Figure 5 is a representative pictograph of spheroid formation. Figure 3 shows the sequential in vitro growth pattern and morphology of the cells before subcutaneous injection to animal, after 1st xenograft formation, and final established cell lines. Final cell lines were able to recapitulate tumors in nude SCID mice. These xenograft tumors matched the histology of the patients' primary tumors by H&E staining (Figure 4).
Summary of the methodological approach used for generating readily available ultra-low passage colon cancer cell lines using the organoid approach.
A tumor formed by CSCL-01-WE cells after 4 weeks.
Phase-contrast microscopy of the cell lines CSCL-01-WE and CSCL-03-BA (100X, Scale bar: 20µm).
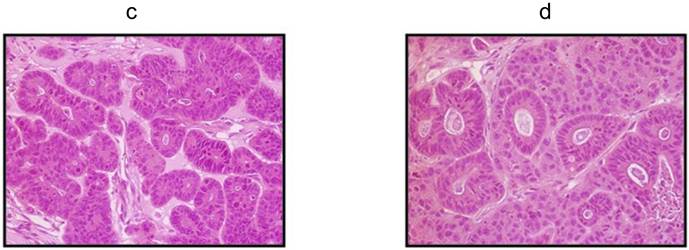
Phase-contrast microscopy of the patients' primary tumors and xenograft tumors by H&E staining. a H&E staining of the CSCL-01-WE patient's primary rectal tumor showing moderately to poorly differentiated adenocarcinoma. b H&E staining of the CSCL-03-BA patient's primary colon tumor showing moderately differentiated adenocarcinoma. c H&E staining of the first xenograft tumor formed by CSCL-01-WE cells showing moderately differentiated adenocarcinoma. d H&E staining of the first xenograft tumor formed by CSCL-03-BA cells.
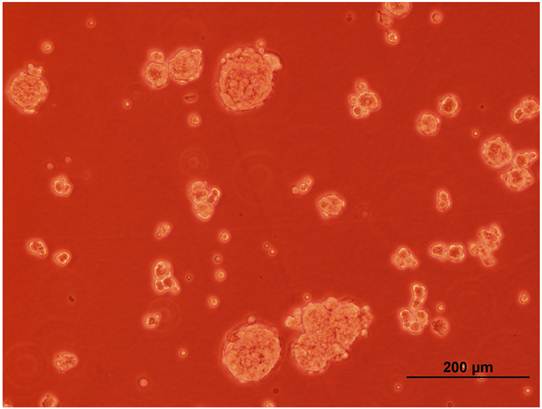
A representative pictograph of spheroid formation.
Validation of the colorectal cancer origins of the ultra-low passage cell lines'
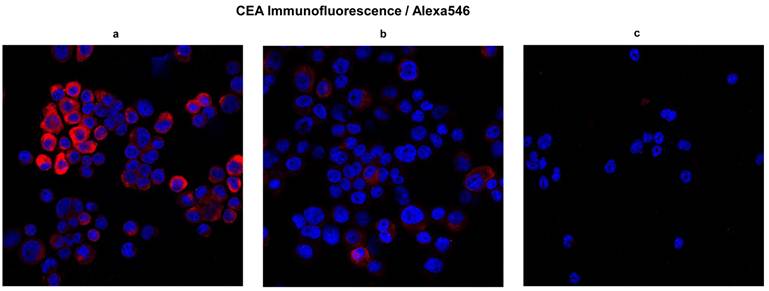
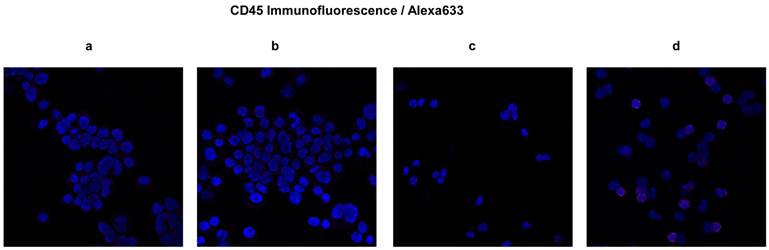
H&E staining can validate morphologically the similarity between the original tumor and the resulting xenograft tumors. However, morphological validation is not sufficient. Immunofluorescence confocal microscopy for CEA and CD45 performed on cells passage 6 and later revealed the presence of CEA and relative absence of CD45 in both established cell lines. We tested CD45 expression to mitigate the argument that hematopoietic derived cells might proliferate in these conditions (spheroids) and falsely resemble tumors. The majority of cells were CEA positive in both the CSCL-01-WE and CSCL-03-BA established cell lines (Figure 6). Both of the cell lines were negative for CD45 expression therefore confirming their non-hematopoietic origin (Figure 7).
CEA expression. To validate CRC origins, here we show CEA expression in cells obtained from final digest. In blue, nuclei stained with DAPI; in red, CEA . a and b Micrographs showing presence of CEA in CSCL-01-WE cells and CSCL-03-BA cells respectively. c Micrograph showing absence of CEA marker in bone marrow of NOD/SCID gamma mouse (negative control).
Expression of CD45 in ultra-low passage cell lines developed as spheroids. Expression of CD45 was studied by immunofluorescence confocal microscopy. In blue, nuclei stained with DAPI; in red, CD45. a, b and c Micrographs showing absence of CD45 marker in CSCL-01-WE cells, CSCL-03-BA cells, and in bone marrow of NOD/SCID gamma mouse (negative control) respectively. d Micrograph showing presence of CD45 marker in the CD45 positive fraction of human whole blood cells (positive control).
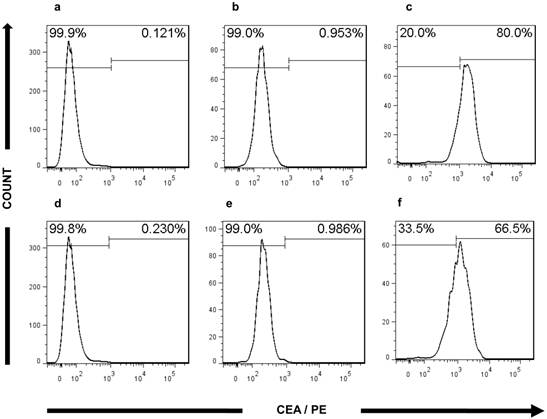
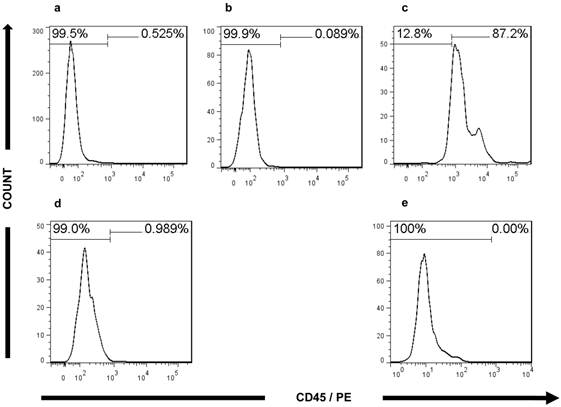
In addition, FACS analysis further validated the CRC characteristics of the ultra-low passage cell lines. The expression of CEA in both cell lines (80% in CSCL-01-WE and 66.5% in CSCL-03-BA) supported the microscopic findings, confirming adenocarcinoma formation (Figure 8). In both of the established cell lines, less than 1% of the cells were CD45 positive (Figure 9), thus confirming the presence of tumor cells, and not a proliferation of inflammatory infiltrate isolated from the original tumor.
Flow cytometry analysis of CEA expression. a, d Histograms of bone marrow from NOD/SCID gamma mouse stained with PE-conjugated mouse monoclonal antibody against Human CEA (negative control). b and e Histograms showing absence of CEA marker in CSCL-01-WE cells, CSCL-03-BA cells respectively, when no antibody was used (technical control). c and f Histograms of CSCL-01-WE cells, CSCL-03-BA cells respectively stained with PE-conjugated mouse monoclonal antibody against Human CEA showing presence of CEA in both cell line.
Flow cytometry analysis of CD45 expression. a Histograms of bone marrow of NOD/SCID gamma mouse stained with PE-conjugated mouse monoclonal antibody against Human CD45 (negative control). b Histograms of CD45 positive fraction of human whole blood cells without any antibody (technical control). c Histograms of CD45 positive fraction of human whole blood cells stained with PE-conjugated mouse monoclonal antibody against Human CD45 (positive control). d, e Histograms of CSCL-01-WE cells, CSCL-03-BA cells respectively stained with PE-conjugated mouse monoclonal antibody against Human CD45 showing absence of CD45 signal.
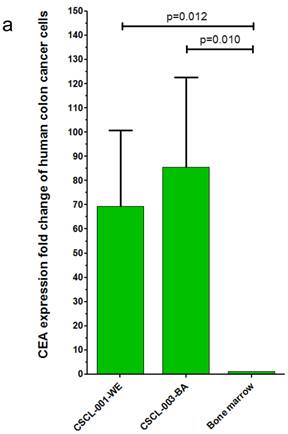
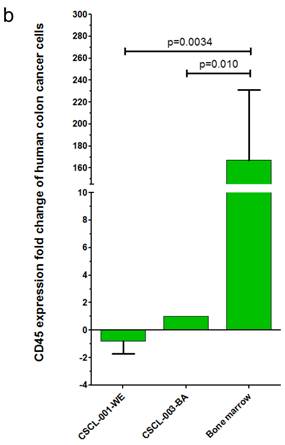
Finally, gene expression analysis using qRT-PCR further validated the CRC origins of the tumors generated by the dual approach: Organoid-Xenografts. These results revealed that CEA was highly expressed in both established cell lines, 69.27 +- 31.39 (p=0.010) folds in CSCL-01-WE and 85.50 +- 37.04 (p=0.012) folds in CSCL-03-BA compared to (negative control) mouse bone marrow cells (Figure 10a There was no significant CD45 expression in these cell lines (Figure 10b).
a) Real-time qRT-PCR analysis, CEA expression. Here we shows that the two cell lines derived from the original patient's tumors express CEA. b) Real-time qRT-PCR analysis, CD45 expression. Here we shows that the two cell lines derived from patient tumors express extremely low levels of the hematopoietic cell marker CD45.
Discussion
The establishment of ultra-low passage CRC cell lines readily available for clinical testing and cancer stem cells investigations is technically challenging. Time invested in establishing CRC cell lines directly from patient samples pays dividends as these lines serve as an optimal in vitro model for downstream testing. Here we show that using a method thought to be conducive for stem cells proliferation results in a reliable method to produce ultra-low passage tumor initiating cell lines that can provide expeditiously enough cells for further investigations.
Because of the importance of the cancer stem cells hypothesis suggesting a role in tumor initiating, metastasis and therapeutic failure for cancer stem cells, [2-4], we hypothesize that generating ultra-low passage cell lines will result in a more accurate depiction of the original tumor. Although intuitive this hypothesis demands further study and beyond the scope of this paper. Thus the purpose of the current study was to establish ultra-low passage human colorectal cancer cell lines directly from surgical specimens.
In this report, we present our optimized protocol using a spheroid enhanced culture with two generation xenografts. This was preferred for two reasons. First, the spheroid formation in this approach permitted us to eliminate fibroblasts and other non tumor cells from the culture. Second, literature suggests spheroid formation maintains stem-like characteristic of the cancer cells [11]. We previously reported a novel methodology to isolate unique population of cancer stem cells i.e. label retaining cancer cells (LRCC) [5]. To test whether LRCC isolated for these cell lines express cancer-stem cells markers, we isolated LRCC as putative cancer stem cells and non- label-retaining cancer cells (non-LRCC) as described previously [5]. Real-time qRT-PCR analysis showed that LRCC express significantly higher levels of aldehyde dehydrogenase 1 family member A1, a colorectal cancer stem cells marker (ALDH1A1, 2.6 +/- 0.3 folds, p=0.0005) and delta-like 1 (DLL1, 139.6 +/- 23.8 folds, p=0.0054) compared to non-LRCC (Supplementary Figure 1). Other investigators reported that ALDH1A1 is a cancer stem cell marker [12] and DLL1 is an important factor in the Notch signaling pathway that is required to maintain colon stem cells phenotype [12, 13]. High levels of ALDH1A1 and DLL1 gene expressions in LRCC suggest that the established cell lines would serve as a good tool for cancer stem cell research.
In summary, we have demonstrated a convenient method for the establishment of ultra-low passage primary human CRC cell lines from fresh surgical specimens. This protocol could be used to establish cell lines from fresh surgical specimens for other histologies. Generation of large quantities of purified ultra-low passage cancer cells will be imperative in the future for “real-time” molecular and sensitivity testing expeditiously available for clinical decision making. The protocol presented in this report is important as it offers an optimal in vitro model for a bone fide method for the production of ultra-low passage cell lines from fresh surgical specimens.
Supplementary Material
Supplementary Figure 1Contributing Author Declaration
We certify that all individuals who qualify as authors have been listed; each has participated in one or more of the following areas: conception and design of this work, the acquisition and/or analysis of data, the writing, and/or critical revision of the document, and supervision of this cooperative research effort. All contributing authors approve of the submission of this version of the manuscript and assert that the document represents valid work. If information derived from another source was used in this manuscript, we obtained all necessary approvals to use it and made appropriate acknowledgements in the document. All contributing authors take public responsibility for this work.
This study was supported by the NIH grant 1ZIABC011005-03.
Copyright protection
Some of the contributing authors are military service members (or employees of the U.S. Government: AS), and this work was prepared as part of their official duties. Title 17 U.S.C. 105 provides the “Copyright protection under this title is not available for any work of the United States Government.” Title 17 U.S.C. 101 defines a U.S. Government work as a work prepared by a military service member or employee of the U.S. Government as part of that person's official duties.
Disclaimer
The views expressed in this manuscript are those of the authors and do not reflect the official policy of the Department of the Army, the Department of Defense or the United States Government.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Siegel R, Ward E, Brawley O, Jemal A. Cancer statistics, 2011: the impact of eliminating socioeconomic and racial disparities on premature cancer deaths. CA Cancer J Clin. 2011;61:212-36
2. O'Brien CA, Pollett A, Gallinger S, Dick JE. A human colon cancer cell capable of initiating tumour growth in immunodeficient mice. Nature. 2007Jan4;445(7123):106-10
3. Ricci-Vitiani L, Lombardi DG, Pilozzi E, Biffoni M, Todaro M, Peschle C, De Maria R. Identification and expansion of human colon-cancer-initiating cells. Nature. 2007Jan4;445(7123):111-5
4. Visvader JE, Lindeman GJ. Cancer stem cells in solid tumours: accumulating evidence and unresolved questions. Nat Rev Cancer. 2008Oct;8(10):755-68
5. Hari D, Xin HW, Jaiswal K, Wiegand G, Kim BK, Ambe C, Burka D, Koizumi T, Ray S, Garfield S, Thorgeirsson S, Avital I. Isolation of Live Label-Retaining Cells and Cells Undergoing Asymmetric Cell Division via Nonrandom Chromosomal Cosegregation from Human Cancers. Stem Cells Dev. 2011 [Epub ahead of print]
6. Gibson PR, van de Pol E, Maxwell LE, Gabriel A, Doe WF. Isolation of colonic crypts that maintain structural and metabolic viability in vitro. Gastroenterology. 1989Feb;96(2 Pt 1):283-91
7. Baten A, Sakamoto K, Shamsuddin AM. Long-term culture of normal human colonic epithelial cells in vitro. FASEB J. 1992Jun;6(9):2726-34
8. Stauffer JS, Manzano LA, Balch GC, Merriman RL, Tanzer LR, Moyer MP. Development and characterization of normal colonic epithelial cell lines derived from normal mucosa of patients with colon cancer. Am J Surg. 1995Feb;169(2):190-5
9. Luca T, Privitera G, Lo Monaco M, Prezzavento C, Castorina S. Validation study of a cell culture model of colorectal cancer. Ital J Anat Embryol. 2007;112(2):81-92
10. Failli A, Consolini R, Legitimo A, Spisni R, Castagna M, Romanini A, Crimaldi G, Miccoli P. The challenge of culturing human colorectal tumor cells: establishment of a cell culture model by the comparison of different methodological approaches. Tumori. 2009;95(3):343-7
11. Gou S, Liu T, Wang C, Yin T, Li K, Yang M, Zhou J. Establishment of clonal colony-forming assay for propagation of pancreatic cancer cells with stem cell properties. Pancreas. 2007;34:429-35
12. Sullivan JP, Spinola M, Dodge M, Raso MG, Behrens C, Gao B, Schuster K, Shao C, Larsen JE, Sullivan LA, Honorio S, Xie Y, Scaglioni PP, DiMaio JM, Gazdar AF, Shay JW, Wistuba II, Minna JD. Aldehyde dehydrogenase activity selects for lung adenocarcinoma stem cells dependent on notch signaling. Cancer Res. 2010Dec1;70(23):9937-48
13. Pellegrinet L, Rodilla V, Liu Z, Chen S, Koch U, Espinosa L, Kaestner KH, Kopan R, Lewis J, Radtke F. Dll1- and dll4-mediated notch signaling are required for homeostasis of intestinal stem cells. Gastroenterology. 2011Apr;140(4):1230-1240
Author contact
![]() Corresponding author: Snorri S.Thorgeirsson, M.D., Ph.D. Head, Center of Excellence in Integrative Cancer Biology and Genomics Chief, Laboratory of Experimental Carcinogenesis, Center for Cancer Research, National Cancer Institute, NIH, 37 Convent Drive MSC 4262, Bethesda, MD 20892-4262, USA. Phone: 1+ (301) 496-1450(O), Phone: 1+ (301) 496-1935(W), Fax: 1+ (301) 496-0734, E-mail: snorri_thorgeirssongov. Or Itzhak Avital, MD, Medical Director, Bon Secours Cancer Institute, Richmond VA. itzhak.avitalcom, Phone: 1+ (804) 285-3225.
Corresponding author: Snorri S.Thorgeirsson, M.D., Ph.D. Head, Center of Excellence in Integrative Cancer Biology and Genomics Chief, Laboratory of Experimental Carcinogenesis, Center for Cancer Research, National Cancer Institute, NIH, 37 Convent Drive MSC 4262, Bethesda, MD 20892-4262, USA. Phone: 1+ (301) 496-1450(O), Phone: 1+ (301) 496-1935(W), Fax: 1+ (301) 496-0734, E-mail: snorri_thorgeirssongov. Or Itzhak Avital, MD, Medical Director, Bon Secours Cancer Institute, Richmond VA. itzhak.avitalcom, Phone: 1+ (804) 285-3225.