Impact Factor ISSN: 1837-9664

 Global reach, higher impact
Global reach, higher impactJ Cancer 2012; 3:354-361. doi:10.7150/jca.4813 This volume Cite
Research Paper
Heterogeneity of p53-pathway Protein Expression in Chemosensitive Chronic Lymphocytic Leukemia: A Pilot Study
Michael J Groves1*, Stephanie F MacCallum1*, Michael T Boylan1, Sally Haydock2, Joan Cunningham2, Keith Gelly3, Duncan Gowans3, Ron Kerr3, Philip J Coates1,4, Sudhir Tauro1,3 ![]()
1. Dundee Cancer Centre, University of Dundee, Ninewells Hospital and Medical School, Dundee, Scotland, United Kingdom. DD19SY;
2. Department of Cytogenetics, Ninewells Hospital and Medical School, Dundee, Scotland, United Kingdom. DD19SY;
3. Department of Haematology, Ninewells Hospital and Medical School, Dundee, Scotland, United Kingdom. DD19SY;
4. Tayside Tissue Bank, Ninewells Hospital and Medical School, Dundee, Scotland, United Kingdom. DD19SY.
* MJG and SFM contributed equally as first authors.
Received 2012-7-3; Accepted 2012-8-16; Published 2012-8-24
Abstract
The presence of p53-pathway dysfunction in chronic lymphocytic leukemia (CLL) can be used to identify patients with chemotherapy-refractory disease. Therapeutic responses are known to vary between patients with chemosensitive CLL and may relate to differences in p53-pathway activity. We hypothesized that the magnitude or type of p53-pathway protein expression is heterogeneous in patients with chemosensitive disease and could associate with white cell responses. In this pilot study, changes in p53 and its transcriptional targets, p21/waf1 and MDM2 were analyzed by immunoblotting and densitometry in CLL cells from 10 patients immediately prior to the start of chemotherapy, and after culture for 24 hours (h) with fludarabine (n=7) or chlorambucil (n=3). The in vitro response was also compared to that in vivo in circulating cells pre-treatment, and at 24h and 96h of chemotherapy. Disease responses were evident in all patients after the first treatment-cycle. Significant p53 induction was observed in CLL cells treated in vitro and in vivo. Greater heterogeneity in the expression-intensity was observed in vivo (σ2=45.15) than in vitro (σ2=1.33) and the results failed to correlate (r2=0.18, p=0.22). p21/waf1 and MDM2 expression-profiles were also dissimilar in vitro and in vivo. Higher in vivo (but not in vitro) responses associated with changes in white cell count (p=0.026). Thus, heterogeneity of p53-pathway activity exists in chemosensitive CLL; in unselected patients, in vivo changes do not correlate with those in vitro, but may associate with post-treatment white cell responses.
Keywords: p53, CLL, in vivo, in vitro.
Introduction
A number of pre-treatment variables are capable of determining prognosis in chronic lymphocytic leukemia (CLL).1,2 Of these, the identification of TP53 mutations is associated with a poor response to chemotherapy3,4 since DNA damage responses are impaired in the presence of mutant TP53. Accordingly, abnormal p53-pathway protein expression has been observed in leukemic cells with mutant TP53 following in vitro radiation.5-7 In these radiation-based assays, it is also possible to identify p53-pathway dysfunction predictive of poorer outcomes, despite the presence of wild-type TP53.7-9 Comparative in vivo and in vitro assays using chemotherapy (instead of radiation) as the genotoxic stimulus have yielded similar results, further indicating differences in p53-pathway activity between patients with chemosensitive and refractory disease.10-12 Thus, the identification of reduced p53-pathway activity due to a variety of genetic aberrations including TP53 or ATM mutations or p21/waf1 polymorphisms is predictive of aggressive disease.3-12
Most CLL patients however have chemosensitive disease that is responsive to alkylating agents and nucleoside analogue drugs,13,14 with the addition of the anti-CD20 monocloncal antibody Rituximab further improving outcomes.15 The speed and depth of responses following standard chemotherapeutic regimens are known to vary amongst patients,14 but underlying mechanisms have not been investigated. Given the central role of wild-type p53 in CLL responses, and the observations that the cellular outcome of p53 activation may relate to the strength of induction,16,17 variation in the magnitude of p53-pathway activation could be responsible for heterogeneity of disease responses. Alternatively, there is evidence for differential induction of pro- or anti-apoptotic downstream targets of p53 under different circumstances, that may also influence the overall response to therapy.18-20 Therefore, in this pilot study, we have investigated whether it is possible to detect variation in the induction of p53 and its downstream proteins in patients with chemosensitive CLL. Simultaneously, we have investigated whether protein expression following exposure to genotoxic chemotherapy in vitro and in vivo correlate, and associates with immediate white cell responses.
Patients and Methods
Patient selection
Ten patients (median age 73 years, range 56-79) starting their first cycle of oral treatment with fludarabine (24mg/m2/day) and cyclophosphamide (150mg/m2/day) for a total of 5 days (n=7), or chlorambucil (10mg/day) for 7 days (n=3) for symptomatic progressive CLL were included in the study. Six patients had received no previous therapy. The study was approved by the Tayside Committee on Medical Research Ethics and written informed consent was obtained from all patients.
CLL cell separation and processing
Peripheral blood mononuclear cells (MNC) were isolated by density gradient centrifugation (Ficoll-PaqueTM Plus) at three time points: immediately prior to therapy (baseline) and at 24 and 96 hours after start of treatment. These time-points were arbitrarily selected, but have been validated in studies on the temporal course of p53-dependent gene-expression in CLL cells following treatment with fludarabine.10-12 The enrichment for neoplastic cells was confirmed by the co-expression of CD5 and CD19 in >92% of immunolabelled cells by flow-cytometry (FACSscan) (not shown). Standard protocols were used to extract genomic DNA (BioRobot EZ1, Qiagen); RNA (Tissue Mini Kit, Qiagen) and proteins using buffer containing 1%Tergitol-type NP-40, 0.5% sodium deoxycholate, 0.1% SDS, protease and phosphatase inhibitors. An aliquot of MNC was also cyropreserved.
TP53 sequence analysis
Exons 5-9 of TP53 gene (common 'hot-spot' for mutations affecting DNA-binding domain) were amplified by PCR using genomic DNA extracted from cells prior to therapy. The purified products were directly sequenced using the ABI PRISM® BigDye™ Terminators V 3.0 sequencing kit and run on an ABI 3130 genetic analyzer (Applied Biosystems).
Fluorescence in situ hybridization
Interphase flourescent in situ hybridisation (FISH) studies were carried out on CLL MNC using the CEP17/TP53 (17p13.1) and ATM (11q22.3) probes (Vysis). Samples were treated with a hypotonic solution (50% potassium chloride) and fixed in methanol/acetic acid (3:1) solution. Fixed material was dropped onto clean slides, dehydrated and denatured for 5 minutes in 70% formamide/saline sodium citrate solution at 72 °C. CEP17/TP53 and ATM probes were denatured at 72°C for 5 minutes and hybridization was performed at 37°C overnight. Slides were washed at 72°C for 2 minutes in 0.4x saline-sodium citrate (SSC)/0.3% Nonidet P40 (NP40) and at room temperature for 1 minute in 2xSSC/0.1% NP40 and mounted in DAPI/Antifade (Vysis). Samples were evaluated using an Olympus BX60 (Olympus UK Ltd, Southend-on-Sea, UK) epifluorescence microscope equipped with the appropriate filter sets. Images were taken with a charge-coupled device camera using the CytoVysion image analysis and capture system (Leica Microsystems, UK). At least 100 cells were scored by two independent scorers (blinded to the identity of the specimen) against a normal control sample.
Cell culture
Cryopreserved MNC (2 x 106 cells/ml) obtained prior to therapy were cultured in RPMI/10% fetal calf serum at 37oC for 24 hours in the presence of the same drug(s) used to treat individual patients. Trypan-Blue exclusion assays suggested that greater than 99% of cells were viable prior to culture. In our hands, studies comparing viability of freshly isolated and cryopreserved cells in paired cultures using terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) or cleaved caspase-3 expression have indicated equivalent levels of apoptosis, not influenced by cryopreservation (r=0.98; p=0.003). In addition, where possible, expression profiles of p53-pathway proteins from cultures established using freshly isolated or cryopreserved cultured CLL cells were compared and found to be identical, in keeping with the previously described concordance of biological phenomena in fresh and cryopreserved CLL cells.10,21
Cells were incubated with 1μg/ml 9-μ-D-arabinosyl-2-fluoroadenine monophosphate (Sigma) or 15μM chlorambucil. Negative controls consisted of cells in medium containing 0.1% DMSO. Cellular protein was extracted after culture.
Intracellular protein expression
Changes in p53-pathway proteins were studied by immunoblotting. The following antibodies were used: anti-p53 (DO1, 1:100), anti-p21/waf1 (clone 118; 1:50) and anti-MDM2 (2A10, 1:50). Anti-p53, p21 and MDM2 antibodies were gifted by Dr B Vojtesek, Masaryk Memorial Cancer Institute, Brno, Czech Republic. Proteins from normal blood MNC cultured with cisplatin and phytohemagglutinin (PHA), and untreated MNC probed with antibodies on the same membrane served as positive and negative controls respectively. To control for protein loading, each membrane was re-probed with a mouse monoclonal antibody to β-actin (AC-74; Sigma).
Immunoreactive bands for p53 were quantified by densitometry in a linear range using calibrated office scanner and ImageJ software, and the ratio of the immunoreactive p53 protein band and β-actin was calculated. Levels of p53 at 24 and 96 hours were expressed as a fraction of pre-treatment values or controls (for in vitro studies) on the same membrane and expressed as the fold-change. Repeat analysis of protein extracts from cells collected before and during treatment, and following cell culture was performed to ensure reproducibility of results.
CD44 cell-surface expression
The expression of CD44, a cellular adhesion molecule expressed at high levels in CLL cells in patients with advanced disease22 is known to be down-regulated by p53 via binding to a noncanonical p53-binding sequence in the CD44 promoter.23 Potential changes in CD44 expression, reflecting functional p53-pathway responses were studied by flow-cytometry following chemotherapy. In brief, 1 x 106 freshly isolated or cultured CLL cells were stained with murine anti-human antibodies labeled with CD19 (FITC) and CD44 (PE) (BD Pharmingen, San Jose, USA). Negative controls were incubated with mouse IgG1 (BD Pharmingen, San Jose, USA). Fluorescence was measured by flow-cytometry using a Becton-Dickinson FACScan immediately after staining
Statistical analysis
The arithmetic mean was used to measure the central tendency of data and the dispersion of values around the mean was expressed as the standard deviation (s.d.) in analysis of raw data, i.e. mean±s.d. The significance of difference between means was tested using Student's t-test or Wilcoxon Two Sample Test (depending on the variances within data). All p-values were two-tailed and statistical-significance was set at the level of p<0.05.
Results
Analysis of TP53 sequence and ATM
Molecular analysis revealed the presence of mutant TP53 (exons 5-9) in one patient (c.736A>G: p.Met246Val mutation in codon 246) and del(17p) was identified by FISH in 20% of circulating cells in this patient. In another patient, monoallelic deletion of ATM was identified in 117/120 cells.
Expression of p53 in vitro and in vivo (Figure 1A)
All data on in vitro p53-pathway activity reported here are from 24 hour assays since longer periods of culture resulted in reduced cell recovery and weaker expression of β-actin making accurate interpretation of p53-expression intensity unreliable. Consistent p53 induction was observed at 24 hours of culture with fludarabine or chlorambucil (Figures 1A and B), indicating integrity of signaling mechanisms that stabilize p53 in all patients. In vivo, a mean 2-fold increase over baseline levels was observed at 24 hours (24h-p53); by 96 hours, the mean-intensity (96h-p53) had increased to 7.8-fold over baseline levels (p=0.003), with the variance (σ2=45.15) indicating greater heterogeneity in the strength of p53-induction than in vitro (σ2=1.33, Figure 1A).
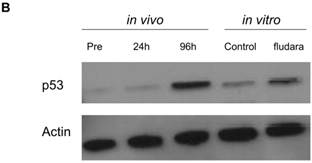
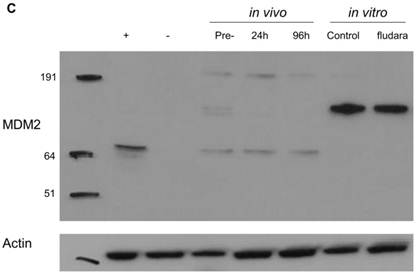
Changes in p53 pathway-protein expression in CLL cells following chemotherapy in vivo and in vitro. p53 levels in circulating CLL cells at 24 hours (24 hours in vivo) and 96 hours (96 hours in vivo) following the start of chemotherapy were quantified and expressed as the fold-change over pre-treatment values (see Patients and Methods for details). In Figure 1 A, the in vivo results from individual patients can be compared with the corresponding p53 induction in cells cultured with chemotherapy, expressed as the fold-difference in expression over untreated cells (in vitro). In Figure 1B, the gel image from Patient 7 is used to exemplify the heterogeneity in the magnitude of p53 response in vivo and in vitro. Here, in vitro assays with fludarabine accurately captured trends of in vivo p53 expression (fludara) over untreated cultures (Control), but the response-magnitude was dissimilar. Figure 1C shows the expression of MDM2 to be discordant in vivo and in vitro (Figure 1C) and different from the expression profile in normal peripheral blood mononuclear cells (MNC) treated with cisplatin (+). No MDM2 was detected in freshly isolated normal MNC (-).
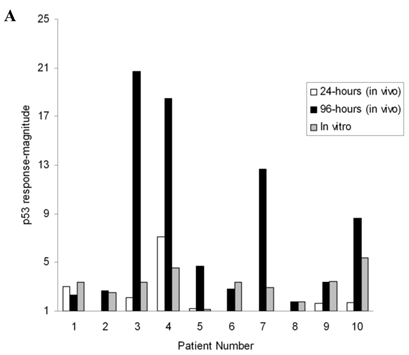
When the in vitro and in vivo expression intensities were compared, the response in cultured cells was greater than the 24h-p53 (p=0.049), but lower than 96h-p53 (p=0.055). In the entire cohort, no correlation was observed between the p53-induction magnitude assayed in vitro and in vivo (r2=0.18, p=0.22), with discordant results evident in 6 at 24 hours and 5 patients at 96 hours (Figure 1A, and exemplified in Figure 1B). In particular, the p53-expression intensity in vivo and in vitro appeared to be dissimilar in 5 out of 7 patients on fludarabine and cyclophosphamide.
Expression of p53-transcriptional target proteins
When downstream protein targets of p53 were analyzed, 4 patients expressed p21/waf1 in vitro. No expression of p21/waf1 was detected in vivo at 24 hours, but was detectable in 4 patients at 96 hours. In vitro p21/waf1 was identified in only one of these patients. Patients with detectable p21/waf1 in vivo had a trend towards a higher p53 response-magnitude (12.6±8-fold increase over baseline) compared to patients with no p21/waf1 induction (4.6±4-fold p=0.07), although this association was not evident in vitro. Dissimilarities of MDM2 expression were also observed in vitro and in vivo (Figure 1C). Complex patterns of MDM2 expression between 40 kDa to 191 kDa were observed in vivo but not in cultured cells, suggesting that post-transcriptional or post-translational modifications in vitro could be different to those occurring in vivo. Despite p53 induction in all 10 patients, MDM2 expression remained unchanged in 7 patients in vitro and in vivo, with an increase observed in only 3 patients.
In contrast to the discordant expression of p21/waf1 and MDM2 in vitro and in vivo, the expression of CD44 (100%) and the mean fluorescence intensity of CD44 expression remained stable in vitro and at 24 and 96 hours despite significant induction of p53.
Post-chemotherapy responses
Following the first cycle of chemotherapy, all patients had chemosensitive disease with 4 partial (PR) and 6 complete remissions (CR).24 The responses include CR in the patient with del(17p) suggesting a low burden of TP53 mutant cells at the time of therapy. PR in the patient with monoallelic loss of chromosome 11q (ATM) suggested likely preservation of p53-pathway signaling through the residual wild-type ATM allele.8 Accordingly, induction of p53 following chemotherapy was evident in both these patients.
Changes in WCC relative to p53-pathway protein expression
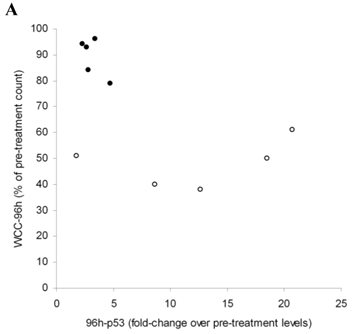
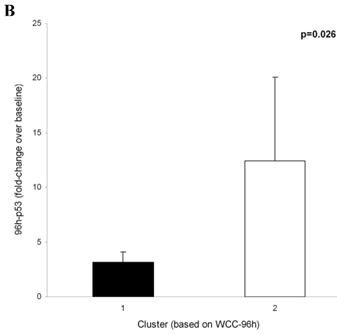
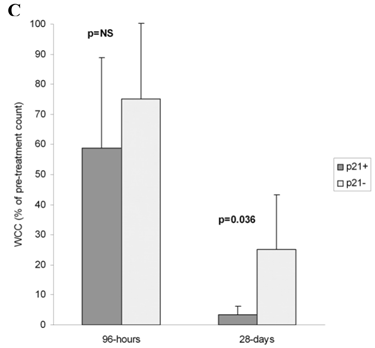
No association was observed between WCC at 24 hours (WCC-24h) or 96 hours (WCC-96h) and the p53 induction-intensity in vitro or at 24 hours in vivo. When the 96-hour p53 response-magnitude in vivo was compared to changes in WCC using two-step cluster analysis and Akaike's information criterion (SPSS), two clusters based on WCC-96h were identified (Figure 2A). The mean WCC-96h in Cluster 1 (89±7.3%of pre-treatment WCC, n=5) was higher than in Cluster 2 (48±9.3%, p<0.0001). Significantly, in Cluster 1, the mean in vivo p53 response-magnitude at 96 hours (3±0.9-fold over baseline levels) was lower than in Cluster 2, where the p53 expression-magnitude registered a mean increase of 12±7.6-fold (p=0.026, Figure 2B). The WCC-28d in Cluster 2 was significantly lower than patients in Cluster 1 (4.6±2.5% and 28.2±17.3% of pre-treatment values respectively, p=0.016). The induction of p21/waf1 in vitro did not associate with WCC-28d, but the expression at 96 hours in vivo associated with lower WCC-28d (3.2± 3.9%, n=4) compared to patients with p53 but no p21/waf1 induction (25± 18%, n=6 p=0.036, Figure 2C).
Changes in white cell counts (WCC) relative to p53-pathway protein expression. The induction-intensity of p53 (relative to baseline values) at 96 hours (96h-p53) was compared with corresponding WCC at 96 hours (WCC-96h). Two-step cluster analysis identified two clusters based on WCC-96h (Figure 2A): Cluster 1 (•) had a higher WCC, and the mean increase in 96h-p53 here was 4-fold lower than in Cluster 2 (ο) (p=0.026, Figure 2B). Co-expression of p21/waf1 with p53 at 96 hours in vivo associated with lower WCC-28d (3.2± 3.9%, n=4) than in patients without p21/waf1 induction (25± 18%, n=6, p=0.036) (Figure 2C), but did not impact on the WCC at 96 hours.
Pre-treatment characteristics of patients treated with chlorambucil (CLB) or fludarabine and cyclophosphamide (FC). Unique numbers can be used to identify individual patients in Figure 1. Clinical and hematological parameters stipulated by IWCLL were used to define partial (PR) or complete remissions (CR) at the end of the first cycle of chemotherapy.
| ID | Gender | Current treatment | Previous treatment | Interval from previous therapy (months) | Response |
|---|---|---|---|---|---|
| 1. | Male | CLB | None | NA | PR |
| 2. | Male | CLB | CLB | 19 | PR |
| 3. | Male | FC | CLB | 0.5 | CR |
| 4. | Female | FC | None | NA | CR |
| 5. | Female | FC | CLB, FC | 18 | PR |
| 6. | Female | FC | None | NA | CR |
| 7. | Male | FC | None | NA | CR |
| 8. | Male* | FC | CLB | 2 | CR |
| 9. | Male** | CLB | None | NA | PR |
| 10. | Female | FC | None | NA | CR |
NA: not applicable.
*patient with del(17p) in 20% of cells and c.736A>G: p.Met246Val mutation in codon 246 of TP53.
**patient with monoallellic deletion of ATM.
Discussion
The inability to activate p53-pathway function in CLL cells following exposure to genotoxic agents in vitro reliably identifies patients with genetic aberrations resulting in a severely compromised p53 pathway and disease that is aggressive or poorly responsive to chemotherapy.3-12 However, few studies have investigated whether quantifiable heterogeneity of p53-pathway function exists in patients responding to chemotherapy. In some cellular models, activation of p53 can lead to distinct outcomes of either growth-arrest or apoptosis through the differential induction of distinct down-stream targets (for a review, see refs. 25 and 26). Evidence that the decision between growth arrest and apoptosis could be dependent on the levels of induced p5316,17 suggests that potential variation in the magnitude of the p53 response to genotoxic stimuli could account for different levels of leukemic control in chemosensitive disease. Alternatively, there are suggestions that activated p53 may preferentially activate apoptotic or growth arrest programs depending on cellular contexts including cell types and genetic background.16,17,19,26 In contrast, human follicular lymphoma cells undergo apoptosis following therapeutic irradiation, despite equivalent induction of apoptotic or growth arrest programs in vivo.27 Thus, the ability to identify differences in p53 stabilization levels and functional activation of specific downstream targets in CLL would enable a correlation with clinical responses and potentially, the development of dose-adapted strategies.
Here, by directly comparing p53-pathway activity in vitro and in vivo in patients responding to chlorambucil or combination therapy with fludarabine and cyclophosphamide, we have demonstrated significant induction of p53 in these patients, in accordance with previous studies.10-12 However, while assays identify heterogeneity of response-magnitude in vitro and in vivo, the results do not correlate, particularly in patients receiving fludarabine and cyclophosphamide. Similar discordance in expression of p21/waf1 and MDM2 expression was also observed. These results contrast with the similarity of p53-downstream target induction observed at the transcription level in cultured cells and from patients receiving fludarabine,10-12 and may be related to differences in post-transcriptional regulation in vitro and in vivo, or differences in assay sensitivity. In particular, MDM2 expression profiles varied widely between patients, with variable dominance of 75-kDa, 90kDa or higher molecular weight proteins in different patients, as reported previously.28
In contrast to p21/waf1 and MDM2, the proportion of cells expressing CD44 and the mean fluorescence intensity of expression remained identical in vitro and in vivo, despite significant expression of p53. Although not commonly studied in p53-pathway function assays in CLL, CD44, the antigenic moniker for a protean ensemble of cell-surface transmembrane glycoproteins is known to promote tumour migration and adhesion,29 and is known to be expressed on CLL cells.22 Since the expression of CD44 is reportedly repressed by p53 in solid organ malignancies,23 we had expected down-regulation of CD44 on CLL cells following chemotherapy and investigated its potential as a measurable marker of p53-pathway activity. The stability of surface CD44 observed even after significant p53-induction in CLL cells suggests that the regulatory relationship between p53 and CD44 may differ in circulating CLL cells compared to other malignancies.
It is important that the clinical correlates of our study are interpreted cautiously. The failure of the in vitro p53-expression-magnitude to associate with early (96 hour) and late (28 day) WCC responses could reflect the inadequacy of current culture techniques to simulate the in vivo microenvironment and migration in CLL.12 p53 expression in vivo at 96 hours associated with early WCC responses and patients co-expressing p21/waf1 in vivo (but not in vitro) experienced a greater reduction in WCC at 28 days, in agreement with previous studies9 indicating the clinical significant of p21/waf1 induction in ex vivo genotoxic assays of p53-pathway function. However, given the size of our patient cohort, and the higher p53-response magnitude in patients co-expressing p21/waf1, it is not possible to establish at present whether p21/waf1 induction is an independent predictor of white cell responses.
In conclusion, our pilot study has indicated that it is feasible to identify variation in p53-pathway activity amongst unselected patients with chemosensitive CLL ex vivo, but assay-results do not corrrelate with the heterogeneity of protein expression observed in vivo. Possible differences between the strength of the genotoxic stimulus in vitro and in vivo in individual patients, and potential selection bias during sampling of circulating cells means that developing chemotherapy-based in vitro culture systems to prospectively predict the magnitude of in vivo p53-stabilization is likely to be challenging. A larger study on uniformly treated patients is now required to confirm the importance of p53-p21/waf1 activity in vivo to immediate and longer-term disease responses.
Abbreviations
ATM: Ataxia Telangiectasia Mutant; CLL: Chronic Lymphocytic Leukemia; CR: Complete remission; p21/waf1: cyclin-dependent kinase inhibitor 1; d: day; FISH: Fluorescence in situ hybridization; h: Hour; MNC: Mononuclear cells; MDM2: Murine double minute; NP40: Nonidet P40; PR: Partial remission; SSC: saline-sodium citrate; TP53: Tumour Protein 53; WCC: White cell count.
Acknowledgements
We thank NHS Tayside Acute Services Division, Dundee for funding the study and Dr Andrew Cassidy, Genetics Core Services Manager, University of Dundee, Ninewells Hospital, Dundee for mutational analysis of TP53.
Conflict of Interest
All authors have no conflicts of interest.
References
1. Zenz T, Fröhling S, Mertens D, Döhner H, Stilgenbauer S. Moving from prognostic to predictive factors in chronic lymphocytic leukaemia (CLL). Best Pract Res Clin Haematol. 2010;23:71-84
2. Gribben JG. How I treat CLL up front. Blood. 2010;115:187-97
3. Zenz T, Kröber A, Scherer K, Häbe S, Bühler A, Benner A. et al. TP53 inactivation is associated with poor prognosis in chronic lymphocytic leukemia: results from a detailed genetic characterization with long-term follow-up. Blood. 2008;112:3322-9
4. Malcikova J, Smardova J, Rocnova L, Tichy B, Kuglik P, Vranova V. et al. Monoallelic and biallelic inactivation of TP53 gene in chronic lymphocytic leukemia: selection, impact on survival, and response to DNA damage. Blood. 2009;114:5307-14
5. Pettitt AR, Sherrington PD, Stewart G, Cawley JC, Taylor AM, Stankovic T. p53 dysfunction in B-cell chronic lymphocytic leukemia: inactivation of ATM as an alternative to TP53 mutation. Blood. 2001;98:814-22
6. Carter A, Lin K, Sherrington PD, Pettitt AR. Detection of p53 dysfunction by flow cytometry in chronic lymphocytic leukaemia. Br J Haematol. 2004;127:425-8
7. Zenz T, Häbe S, Denzel T, Mohr J, Winkler D, Bühler A. et al. Detailed analysis of p53 pathway defects in fludarabine-refractory chronic lymphocytic leukemia (CLL): dissecting the contribution of 17p deletion, TP53 mutation, p53-p21 dysfunction, and miR34a in a prospective clinical trial. Blood. 2009;114:2589-97
8. Austen B, Skowronska A, Baker C, Powell JE, Gardiner A, Oscier D. et al. Mutation status of the residual ATM allele is an important determinant of the cellular response to chemotherapy and survival in patients with chronic lymphocytic leukemia containing an 11q deletion. J Clin Oncol. 2007;25:5448-57
9. Johnson GG, Sherrington PD, Carter A, Lin K, Liloglou T, Field JK. et al. A novel type of p53 pathway dysfunction in chronic lymphocytic leukemia resulting from two interacting single nucleotide polymorphisms within the p21 gene. Cancer Res. 2009;69:5210-17
10. Rosenwald A, Chuang EY, Davis RE, Wiestner A, Alizadeh AA, Arthur DC. et al. Fludarabine treatment of patients with chronic lymphocytic leukemia induces a p53-dependent gene expression response. Blood. 2004;104:1428-34
11. Moussay E, Palissot V, Vallar L, Moussay E, Palissot V, Vallar L. et al. Determination of genes and microRNAs involved in the resistance to fludarabine in vivo in chronic lymphocytic leukemia. Mol Cancer. 2010;9:115
12. Ferracin M, Zagatti B, Rizzotto L, Cavazzini F, Veronese A, Ciccone M. et al. MicroRNAs involvement in fludarabine refractory chronic lymphocytic leukemia. Mol Cancer. 2010;9:123
13. Oscier D, Fegan C, Hillmen P, Illidge T, Johnson S, Maguire P. et al. Guidelines on the diagnosis and management of chronic lymphocytic leukaemia. Br J Haematol. 2004;125:294-317
14. Catovsky D, Richards S, Matutes E, Oscier D, Dyer MJ, Bezares RF. et al. Assessment of fludarabine plus cyclophosphamide for patients with chronic lymphocytic leukaemia (the LRF CLL4 Trial): a randomised controlled trial. Lancet. 2007;370:230-9
15. Hallek M, Fischer K, Fingerle-Rowson G, Fink AM, Busch R, Mayer J. et al. Addition of rituximab to fludarabine and cyclophosphamide in patients with chronic lymphocytic leukaemia: a randomised, open-label, phase 3 trial.International Group of Investigators; German Chronic Lymphocytic Leukaemia Study Group. Lancet. 2010;376:1164-74
16. Lassus P, Ferlin M, Piette J, Hibner U. Anti-apoptotic activity of low levels of wild-type p53. Embo J. 1996;15:4566-4573
17. Chen X, Ko LJ, Jayaraman L, Prives C. p53 levels, functional domains, and DNA damage determine the extent of the apoptotic response of tumor cells. Genes Dev. 1996;10:2438-51
18. Coates PJ, Lorimore SA, Lindsay KJ, Wright EG. Tissue-specific p53 responses to ionizing radiation and their genetic modification: the key to tissue-specific tumour susceptibility? J Pathol. 2003;201:377-88
19. Coates PJ, Lorimore SA, Wright EG. Cell and tissue responses to genotoxic stress. J Pathol. 2005;205:221-35
20. Coates PJ, Appleyard MV, Murray K, Ackland C, Gardner J, Brown DC. et al. Differential contextual responses of normal human breast epithelium to ionizing radiation in a mouse xenograft model. Cancer Res. 2010;70:9808-915
21. Orchard JA, Ibbotson RE, Davis Z, Wiestner A, Rosenwald A, Thomas PW. et al. ZAP-70 expression and prognosis in chronic lymphocytic leukaemia. Lancet. 2004;363:105-11
22. De Rossi G, Tenca C, Cerruti G, Favre A, Zarcone D, Tabilio A. et al. Adhesion molecule expression on B-cells from acute and chronic lymphoid leukemias. Leuk Lymphoma. 1994;16:31-6
23. Godar S, Ince TA, Bell GW, Feldser D, Donaher JL, Bergh J. et al. Growth-inhibitory and tumor- suppressive functions of p53 depend on its repression of CD44 expression. Cell. 2008;134:62-73
24. Hallek M, Cheson BD, Catovsky D, Caligaris-Cappio F, Dighiero G, Döhner H. et al. International Workshop on Chronic Lymphocytic Leukemia. Guidelines for the diagnosis and treatment of chronic lymphocytic leukemia: a report from the International Workshop on Chronic Lymphocytic Leukemia updating the National Cancer Institute-Working Group 1996 guidelines. Blood. 2008;111:5446-56
25. Vogelstein B, Lane D, Levine AJ. Surfing the p53 network. Nature. 2000;408:307-10
26. Zilfou JT, Lowe SW. Tumor suppressive functions of p53. Cold Spring Harb Perspect Biol. 2009;00:aoo1883
27. Knoops L, Haas R, de Kemp S, Majoor D, Broeks A, Eldering E. et al. In vivo p53 response and immune reaction underlie highly effective low-dose radiotherapy in follicular lymphoma. Blood. 2007;110:1116-22
28. Haidar MA, El-Hajj H, Bueso-Ramos CE, Manshouri T, Glassman A, Keating MJ. et al. Expression profile of MDM-2 proteins in chronic lymphocytic leukemia and their clinical relevance. Am J Hematol. 1997;54:189-95
29. Ponta H, Sherman L, Herrlich PA. CD44: from adhesion molecules to signalling regulators. Nat Rev Mol Cell Biol. 2003;4:33-45
Author contact
![]() Corresponding author: Dr Sudhir Tauro PhD, FRCPath, Dundee Cancer Centre, University of Dundee, Dundee DD1 9SY, Scotland, United Kingdom. Tel: 44(0)1382 740403, Fax: 44(0)1382 632492, Email: s.tauroac.uk.
Corresponding author: Dr Sudhir Tauro PhD, FRCPath, Dundee Cancer Centre, University of Dundee, Dundee DD1 9SY, Scotland, United Kingdom. Tel: 44(0)1382 740403, Fax: 44(0)1382 632492, Email: s.tauroac.uk.