Impact Factor ISSN: 1837-9664

 Global reach, higher impact
Global reach, higher impactJ Cancer 2013; 4(5):383-390. doi:10.7150/jca.6545 This issue Cite
Research Paper
Plasma Fibronectin Promotes Tumor Cell Survival and Invasion through Regulation of Tie2
Lynn M. Knowles1, Gunjan Malik1, Jan Pilch1,2 ![]()
1. Department of Urology, University of Pittsburgh School of Medicine, Shadyside Medical Center, 5200 Centre Avenue, Pittsburgh, PA 15232, USA.
2. Prostate and Urological Cancers Program, University of Pittsburgh Cancer Institute, 5150 Centre Avenue, Pittsburgh, PA 15232, USA.
Received 2013-4-23; Accepted 2013-6-12; Published 2013-6-21
Abstract
Our previous research has shown that plasma fibronectin promotes lung metastasis by facilitating tumor cell invasion in clotted plasma. To evaluate the role of clotted plasma for tumor cell survival, we treated B16F1 cells embedded in a 3-dimensional matrix of fibrin with tumor necrosis factor α (TNFα), a cytokine with anti-tumor activity. Under these conditions, TNFα caused significant cytotoxicity, which was prevented when we added plasma fibronectin to the fibrin clot. Fibronectin-mediated TNFα resistance was dependent on PI3-kinase, which also mediated the pro-adhesive and pro-invasive effects of plasma fibronectin on tumor cells. To further investigate the role of plasma fibronectin in tumor cell signaling, we performed a gene array that showed specific upregulation of Tie2 in B16F1 cells embedded in fibrin-fibronectin compared to fibrin. Importantly, inhibition of Tie2 resulted in decreased tumor cell invasion, reduced colony formation and increased tumor cell death in response to TNFα. Together, our findings indicate that plasma fibronectin induces tumor cell invasion and protects tumor cells from the cytotoxic effects of inflammatory mediators through up-regulation of Tie2.
Keywords: Plasma fibronectin, Metastasis, Fibrin, Tie2.
Introduction
Localized tumors are potentially curable, but the prognosis of cancer deteriorates once tumors have generated metastases (1). The initial step of metastasis begins with the intravasation of tumor cells into the blood circulation (1). After entering the blood stream, tumor cells are carried into the vasculature of distant organs, where they attach, extravasate and begin to form colonies. Metastatic seeding is aided by the capacity of circulating tumor cells to activate the clotting cascade and surround themselves with a fibrin-platelet thrombus, which facilitates tumor cell survival and extravasation in the target organ (2, 3). This process plays a critical role for metastasis because knocking down clotting factors in transgenic mice or inhibiting clot formation with anti-coagulants has a marked anti-metastatic effect (4-7).
An important function of clotting is to protect tumor cells from the cytotoxic activity of natural killer (NK) cells by generating a physical barrier that blocks interactions between activating NK cell receptors and their ligands on tumor cells (3, 7). In addition, clotted plasma provides circulating tumor cells with a provisional extracellular matrix that promotes tumor cell adhesion and invasion (2, 8). Adhesive interactions of tumor cells with clotted plasma are mediated by integrins, which bind to fibrin or plasma adhesion proteins cross-linked to fibrin and, consequently activate important intracellular signaling cascades such as the PI3-kinase pathway (9). Moreover, signaling through integrins has been shown to protect tumor cells from the cytotoxic activity of TRAIL and tumor necrosis factor α (TNFα), two cytokines secreted by NK cells, suggesting that adhesive interactions with clotted plasma could be relevant for the survival of circulating tumor cells (10, 11).
Clotted plasma consists predominantly of fibrin and plasma fibronectin, which are covalently cross-linked by activated coagulation factor XIII, a plasma transglutaminase (12, 13). This is a significant modification of blood clot as fibronectin has been shown to support tumor cell survival, invasion and proliferation (14, 15). In line with this, we recently demonstrated that lung metastasis is reduced in transgenic plasma fibronectin-deficient mice and attributed this finding to the lack of plasma fibronectin in clotted plasma (2). In addition, we found that plasma fibronectin promoted invadopodia formation of tumor cells embedded in a three-dimensional matrix of cross-linked fibrin-fibronectin. Here, we show that the presence of plasma fibronectin protects fibrin-embedded tumor cells from the cytotoxic activity of TNFα through a novel mechanism that depends on the receptor tyrosine kinase Tie2.
Materials and Methods
Cytotoxicity Assay. To determine cell death, 5x104 B16F1 cells (American Type Culture Collection) were suspended in a solution of 2 mg/ml fibrinogen (Enzyme Research Laboratories, Inc.), 2 mM CaCl2 and 25 µg/ml FXIII (Enzyme Research Laboratories, Inc.). To produce fibrin, 2.5 U/ml thrombin (Sigma) was added to the fibrinogen/tumor cell mixture. For the generation of fibrin-fibronectin, 200 µg/ml pFN (Sigma) was added to the fibrinogen solution. After the clot solidified, cells were treated with TNFα, interferon γ (IFNγ) (R&D Systems) or vehicle (DMEM supplemented with 10% fetal bovine serum and gentamicin) for at least 16 hours at 37ºC under a humidified, 5% CO2 atmosphere. At indicated times, clots were incubated with 5 µg/ml propidium iodide (PI; Roche Applied Science) for 15 minutes and transferred to an inverted fluorescence microscope connected to a camera to assess the number of PI-positive cells in random optical fields (Nikon Eclipse TE2000-U; magnification 10x). Cell death was also assessed with commercially available reagents for the detection of LDH, DNA fragments and genomic DNA as previously described (16).
Cell Invasion and colony formation. To determine cell invasion, fibrin-fibronectin-embedded B16F1 cells were analyzed for invadopodia formation as previously described (2, 17). To this end, 5×104 B16F1 cells were suspended in fibrinogen (2 mg/ml), pFN (200 µg/ml), FXIII (25 μg/ml) and CaCl2 (2 mM) and 2.5 U/ml thrombin was added to induce clotting. Clot-embedded cells were incubated in 10% fetal bovine serum overnight and then recorded for invadopodia formation at designated areas with a phase contrast microscope and attached camera (Nikon Eclipse TS100; 10× magnification). Invadopodia formation was classified as complete when cells had a spindle or stellate shape, which was mediated by the proteolytic activity of matrix metalloproteinases (MMPs) (2). B16F1 cells treated with a broad spectrum MMP inhibitor or siRNA against membrane-type 1 matrix metalloproteinase remained round (with or without rudimentary invadopodia) and, therefore, where classified as non-invasive (2). Invasion was analyzed by assessing invadopodia-positive tumor cells as percent of all tumor cells. Colony formation was monitored by measuring the diameter of B16F1 cell spheres at indicated times. Where indicated, clots were incubated in the presence of the PI3-kinase inhibitor LY294002 (10 µM), the protein kinase C (PKC) inhibitor Gö6976 (1 µM), the JNK inhibitor SP600125 (10 µM), the MEK inhibitor U0126 (10 µM) (Cell Signaling) or the Tie2 inhibitor 4-(6-Methoxy-2-naphthyl)-2-(4-methylsulfinylphenyl)-5-(4-pyridyl)-1H-imidazole (Santa Cruz Biotechnology) (18, 19).
Cell Adhesion Assay. To assess adhesion of B16F1 cells, 48-well plates (Costar, polystyrene, non-tissue culture treated) were incubated with 10 µg/ml of fibrinogen or solubilized clot material from fibrin-fibronectin (4°C overnight) (20), then washed with PBS and blocked with 1 % BSA (1 hour, 37°C). 2 × 105 cells suspended in HEPES-Tyrode's buffer (+ 0.1% BSA and 2 mM CaCl2) were added to the plate and allowed to attach for 45-60 minutes (37°C, 5% CO2). To induce adhesion to fibrinogen, solubilized fibrin-fibronectin complexes were added to the cell binding buffer at the time of plating (2). Plates were washed to remove floating cells. Attached cells were incubated with para-nitrophenol phosphate for 30 minutes and quantified at 405 nm after adding 0.3 M sodium hydroxide (21).
RNA analysis. B16F1 cells were monitored for changes in gene expression following embedding in fibrin-fibronectin versus fibrin after 16 hours. Total RNA was isolated from embedded cells using the Qiagen RNeasy kit (Qiagen) and hybridized to the Mouse Cancer PathwayFinder PCR Array (SABiosciences) according to the manufacturer's instructions. RNA expression was measured using the iQ5 Real-Time PCR Detection System (Bio-Rad Laboratories) and values were normalized to housekeeping genes present on the array. Genes that changed by at least 1.5 fold in response to fibrin-fibronectin compared to fibrin were identified.
siRNA Mediated Gene Silencing. B16F1 cells were transfected with 25 nM Tie2 (Dharmacon On-TARGETplus SMARTpool L-045325-00), uPA (Dharmacon On-TARGETplus SMARTpool L-060180-01), TNFα (Dharmacon On-TARGETplus SMARTpool L042302-00-0010) or non-targeting control (Dharmacon On-TARGETplus D-001810-10-20) siRNA in Opti-MEM medium (Invitrogen) using LipofectAMINE 2000 reagent (Invitrogen) for 5 hours according to manufacturer instructions. Then cells were grown for an additional 43 hours in normal culture medium. Cells were then harvested using 5 mM EDTA, washed twice with PBS and embedded in fibrin-fibronectin as described above.
Statistical Analysis. Data were analyzed using unpaired two-tailed Student's t test or one-way ANOVA followed by the posthoc Tukey's multiple comparisons test (GraphPad Prism 5). Treatment differences with a two-sided p value < 0.05 were considered significantly different. Error bars show mean ± SEM.
Results
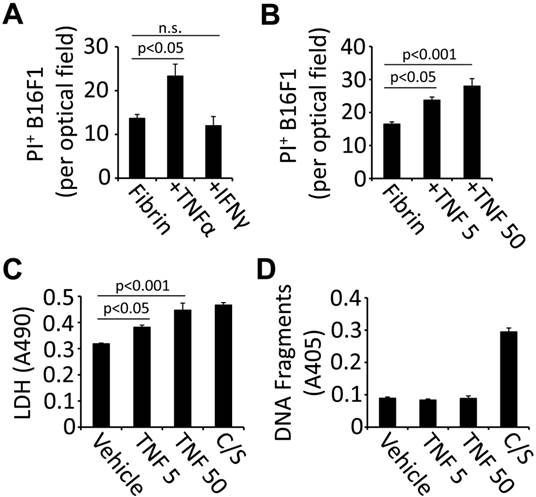
TNFα induces cell death in fibrin-embedded tumor cells. Circulating tumor cells can co-opt the coagulation system to generate a provisional extracellular matrix that provides protection from immune cells and their secreted effectors (6, 7). To analyze the role of blood clotting for tumor cell fate in response to inflammatory cytokines, we treated fibrin-embedded B16F1 with TNFα or IFN-γ and assessed cell death by measuring PI uptake. Using this method, we detected a significant increase of cell death 16 hours after treatment with TNFα (50 ng/ml) while IFNγ (14,000 U/ml) had no effect (Fig. 1A). TNFα-induced cell death was detectable at 5 ng/ml and further increased at 50 ng/ml as assessed by PI uptake as well as LDH release (Fig. 1B-C). However, we did not detect apoptosis-specific DNA fragments in response to TNFα indicating that PI uptake and LDH release are the result of necrotic cell death (Fig. 1D). Taken together, our results show that TNFα is a potent inducer of cell death in fibrin-embedded B16F1 tumor cells.
TNFα is cytotoxic for fibrin-embedded tumor cells. (A), B16F1 cells were analyzed for PI uptake by fluorescence microscopy 16 hours after embedding in fibrin and treatment with 50 ng/ml TNFα, 14,000U/ml IFNγ or vehicle (n.s., not significant). (B-D), B16F1 cells embedded in fibrin were analyzed for PI uptake (B), LDH release (C) or DNA fragmentation (D) after treatment with TNFα (5 or 50 ng/ml) compared to vehicle (Veh). Camptothecin and staurosporin (C/S) were used as a positive control.
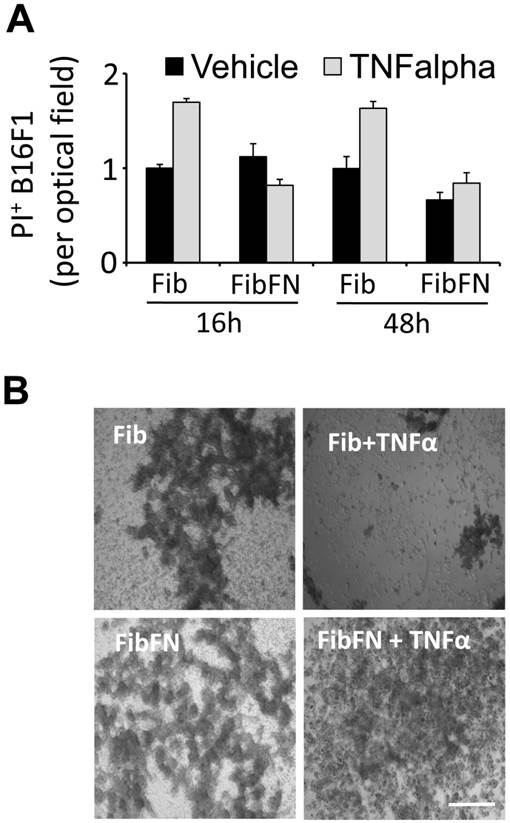
Plasma fibronectin protects tumor cells from TNFα-induced cell death. Fibronectin has been shown to promote tumor cell survival and proliferation (14, 15). To determine if fibronectin protects tumor cells from the cytotoxic activity of TNFα, we embedded B16F1 cells in fibrin that has been cross-linked to plasma fibronectin. Analysis of the clots for PI+ cells after 16 and 48 hours revealed that the addition of plasma fibronectin to fibrin had no effect on the cell fate (Fig. 2A). However, while TNFα was able to induce B16F1 cell death in fibrin clots, TNFα-induced cell death was effectively suppressed in B16F1 cells embedded in fibrin-fibronectin (Fig. 2A). The protective effect of fibrin-fibronectin is lasting and allows B16F1 cells to colonize the clot within 96 hours at concentrations of TNFα that severely impact the overall survival of fibrin-embedded B16F1 cells in absence of plasma fibronectin (Fig. 2B). Together, our results indicate that plasma fibronectin protects fibrin-embedded B16F1 cells from the cytotoxic activity of TNFα.
Fibrin-fibronectin protects B16F1 cells from TNFα. (A), PI+ B16F1 cells embedded in fibrin (Fib) or fibrin-fibronectin (FibFN) were counted in random optical field after treatment with TNFα (50 ng/ml) for 24 and 48 hours compared to vehicle. PI uptake in fibrin-embedded cells treated with vehicle is set to 1. (B), representative phase contrast microscopy images of B16F1 melanoma cells 96 hours after embedding in fibrin or fibrin-fibronectin (FibFN) and treatment with 50 ng/ml TNFα. Scale bar, 500 µm.
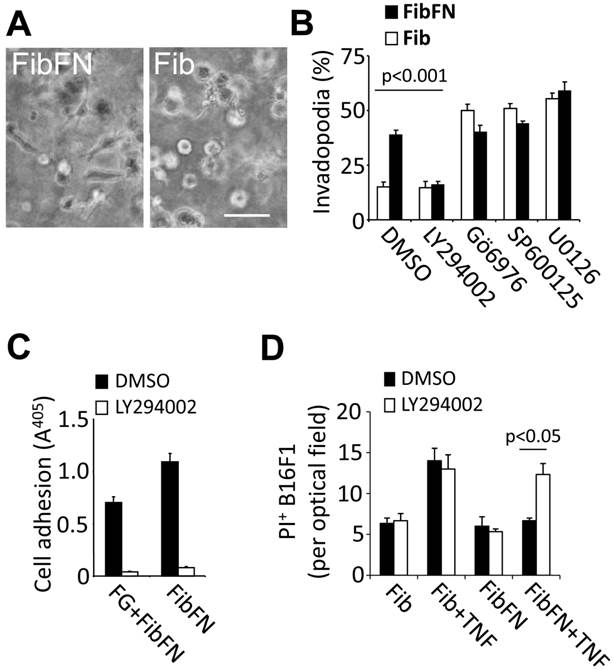
PI3-kinase promotes plasma fibronectin-mediated resistance against tumor cell death. We previously demonstrated that plasma fibronectin promotes tumor cell adhesion and this effect correlates with increased invadopodia formation in B16F1 cells embedded in fibrin-fibronectin compared to fibrin (Fig. 3A) (2). To determine if there is an overlap between adhesive tumor cell functions and the TNFα-protective effect of plasma fibronectin, we sought to identify pathways that mediate invadopodia formation of fibrin-fibronectin embedded B16F1 cells (Fig. 3B-C). While inhibition of JNK, MEK and PKC with SP600125 (10 µM), U0126 (10 µM) or Gö6976 (1 µM), respectively, had no effect on fibrin-fibronectin-mediated invasion and even promoted fibrin invasion in absence of plasma fibronectin, we found a marked decrease of invasion as well as adhesion after treatment with 10 µM of the PI3-kinase inhibitor LY294002 (Fig. 3B-C). In addition, LY294002 sensitized fibrin-fibronectin-embedded tumor cells for TNFα-induced cell death indicating that PI3-kinase cooperates with plasma fibronectin to mediate protection of B16F1 cells against the cytotoxic effects of TNFα (Fig. 3D). Together, our results indicate that PI3-kinase plays a prominent role in mediating critical fibrin-fibronectin-induced functions including tumor cell adhesion, invasion and survival.
Fibrin-fibronectin mediated B16F1 invadopodia formation, cell adhesion and TNFα-resistance depends on PI3-kinase. (A), representative phase contrast images of B16F1 cells 16 hours after embedding in a 3-dimensional matrix of fibrin (Fib; right image) or fibrin-fibronectin (FibFN; left image). Scale bar, 50 µm. (B), B16F1 cells were analyzed for invadopodia formation by phase contrast microscopy in the presence of inhibitors against PI3-kinase (LY294002, 10 µM), PKC (Gö 6976, 1 µM), JNK (SP600125, 10 µM), and MEK1/2 (U0126, 10 µM) compared to vehicle (DMSO) 16 hours after embedding in Fib or FibFN. (C), B16F1 cells treated with 10 µM LY294002 or vehicle were allowed to attach to plates coated with fibrinogen (FG) or fibrin-fibronectin (FibFN) (ea. 10 µg/ml). Adhesion to FG was induced by adding solubilized fibrin-fibronectin complexes to the binding buffer. (D), PI+ B16F1 cells embedded in fibrin (Fib) or fibrin-fibronectin (+FN) were counted in random optical field after treatment with TNFα (50 ng/ml) and 10 µM LY294002 compared to vehicle.
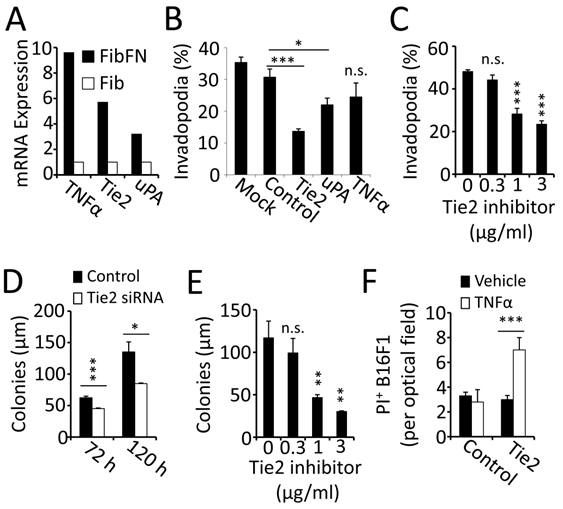
Fibrin-fibronectin mediates tumor cell invasion and survival through Tie-2. To identify mechanisms that contribute to invadopodia formation, we analyzed gene expression of cancer-relevant pathways in B16F1 cells embedded in fibrin-fibronectin compared to fibrin alone. This study led to the identification of TNFα, Tie-2 and uPA, which were found to be specifically upregulated in the presence of fibronectin (Fig. 4A). To determine the functional relevance of the gene expression analysis, we treated B16F1 cells with siRNA, which demonstrated an important role of Tie2 for invadopodia formation (Fig. 4B). Knocking down uPA or TNFα, on the other hand, had only minor effects on tumor cell invasion into fibrin-fibronectin. In addition, Tie2 knockdown reduced B16F1 colony formation and reversed the protective effect of fibrin-fibronectin against TNFα (Fig. 4D, F). To further analyze the function of Tie2, we treated B16F1 cells with a Tie2 kinase inhibitor. Paralleling our results with Tie 2 siRNA, treatment with Tie2 kinase inhibitor resulted in significant reduction of both invadopodia and colony formation in fibrin-fibronectin embedded B16F1 cells indicating that the tyrosine kinase function of Tie2 is required for these processes (Fig. 4C, E). Together, our results indicate that plasma fibronectin promotes invasion, colony formation and TNFα-resistance in fibrin-embedded tumor cells through upregulation of Tie2.
Tie2 is upregulated in fibrin-fibronectin-embedded B16F1 cells. (A), gene expression analysis showed upregulation of mRNA for TNFα, Tie2 and uPA in FibFN- over Fib-embedded B16F1 cells. (B), invadopodia formation after 16 hours of FibFN-embedded B16F1 cells treated with siRNA against Tie2, uPA and TNFα compared to non-targeted siRNA. (C), invadopodia formation of FibFN-embedded B16F1 cells 16 hours after treatment with a Tie2 kinase inhibitor. (D-E), colony size was analyzed in fibrin-fibronectin embedded B16F1 cells treated with Tie2 and control siRNA for 72 and 120 hours (D) or with a Tie2 kinase inhibitor after 120 hours (E). (F), PI+ B16F1 cells embedded in fibrin-fibronectin were counted in random optical field after treatment with Tie2 siRNA and TNFα (50 ng/ml). *, P<0.05; **, P<0.01; ***, P<0.001; n.s., not significant; versus control (vehicle or scrambled siRNA).
Discussion
We previously showed that plasma fibronectin supports lung metastasis by facilitating tumor cell invasion in blood clot (16). Here we demonstrate that plasma fibronectin protects fibrin-embedded tumor cells from the cytotoxic activity of TNFα. The protective effect of plasma fibronectin depends on PI3-kinase as well as Tie2, which is upregulated in fibrin-fibronectin-embedded tumor cells. In addition, Tie2 promotes invadopodia formation and colonization of tumor cells in fibrin-fibronectin, suggesting that Tie2 controls critical tumor cell functions involved in metastasis.
Adhesive interactions with the extracellular matrix provide cells with important cues about their placement in a given environment. These cues are detected by integrins, which activate intracellular growth and survival signals upon binding to specific extracellular matrix proteins (9). Whereas non-transformed cells die upon loss of extracellular matrix attachment, metastatic tumor cells have acquired the capacity to survive in absence of interactions with an extracellular matrix (22). Instead, metastatic tumor cells depend on integrin-mediated interactions with the extracellular matrix to mediate migration and invasion as a means to leave the primary tumor, attach in the metastatic target organ and extravasate into the perivascular tissue (15). Interestingly, intracellular signals that mediate cell motility such as activation of PI3-kinase are also crucial for anchorage-independence and resistance against cytotoxic agents (11, 23, 24). Previously, we demonstrated that plasma fibronectin promotes tumor cell invasion into blood clot in vitro and lung metastasis in vivo (2). Here, we show that plasma fibronectin protects fibrin-embedded B16F1 melanoma cells from the cytotoxic activity of TNFα.
Our results indicate that PI3-kinase is at the center of plasma fibronectin-mediated functions by demonstrating inhibition of B16F1 cell adhesion and invasion into fibrin-fibronectin matrix after treatment with the PI3-kinase inhibitor LY294002. This is in line with previous reports showing that PI3-kinase is capable of activating integrin αvβ3, which is critical for tumor cell adhesion to fibrin(ogen) (25). It is also in line with our previous observation that fibrin-fibronectin complexes can activate integrin αvβ3 (2). In addition, PI3-kinase plays an important role in cell adhesion and invadopodia biogenesis downstream of integrins by regulating actin filament dynamics through activation of Rac1, Cdc42 and PAK (26-28). While each of these factors can suppress pro-apoptotic signals, they also act as a positive feedback loop to increase activation of PI3-kinase and Akt as a result of increased cell-ECM interactions and focal adhesion maturation (28-31).
An important function of PI3-kinase/Akt is to inactivate the proapoptotic peptide Bad and, thus, promote cell survival (32). However, this mechanism is thought to specifically prevent apoptotic cell death whereas we found that TNFα caused non-apoptotic cell death in fibrin-embedded B16F1 cells, which is in line with previous studies demonstrating that TNFα, depending on cell type and context, can induce apoptosis as well as necrosis (33). TNFα-induced necrosis depends on Rip1 kinase and occurs in cells that are unable to undergo apoptosis (34). In these cells, Rip1 forms a complex with Rip3 kinase, which leads to increased production of reactive oxygen species as a prerequisite of necrotic cell death (35, 36). Importantly, the ability of Rip1 to induce necrotic cell death has been shown to be inhibited following activation of NF-κB, which prevents TNFα from inducing cell death by mediating polyubiquitination of Rip1 through cIAP1 and 2 (37, 38). Polyubiquinated Rip1 in turn presents a scaffold for the assembly of a protein complex that mediates NF-κB activation and cell survival in reponse to TNFα (39). Considering the capacity of PI3-kinase to activate NF-κB, it is conceivable that this mechanism could lead to cIAP expression and protection of tumor cells from TNFα-induced cell death (40, 41).
To further elucidate the effect of plasma fibronectin on tumor cell survival and invasion, we performed a comparative gene expression analysis between fibrin and fibrin-fibronectin embedded B16F1 cells. Our study showed significant up-regulation of the NFκB-target gene TNFα, suggesting that fibrin-fibronectin complexes can activate inflammatory pathways (42). In addition, we detected a significant increase of Tie2 in fibrin-fibronectin-embedded B16F1 cells, which is surprising as Tie2 has been shown to be predominantly expressed in endothelial cells and angiogenic macrophages (43, 44). Tie2 is a receptor tyrosine kinase, which upon binding to its ligand angiopoietin 1 induces activation of PI3-kinase and PAK (45, 46). In line with this function, we found that knockdown of Tie2 or treatment with a Tie2 kinase inhibitor significantly inhibited invadopodia and sphere formation. In addition, we found that Tie2 provided protection from TNFα-mediated cytotoxicity in fibrin-fibronectin embedded cells suggesting that Tie2 acts in concert with plasma fibronectin and PI3-kinase. Moreover, the role of Tie2 for B16F1 clot colonization is interesting considering that B16F1 cells raised in 3-dimensional fibrin take on characteristics of tumor stem cells (47).
Based on our results, we propose a mechanism where the incorporation of plasma fibronectin into a 3-dimensional matrix of fibrin induces increased expression of the receptor tyrosine kinase Tie2 in B16F1 cells. Tie2, which has been shown to activate PI3-kinase and PAK (45, 46), promotes invadopodia and colony formation in fibrin-fibronectin embedded B16F1 cells. Inhibiting these pro-metastatic functions and sensitizing tumor cells for TNFα with Tie2 inhibitors could be an attractive strategy for the treatment of metastatic cancer.
Acknowledgements
This work was supported by National Institutes of Health grants CA134330 (JP) and P30CA047904 (UPCI CCSG).
Conflict of Interest
None.
References
1. Chaffer CL, Weinberg RA. A perspective on cancer cell metastasis. Science. 2011;331:1559-64
2. Malik G, Knowles LM, Dhir R, Xu S, Yang S, Ruoslahti E. et al. Plasma fibronectin promotes lung metastasis by contributions to fibrin clots and tumor cell invasion. Cancer Res. 2010;70:4327-34
3. Gorelik E, Bere WW, Herberman RB. Role of NK cells in the antimetastatic effect of anticoagulant drugs. Int J Cancer. 1984;33:87-94
4. Esumi N, Fan D, Fidler IJ. Inhibition of murine melanoma experimental metastasis by recombinant desulfatohirudin, a highly specific thrombin inhibitor. Cancer research. 1991;51:4549-56
5. Palumbo JS, Barney KA, Blevins EA, Shaw MA, Mishra A, Flick MJ. et al. Factor XIII transglutaminase supports hematogenous tumor cell metastasis through a mechanism dependent on natural killer cell function. J Thromb Haemost. 2008;6:812-9
6. Palumbo JS, Kombrinck KW, Drew AF, Grimes TS, Kiser JH, Degen JL. et al. Fibrinogen is an important determinant of the metastatic potential of circulating tumor cells. Blood. 2000;96:3302-9
7. Palumbo JS, Talmage KE, Massari JV, La Jeunesse CM, Flick MJ, Kombrinck KW. et al. Platelets and fibrin(ogen) increase metastatic potential by impeding natural killer cell-mediated elimination of tumor cells. Blood. 2005;105:178-85
8. Im JH, Fu W, Wang H, Bhatia SK, Hammer DA, Kowalska MA. et al. Coagulation facilitates tumor cell spreading in the pulmonary vasculature during early metastatic colony formation. Cancer Res. 2004;64:8613-9
9. Giancotti FG, Ruoslahti E. Integrin signaling. Science. 1999;285:1028-32
10. Phipps LE, Hino S, Muschel RJ. Targeting Cell Spreading: A Method of Sensitizing Metastatic Tumor Cells to TRAIL-Induced Apoptosis. Molecular Cancer Research. 2011;9:249-58
11. Chekenya M, Krakstad C, Svendsen A, Netland IA, Staalesen V, Tysnes BB. et al. The progenitor cell marker NG2/MPG promotes chemoresistance by activation of integrin-dependent PI3K/Akt signaling. Oncogene. 2008;27:5182-94
12. Aeschlimann D, Mosher D, Paulsson M. Tissue transglutaminase and factor XIII in cartilage and bone remodeling. Seminars in thrombosis and hemostasis. 1996;22:437-43
13. Ruoslahti E. Fibronectin in cell adhesion and invasion. Cancer Metastasis Rev. 1984;3:43-51
14. Barkan D, Kleinman H, Simmons JL, Asmussen H, Kamaraju AK, Hoenorhoff MJ. et al. Inhibition of metastatic outgrowth from single dormant tumor cells by targeting the cytoskeleton. Cancer Res. 2008;68:6241-50
15. Mehrotra S, Languino LR, Raskett CM, Mercurio AM, Dohi T, Altieri DC. IAP regulation of metastasis. Cancer Cell. 2010;17:53-64
16. Knowles LM, Malik G, Hood BL, Conrads TP, Pilch J. CLT1 targets angiogenic endothelium through CLIC1 and fibronectin. Angiogenesis. 2012;15:115-29
17. Hiraoka N, Allen E, Apel IJ, Gyetko MR, Weiss SJ. Matrix metalloproteinases regulate neovascularization by acting as pericellular fibrinolysins. Cell. 1998;95:365-77
18. Hasenstein JR, Kasmerchak K, Buehler D, Hafez GR, Cleary K, Moody JS. et al. Efficacy of Tie2 receptor antagonism in angiosarcoma. Neoplasia. 2012;14:131-40
19. Semones M, Feng Y, Johnson N, Adams JL, Winkler J, Hansbury M. Pyridinylimidazole inhibitors of Tie2 kinase. Bioorg Med Chem Lett. 2007;17:4756-60
20. Corbett SA, Wilson CL, Schwarzbauer JE. Changes in cell spreading and cytoskeletal organization are induced by adhesion to a fibronectin-fibrin matrix. Blood. 1996;88:158-66
21. Pilch J, Habermann R, Felding-Habermann B. Unique ability of integrin alpha(v)beta 3 to support tumor cell arrest under dynamic flow conditions. J Biol Chem. 2002;277:21930-8
22. Frisch SM, Vuori K, Ruoslahti E, Chan-Hui PY. Control of adhesion-dependent cell survival by focal adhesion kinase. J Cell Biol. 1996;134:793-9
23. Moore SM, Rintoul RC, Walker TR, Chilvers ER, Haslett C, Sethi T. The presence of a constitutively active phosphoinositide 3-kinase in small cell lung cancer cells mediates anchorage-independent proliferation via a protein kinase B and p70s6k-dependent pathway. Cancer Res. 1998;58:5239-47
24. Levental KR, Yu H, Kass L, Lakins JN, Egeblad M, Erler JT. et al. Matrix crosslinking forces tumor progression by enhancing integrin signaling. Cell. 2009;139:891-906
25. Byzova TV, Goldman CK, Pampori N, Thomas KA, Bett A, Shattil SJ. et al. A mechanism for modulation of cellular responses to VEGF: activation of the integrins. Mol Cell. 2000;6:851-60
26. Rodriguez-Viciana P, Warne PH, Khwaja A, Marte BM, Pappin D, Das P. et al. Role of phosphoinositide 3-OH kinase in cell transformation and control of the actin cytoskeleton by Ras. Cell. 1997;89:457-67
27. Guo FK, Debidda M, Yang L, Williams DA, Zheng Y. Genetic deletion of Rac1 GTPase reveals its critical role in actin stress fiber formation and focal adhesion complex assembly. Journal of Biological Chemistry. 2006;281:18652-9
28. Clark EA, King WG, Brugge JS, Symons M, Hynes RO. Integrin-mediated signals regulated by members of the rho family of GTPases. J Cell Biol. 1998;142:573-86
29. Schurmann A, Mooney AF, Sanders LC, Sells MA, Wang HG, Reed JC. et al. p21-activated kinase 1 phosphorylates the death agonist bad and protects cells from apoptosis. Mol Cell Biol. 2000;20:453-61
30. Coniglio SJ, Jou TS, Symons M. Rac1 protects epithelial cells against anoikis. J Biol Chem. 2001;276:28113-20
31. Zugasti O, Rul W, Roux P, Peyssonnaux C, Eychene A, Franke TF. et al. Raf-MEK-Erk cascade in anoikis is controlled by Rac1 and Cdc42 via Akt. Mol Cell Biol. 2001;21:6706-17
32. Datta SR, Dudek H, Tao X, Masters S, Fu H, Gotoh Y. et al. Akt phosphorylation of BAD couples survival signals to the cell-intrinsic death machinery. Cell. 1997;91:231-41
33. Vandenabeele P, Declercq W, Van Herreweghe F, Vanden Berghe T. The role of the kinases RIP1 and RIP3 in TNF-induced necrosis. Sci Signal. 2010;3:re4
34. Vercammen D, Beyaert R, Denecker G, Goossens V, Van Loo G, Declercq W. et al. Inhibition of caspases increases the sensitivity of L929 cells to necrosis mediated by tumor necrosis factor. J Exp Med. 1998;187:1477-85
35. He S, Wang L, Miao L, Wang T, Du F, Zhao L. et al. Receptor interacting protein kinase-3 determines cellular necrotic response to TNF-alpha. Cell. 2009;137:1100-11
36. Lin Y, Choksi S, Shen HM, Yang QF, Hur GM, Kim YS. et al. Tumor necrosis factor-induced nonapoptotic cell death requires receptor-interacting protein-mediated cellular reactive oxygen species accumulation. J Biol Chem. 2004;279:10822-8
37. Thapa RJ, Basagoudanavar SH, Nogusa S, Irrinki K, Mallilankaraman K, Slifker MJ. et al. NF-kappaB protects cells from gamma interferon-induced RIP1-dependent necroptosis. Mol Cell Biol. 2011;31:2934-46
38. Bertrand MJ, Milutinovic S, Dickson KM, Ho WC, Boudreault A, Durkin J. et al. cIAP1 and cIAP2 facilitate cancer cell survival by functioning as E3 ligases that promote RIP1 ubiquitination. Mol Cell. 2008;30:689-700
39. Ea CK, Deng L, Xia ZP, Pineda G, Chen ZJ. Activation of IKK by TNFalpha requires site-specific ubiquitination of RIP1 and polyubiquitin binding by NEMO. Mol Cell. 2006;22:245-57
40. Dan HC, Cooper MJ, Cogswell PC, Duncan JA, Ting JP, Baldwin AS. Akt-dependent regulation of NF-{kappa}B is controlled by mTOR and Raptor in association with IKK. Genes Dev. 2008;22:1490-500
41. Ozes ON, Mayo LD, Gustin JA, Pfeffer SR, Pfeffer LM, Donner DB. NF-kappaB activation by tumour necrosis factor requires the Akt serine-threonine kinase. Nature. 1999;401:82-5
42. Collart MA, Baeuerle P, Vassalli P. Regulation of tumor necrosis factor alpha transcription in macrophages: involvement of four kappa B-like motifs and of constitutive and inducible forms of NF-kappa B. Mol Cell Biol. 1990;10:1498-506
43. Schlaeger TM, Bartunkova S, Lawitts JA, Teichmann G, Risau W, Deutsch U. et al. Uniform vascular-endothelial-cell-specific gene expression in both embryonic and adult transgenic mice. Proc Natl Acad Sci U S A. 1997;94:3058-63
44. Venneri MA, De Palma M, Ponzoni M, Pucci F, Scielzo C, Zonari E. et al. Identification of proangiogenic TIE2-expressing monocytes (TEMs) in human peripheral blood and cancer. Blood. 2007;109:5276-85
45. Master Z, Jones N, Tran J, Jones J, Kerbel RS, Dumont DJ. Dok-R plays a pivotal role in angiopoietin-1-dependent cell migration through recruitment and activation of Pak. EMBO J. 2001;20:5919-28
46. Kontos CD, Stauffer TP, Yang WP, York JD, Huang L, Blanar MA. et al. Tyrosine 1101 of Tie2 is the major site of association of p85 and is required for activation of phosphatidylinositol 3-kinase and Akt. Mol Cell Biol. 1998;18:4131-40
47. Liu J, Tan Y, Zhang H, Zhang Y, Xu P, Chen J. et al. Soft fibrin gels promote selection and growth of tumorigenic cells. Nat Mater. 2012;11:734-41
Author contact
![]() Corresponding author: Shadyside Medical Center, Suite G33, 5200 Centre Avenue, Pittsburgh, PA 15232, Phone: 412-623-3917, Fax: 412-623-3907, Email: pilchjedu.
Corresponding author: Shadyside Medical Center, Suite G33, 5200 Centre Avenue, Pittsburgh, PA 15232, Phone: 412-623-3917, Fax: 412-623-3907, Email: pilchjedu.