Impact Factor ISSN: 1837-9664

 Global reach, higher impact
Global reach, higher impactJ Cancer 2013; 4(5):402-415. doi:10.7150/jca.6780 This issue Cite
Research Paper
Novel Phenotypic Fluorescent Three-Dimensional Platforms for High-throughput Drug Screening and Personalized Chemotherapy
Changge Fang1,2, ![]() , Ingalill Avis2, David Salomon3, Frank Cuttitta2
, Ingalill Avis2, David Salomon3, Frank Cuttitta2
1. Advanced Personalized Diagnostics LLC, 6006 Bangor Drive, Alexandria, VA 22303, USA.
2. Angiogenesis Core Facility, Advanced Technology Center, Room 115, Center for Cancer Research, National Cancer Institute, 8717 Grovemont Circle, Bethesda, MD 20892-4605, USA.
3. Tumor Growth Factor Section/Laboratory of Cancer Prevention, National Cancer Institute, Frederick, MD 21702-1201.
Received 2013-5-28; Accepted 2013-6-13; Published 2013-6-26
Abstract
We have developed novel phenotypic fluorescent three-dimensional co-culture platforms that efficiently and economically screen anti-angiogenic/anti-metastatic drugs on a high-throughput scale. Individual cell populations can be identified and isolated for protein/gene expression profiling studies and cellular movement/interactions can be tracked by time-lapse cinematography. More importantly, these platforms closely parallel the in vivo angiogenic and metastatic outcomes of a given tumor xenograft in the nude mouse model but, unlike in vivo models, our co-culture platforms produce comparable results in five to nine days. Potentially, by incorporating cancer patient biopsies, the co-culture platforms should greatly improve the effectiveness and efficiency of personalized chemotherapy.
Keywords: Three-dimensional, Fluorescent, Angiogenesis, Metastasis, High-throughput, Drug screening, Personalized chemotherapy.
Introduction
Although the development of anti-angiogenic drugs has been prolific, patient survival rates are relatively low and uneven, many of the drugs are prohibitively expensive, and some of them have unfavorable side effects [1]. These adverse characteristics have prompted critical inquiries regarding the development of more efficient anti-angiogenic and anti-metastatic drugs, tailored chemotherapy, and advanced technologies for monitoring and avoiding drug-resistance in cancer patients. The in vitro traditional two-dimensional (2D) cell culture systems have notably contributed to the understanding of the basic cell biology of tumors and the development of anti-angiogenic drugs [2]. However, 2D systems tend to have distorted signaling and cellular functions distinctly different from their in vivo counterparts. Thus, the predictability of drug efficacy data derived from 2D systems has not proven itself helpful for in vivo studies and clinical outcomes [3, 4]. Given that cancerous tumors reside within a three-dimensional (3D) microenvironment in vivo and require a nutritional source and effective waste removal through angiogenesis/lymphangiogenesis for expanded growth and metastasis [5-7], 3D cell culture systems are the logical mimetic platforms. Also, as a result of its recapitulating physiologically, geometrically, and relevantly accurate signaling environment, 3D cell culture systems, as opposed to 2D systems, better replicate the in vivo drug efficacy data [3, 5-7].
The development of novel in vitro model systems that closely mimic the tumor in vivo 3D microenvironment and are able to predict a drug's clinical outcome in a timely and cost-effective manner has been pursued for nearly three decades. A spatial organizational approach to drug sensitivity was first attempted in the mid-1980's using soft-agar clonogenic assays to predict a lung cancer response base for mimicking solid tumor in vivo 3D features [8]. However, because of its single cell type construction, these assays do not reflect the collective component cell interactions commonly associated with the in vivo microenvironment of the tumor. Clinical tumors grow and progress in a complex 3D infrastructure consisting of a dynamically changing extracellular matrix (ECM) and many other non-malignant cell components that include vascular, inflammatory, and stromal cells in vivo [9-13]. ECM is an important facilitator for most cells in vivo to communicate and function. 3D cultures in ECM have been successfully used to study prostate and breast cancers [14, 15]. Currently, 3D co-culture systems are not being broadly utilized for drug screening due to a lack of a simple, reproducible, multicolored, and well-defined in vitro model [2]. In addition, there is no efficient manner to determine drug efficiency prior to treatment or to detect/monitor and avoid drug resistance to modify chemotherapy before the patient's symptoms worsen. Accordingly, the quest for novel in vitro model systems for anticancer drug screening and personalized chemotherapy that are more efficient and cost-effective has been a major pharmaceutical endeavor.
In this study, we present novel phenotypic fluorescent 3D co-culture systems (3D platforms) to study anticancer drug sensitivity. These systems incorporate a fluorescent tumor colony or mouse human tumor xenograft biopsy (xeno-biopsy) tissue surrounded by evenly dispersed fluorescent endothelial cells with/without other fluorescent component cells in ECM. In order to demonstrate that our 3D platforms closely recapitulate in vivo tumor angiogenic and metastatic features, data derived from 2D models and animal models were compared. To further demonstrate the anticancer drug high-throughput screening potential and efficacy, we assessed drug responses in nude mouse xenograft models and in our 3D platforms. Initial studies were done with three FDA approved anti-angiogenic compounds (Avastin, sunitinib, and thalidomide) along with another angiogenic inhibitor, fumagillin. Utilizing different fluorescent colors, we were able to easily track cell-to-cell interactions and differentiate the specific geographic areas involved with tumor cell migration. As a “Proof-of-Principle” approach, a xeno-biopsy can be used in lieu of a cancer patient biopsy to evaluate the potential application of our 3D platforms for personalized cancer patient chemotherapy. Hence, these novel 3D platforms are not only meaningful as effective models for cancer researchers to conduct and understand basic tumor biology, but also for pharmaceutical companies to engage in high-throughput preclinical anti-angiogenic/anti-metastatic drug screening. More importantly, these are ideal systems for identifying personalized chemotherapy regimens and monitoring drug resistance for individual cancer patients.
Materials and Methods
Stable Fluorescent Cell Lines
A rat pheochromocytoma cell line (PC-12), a human lung adenocarcinoma cell line (A549), a human ocular melanoma cell line (92-1, obtained from Dr. Martin J. Jager in Department of Pathology, University of Hospital Nijmegen, The Netherlands), a human glioblastoma cell line (U-87), a human leiomyosarcoma cell line (SK-LMS-1), an immortalized porcine aortic endothelial cell line (PAE, a gift from Dr. Carl-Henrik Heldin in Ludwig Institute for Cancer Research, Uppsala University in Sweden), an immortalized human microvascular endothelial cell line (HMEC-1, obtained from the Center for Disease Control with a material transfer agreement (MTA)), a telomerase-immortalized human microvascular endothelial cell line (TIME), an immortalized monkey endothelial cell line (RF/6A) with positive pericyte markers (CD31, TIMP3, NG2, α-SMA, DESMIN and 3G5), an immortalized human mast cell line (HMC-1, a kind gift from Dr. Joseph H. Butterfield, Mayo Clinic), and any other tumor cell lines used in this research were harvested when they reached 70~80% confluence and were then washed in phosphate buffer saline (PBS). All cell lines, unless otherwise mentioned, were purchased from the American Type Culture Collection with MTAs. 2~3 million cells were suspended in 100μl nucleofector solution (Lonza, VCA-1002 and VCA-1003) and 2µg plasmids. Three types of plasmids were used, i.e., pAmCyan1-C1 (Clontech, 632441), Amaxa pMAX-YFP (Lonza, VDF-1021), and pDsRed-Express-C1 (DsRed 2) (Clontech, Cat. 632430). The cells were then transfected according to the protocol (Lonza). The stably transfected cells were selected using 500~800μg/ml of G418 (Invitrogen, 10131-027) and enriched using flow cytometry ARIA II (BD). Permanent fluorescent cell lines were maintained in a relevant medium without G418 for a long-term culture.
Two-dimensional and Modified 2D Endothelial Cell Tubule Formation Model Systems
We performed endothelial cell tubule formation assays (2D assays) in accordance with the previous report [16]. To incorporate a monolayer of tumor cells, we modified the 2D assays in the following manner. Different tumor cell lines were cultured in suitable medium and were harvested when they reached 70~80% confluence. Tumor cells were resuspended at 200,000 cells/ml in complete growth medium and a 100μl of tumor cell suspension was added to each well of a 96-well plate. After the cells reached about 70% confluence, the medium was aspirated and the wells were washed three times in PBS. 50μl of gel matrix (Geltrex, Invitrogen) was added onto the monolayer of tumor cells of each well. Fluorescent endothelial cells were harvested at 50~70% confluence and washed three times in PBS and resuspended at 200,000-250,000 cells/ml in EBM2 basic medium (Lonza). 100Μl of endothelial cell suspension was added onto the solidified Geltrex of each well. The plates were incubated at 37°C and 5% CO2 for 6 hours. Images were taken at 4x magnification using a SPOT camera (Model 7.4 Slider) with SPOT software (version 4.6) under an inverted fluorescent microscope (Olympus IX70).
Tumor Spheroids
Tumor spheroid colonies were prepared according to the following modified protocol based on Hamburger et al. [17]. Appropriate volumes of deionized water, 100X Anti-Anti (Invitrogen), fetal bovine serum (FBS, Gibco), 10X RPMI 1640 medium (Invitrogen), and 2% agarose (SeaPaque, FMC) were mixed to make a final concentration of 1% agarose with 20% FBS, 2X Anti-Anti, and 1X RPMI 1640. 2ml of the mixture was added to each well of a 6-well plate and set aside to solidify for 20 minutes. Tumor cells at approximately 70% confluence were harvested and suspended at 1,500 cells/ml in 0.22% of agarose, 2X Anti-Anti, 20% FBS, and 1X RPMI 1640. 3ml of the cell suspension was added to each well of the 6-well plate with a solidified layer of 1% agarose. The plate was incubated at 100% humidity for 14-20 days. The well-formed colonies were harvested and washed three times PBS, pH 7.4, to get rid of the agarose residue. The colonies were suspended in PBS with 1% glucose, 0.3mM ethylenediaminetetraacetic acid (EDTA, Sigma), 0.5 % bovine serum albumin (BSA, Sigma), 20mM 4-(2-hydroxyethlyl)-1-piperazineethanesulfonic acid (HEPES, Sigma), and 1X Anti-Anti. The colonies of similar sizes (~400 cells) were selected using a microdispenser (Drummond) and an Olympus inverted fluorescent microscope (Olympus IX70) for our 3D platforms.
Tumor Nude Mouse Xenografts
The tumor nude mouse xenograft experiments have been performed under the supervision and guidelines of the NIH Institutional Animal Care and Use Committee. 1 X 106 or 1X 107 tumor cells were injected subcutaneously in the hind flank of a nude mouse. With a microchip embedded in the back of each mouse and the mice were randomized into groups of 10 mice/group having a tumor volume of ~100mm3 and drug treatment was started at this time. Avastin 5mg/kg (Roche/NIH DVR Pharm.), fumagillin 100mg/kg, and thalidomide 12.5mg/kg (A. G. Scientific Inc.) were injected intraperitoneally three times a week. Sunitinib 10mg/kg (Pfizer/NIH DVR Pharm.) was administrated orally five times a week. The vehicle is PBS with 0.1% dimethyl sulfoxide (Sigma). Two control groups of mice were administrated the vehicle orally and intraperitoneally. Treatment was continued for an additional 2-3 weeks or until the tumors reached a maximum volume of 2000mm3. The tumor sizes were recorded using Studylog software (Studylog Inc.). The two controls were combined for the final statistical analyses.
Xenograft Biopsy
Tumor xenografts were dissected when they reached about 1000mm3 and stored in 50ml tubes with RPMI 1640 medium supplemented with 10% FBS, 1% glucose, and 4X Anti-Anti. The xenografts were rinsed three times in PBS and three times in 70% ethanol. A core biopsy was performed using a biopsy punch (Miltex) and placed in a 100cm petri dish (Corning). Using a disposable scalpel (Feather Safety Razor), the core biopsy was dissected for the 3D co-cultures. 1 mm of both ends and the center of the biopsy tissue were carefully collected and separately transferred into a 4-well plate (Nunc). A drop of PBS was added to the samples to keep them moist. Using a disposable scalpel, each section was cut into 10 pieces under a dissection microscope (MZ125, Leica, Germany) and all of the pieces were stored on ice for our 3D co-cultures. See 3D Platforms for more details.
3D Platforms
Our phenotypic fluorescent 3D platforms were prepared in three layers. The first layer, which is in contact with the bottom of a 96-well plate, is 50Μl of solidified 1.5% agarose. The second layer consists of 50Μl of a mixture of two or more types of cells evenly dispersed in Geltrex. The third layer consists of EBM2 medium supplemented with 1/10 of a SingleQuots supplement kit and may contain the testing agent(s).
To create the second layer, fluorescent endothelial cells and/or a third cell type (mast cells or pericytes) were harvested at 50~70% confluence and resuspended in EBM2 complete medium. Endothelial cells then were suspended at 140,000-280,000 cells/ml in Geltrex and 50Μl of the mixture was added onto the first layer. If a third cell population was used, it was mixed with the endothelial cells at 1:20-1:10 ratio. A spheroid tumor colony (~400 cells) or a xeno-biopsy (prepared as described in Tumor Spheroids and Tumor Nude Mouse Xenografts) was transplanted into the second layer. This complex was solidified at 37°C and cultured in 100Μl/well of EBM2 growth medium to make a final concentration of 0.5% FBS. The complex was cultured for 5-9 days before imaging (Zen2007, LSM 510, Zeiss). The high-resolution 3D z-stack images were rendered using the iso-surface 3D mode and their capillary density was quantified by using the Imaris 7.4.0 software (Biplane) [13]. The 360° view movies were generated using the maximum intensity panoramic projection (Zen2007). A time-lapse movie was recorded every 10 minutes for about 72 hours using a Nikon confocal microscope (LiveScan SFC) and rendered for a video file using a snapshot maximum intensity projection. All of the videos were converted into a MP4 format using MPEG Streamclip software (Squared 5).
3D Co-Culture Cryosection
After the medium was removed, the 3D co-cultures were washed three times in cold PBS. Each co-culture was fixed in 100μl of pre-chilled 0.5% glutaraldehyde in PBS, pH 7.4, at 4°C for 24 hours and embedded in O.C.T. (Tissue-Tek) following a brief wash in cold PBS. The co-cultures were sectioned at 5μm using a cryostat (Leica). The sections were mounted in an anti-fading fluorescent mounting medium without DAPI (Dako) for imaging (LSM510, Zeiss). Images were processed using Imaris 7.4.0 (Bitplane). The cryosection protocol was modified from a previous report [18].
Statistical Analysis
Statistical analysis was carried out using a two-tailed Student's t-test. For our collective studies, a minimal “n” value of 3 was routinely used for any given experimental group examined and all of the in vitro studies were repeated three times. P values less than 0.05 were considered statistically significant.
Results
Development of High-Resolution Phenotypic Fluorescent 3D Platforms
Angiogenesis is a fundamental step required by dormant microscopic tumors to grow beyond the size of 2mm and to progress down a metastatic pathway [19-21]. This vascular network process consists of a multi-stage event involving diverse cellular components and ECM [9-12, 20] (Figure 1a). Since 1971, tumor angiogenesis has become a major investigative focus regarding tumor growth, metastasis, and anticancer drug discovery [22, 23]. Additionally, capillary endothelial cells cultured with tumor cell conditional medium in in vitro models recapitulate some of in vivo angiogenic events including elongating, branching, and forming capillary vessels [24, 25]. Using traditional endothelial cell 2D in vitro models (EC Tubule Formation, Figure 1b), we have begun to understand the progressional events involved in tumor induced angiogenesis.
Tumor Microenvironment and Development/Validation of the Phenotypic Fluorescent 3D Platforms. (a), schematic of the tumor microenvironment consisting of malignant cells, non-malignant cells, and ECM. (b), flowchart showing the development of phenotypic fluorescent 3D platforms. 2D Assays: EC tubule formation - the light blue is a solidified layer of gel matrix (Geltrex) and the red objects with dark blue spheres are endothelial cells. Modified EC tubule formation/2D co-culture - the dark blue with red spheres in the center is a monolayer of tumor cells at 70-80% confluence; the light blue is a solidified layer of Geltrex, and the red objects with dark blue spheres in the center are endothelial cells. EC, endothelial cells and TC, tumor cells. Basic 3D Platforms are basic phenotypic fluorescent 3D platforms. The black at the bottom is a solidified layer of 1% agarose; the light blue is a layer of Geltrex with evenly dispersed endothelial cells (red objects with dark blue spheres in center) and a tumor spheroid (dark blue sphere cluster). Advanced 3D Platforms are advanced phenotypic fluorescent 3D platforms. The black at the bottom is a solidified layer of 1% agarose and the light blue is a layer of Geltrex with evenly dispersed endothelial cells (red objects with dark blue sphere in center) with either a tumor colony (dark blue sphere cluster) and a third cell component (yellow) or xeno-biopsy tissue (black sphere cluster). Xeno-Biopsy, a biopsy from a mouse human tumor xenograft. (c), the vascularization of freshly dissected nude mouse tumor xenografts. A549, a human lung adenocarcinoma cell line; SK-LMS-1, a human leiomyosarcoma cell line; PC-12, a rat pheochromocytoma cell line; and 92.1, a human ocular melanoma cell line. (d), vessel formation in 2D and modified 2D endothelial cell tubule formation assays at 6 hours of incubation. On the top row, the left and middle images were generated from the traditional 2D endothelial cell (PAE, an immortalized porcine aortic endothelial cell line in red) tubule formation assays with/without FBS supplement. The remaining images were taken from the tumor monolayer induced endothelial cell tubule formation assays, described as “modified 2D endothelial cell tubule formation assays”. (e), the vascularization in our fluorescent basic 3D platforms of PAE cells (red) and a tumor colony (dark blue) at 9 days in culture. 3D-rendered images of our 3D platforms demonstrate that endothelial cell vascularization clearly depends on the type of tumor cell colonies used in the assays and reveal how these two component cell types interact with each other. See Additional file 1: Supplementary Figure 1 for a variety of tumor colony induced vascularization in the 3D platforms. Also see Additional file 2: Movie 1, Additional file 3: Movie 2, and Figure 2 for intensive angiogenesis and the interactions between the tumor and endothelial cells. Scale bar = 100μm.
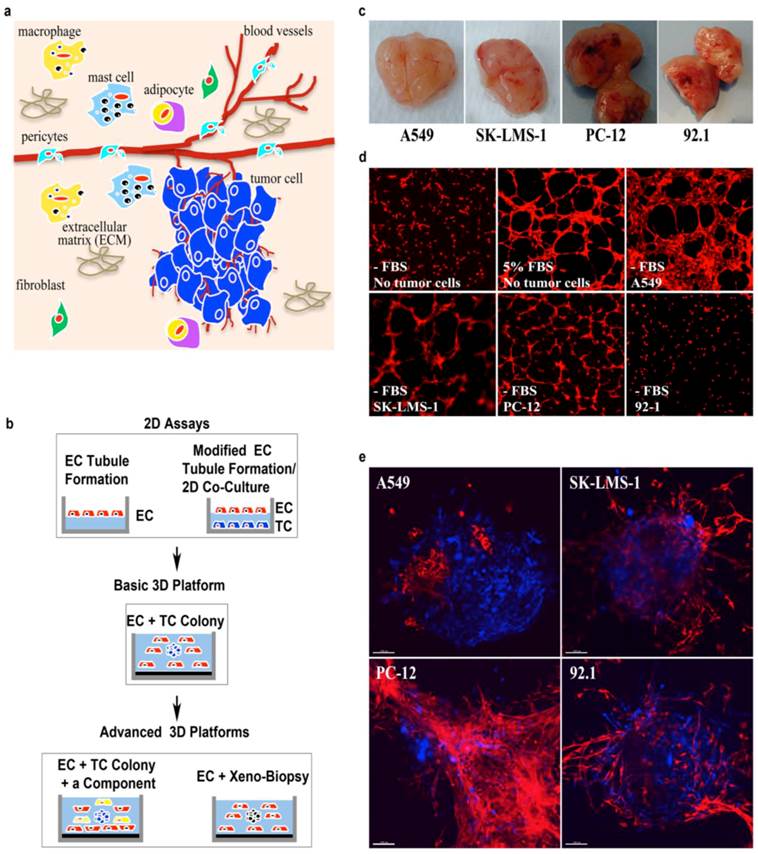
Given that tumor cell conditional medium was able to maintain and induce endothelial cells angiogenesis in vitro [24, 25], we evaluated whether a monolayer of tumor cells could have similar effects (Figure 1b). This method is referred to as the modified endothelial cell tubule formation (Modified EC Tubule Formation/2D Co-Culture, Figure 1b). Furthermore, considering that a tumor grows in a 3D microenvironment and interacts with multiple types of cells and ECM in vivo, we developed basic and advanced 3D platforms to mimic the tumor in vivo status (Figure 1b). In the basic 3D platform, there are only two different types of cell components - endothelial cells and a tumor cell colony (Figure 1b). In the advanced 3D platforms, endothelial cells are co-cultured with several types of cell components: (1) a tumor colony (blue sphere cluster on the left, Figure 1b) with a third type of cells (yellow on the left, Figure 1b); and (2) xeno-biopsy tissue (black sphere cluster on the right, Figure 1b) containing multiple types of cells. Introducing discriminatory fluorescent proteins into different types of cells in our 3D platforms, we can observe, identify, and track the interactions between individual cell populations in real-time. However, because we have observed varied tumor induced angiogenesis for a given tumor type in our 2D and 3D co-cultures, the pending question is which assay system best replicates the tumor in vivo status?
Tumor Angiogenic Activities in Phenotypic Fluorescent 3D Platforms Mimic in vivo Tumor Behaviors
It is well known that cells cultured in a 3D microenvironment change their morphology, growth patterns, gene expression profile, protein repertoire, biological function, and signaling pathways compared to cells cultured in 2D assays [7, 26]. Given the anatomical orientation of tumors in their microenvironment, we theorized that a 3D platform approach would be more consistent with the growth constraints observed in vivo. To prove this theory, we compared vascularization results in nude mouse tumor xenografts (Figure 1c) to the results obtained from 2D assays (Figure 1d) and our 3D platforms (Figure 1e). Figure 1c shows the freshly excised xenografts of a human lung adenocarcinoma (A549), a human leiomyosarcoma (SK-LMS-1), a rat pheochromocytoma (PC-12), and a human ocular melanoma (92.1). The A549 xenograft had moderate peripheral vasculature and little interior vessel formation, while the SK-LMS-1, PC-12, and 92.1 xenografts had highly angiogenic activities. However, in the 2D co-cultures, the A549 cells induced dramatic tubule formation of PAE cells compared to the controls (2D endothelial cell tubule formation with and without FBS), while the SK-LMS-1 and PC-12 induced moderate vessel formation and 92.1 was devoid of tubules (Figure 1d) at 6 hours of incubation. Thus, tumor cell induced endothelial cell angiogenesis in 2D co-cultures failed to consistently correlate with angiogenesis observed in vivo.
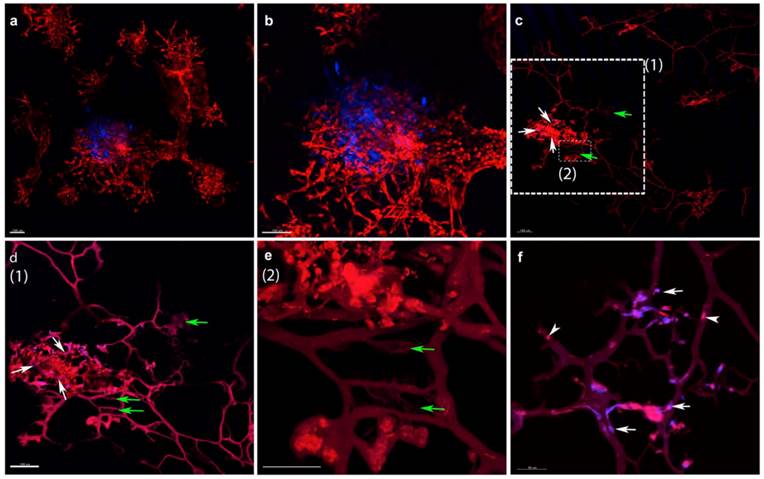
Contrastingly, when we assessed our 3D platforms for tumor cell induced angiogenesis using the same interactive cell populations, a correlative mimetic response to the in vivo environment was observed (Figure 1c versus Figure 1e). The high-resolution phenotypic confocal microscopy z-stack iso-surface 3D rendered images show that the A549 colony has peripheral vessels, while the SK-LMS-1, PC-12, and 92.1 colonies stimulated dramatic capillary formation peripherally and internally (Figure 1e). Detailed neovascularization characteristics of the 3D platforms are showed in Movies 1 and 2, as well as in Figure 2. A representative 3D panoramic video projection from a live 9-day culture of 92.1 induced PAE vascularization is illustrated in Movie 1. This video clearly shows that vessels surround the tumor colony. The real-time cell interactions were displayed in a time-lapse 72-hour-movie (Movie 2). To validate that vessels infiltrated into the tumor colony, 3D rendered images were taken from the frozen sections of PAE and 92.1 3D co-cultures after 9 days. As exemplified in Figure 2, our 3D live sample demonstrated endothelial cell (red) lattice formation around the tumor spheroid colony (blue) (Figure 2a and 2b) and more detailed observations were made on vessel structural complexities using frozen sections (Figure 2c-f). Endothelial network with some microvessels (green arrows, Figure 2, c-e) surround and infiltrated into the tumor colony (tumor cells indicated by white arrows, Figure 2c, d, and f). Along the vessels, we can see some endothelial nuclei (arrowheads) and tumor cells (arrows) (Figure 2f). For panoramic and real-time observations of the tumor induced vascularization in our 3D platforms, see Movies 1 and 2, respectively. Our 3D platforms have been applied to a variety of anatomically distinct cancer cell lines and have proven to represent similar tumor induced vascularization as observed in their in vivo counterparts, see Additional file 1: Supplementary Figure 1.
Vascularization Observed in 3D Rendered Images of a Live and Fixed Fluorescent 3D Co-Culture. (a) and (b): images of the live sample at different magnifications. Red cells are endothelial cells (PAE) and blue cells are tumor cells (92.1). PAE, an immortalized porcine aortic endothelial cell line and 92.1, a human ocular melanoma cell line. Scale bar = 100μm.(c) - (f): images of cryosections from the live sample in (a) and (b). (c): an image showing that vessels (red) not only surrounded but also penetrated into the tumor colony (blue). White arrows indicate individual tumor cells and green arrows indicate the microvessels. White dotted-framed areas (1) and (2) in image (c) were magnified and are shown in (d)(1) and (e)(2). (d): a magnified tumor-endothelial complex. White arrows indicate individual tumor cells and green arrows indicate the microvessels. Scale bar = 100μm. (e): magnified microvessels indicated by green arrows. Scale bar = 50μm. (f): the interactions between endothelial and tumor cells. White arrows indicate individual tumor cells along endothelial vascular highways and white arrowheads are individual endothelial cell nuclei. Scale bar = 50μm. For more details regarding tumor induced vascularization, see Movies 1 and 2.
Quantification of Capillary Density and Metastasis Phenomenon in Phenotypic Fluorescent 3D Platforms
An in vivo vascular system serves as a nutritional blood supply to tissues and as tumor neovascularization increases so does the angiogenesis dependent tumor growth. We have demonstrated that 3D platforms can recapitulate the tumor in vivo angiogenic status. However, it would be difficult to accurately determine an anti-angiogenic drug response without a quantitative assessment of the tumor induced vascular density for a given tumor cell line or an anatomical neoplastic biopsy. Fortunately, using 3D rendered images (Figure 3a), applying fluorescent cell discrimination techniques, and incorporating Imaris MeasurementPro software (7.4.0, Bitplane), we were able to accurately quantify tumor induced vascular density and establish a mechanism to statistically assess an anti-angiogenic drug sensitivity hierarchy [13] (Figure 3b). As illustrated in Figure 3b, the quantified capillary density data shows that A549 induced minimum vessel formation while PC-12, SK-LMS-1, 92.1, and U-87 had significant more angiogenesis, all of which correlates with their in vivo state (Figure 1c). Therefore, our 3D platforms appear to be an ideal surrogate for the nude mouse xenograft model.
Quantification of Tumor Induced Vacularization and Metastatic Phenomena. (a): 3D-rendered images of vascularization (red) with five different tumor cell colonies (blue). PAE, an immortalized porcine aortic endothelial cell line, A549, a human lung adenocarcinoma cell line, PC-12, a rat pheochromocytoma cell line, SK-LMS-1, a human leiomyosarcoma cell line, 92.1, a human ocular melanoma cell line, and U87, a human glioblastoma cell line. See Movies 1 and 2 regarding a 360° view of tumor/endothelial cell interaction and the observation of early events in tumor induced angiogenesis. Scale bar = 100μm. (b): quantified capillary density. N=3; Mean ±STD; and * p < 0.05. (c): phenomena of tumor vascular highway mediated metastasis or angiotrophism. The 3D platforms also recapitulate the tumor migration status in vitro. The four tumor cell lines (blue) in (a) were evaluated against three distinct endothelial cell lines (red). HMEC-1, an immortalized human microvascular endothelial cell line and TIME, a telomerase-immortalized human microvascular endothelial cell line. White arrows indicate the primary tumor colonies; white arrowheads indicate migrated tumor cells; and green arrows indicate the secondary tumor colonies. Scale bar =100 μm.
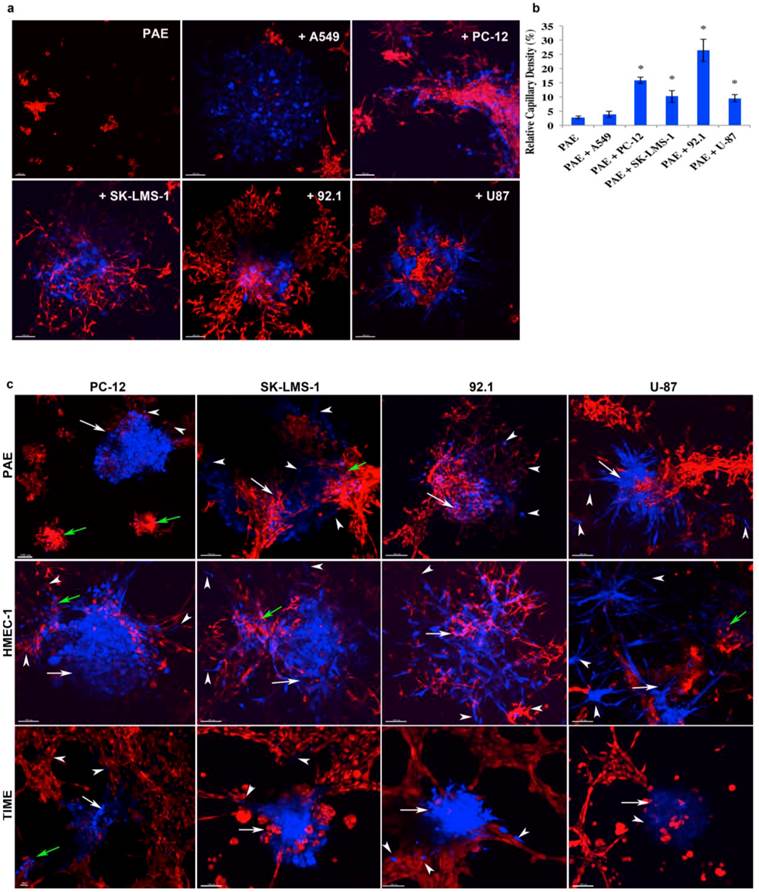
Although metastasis accounts for approximately 90% of cancer-related deaths [27], to date few existing agents specifically block primary tumor cell migration to distal anatomical sites. Moreover, because there are no known effective in vitro metastatic models, pharmaceutical companies have done limited work in pursuing the development of anti-metastatic drugs. An efficient high-throughput in vitro assay mimicking the tumor metastatic process would augment the identification of effective drugs that block neoplastic cell migration to distal loci. Primary tumor cells can travel to secondary anatomical sites through established routes such as the blood stream or the lymphatic system [28]. Also, a lesser known mechanism, perivascular migration or angiotrophism, involves neoplastic cells migrating along vessels externally to remote sites of the body [29]. With the use of our 3D platforms, however, it is now possible to assess not only metastasis and secondary tumor loci development but also the angiotrophic migration of tumor cells in vitro. In Figure 3c, we tested tumor colonies (blue) from PC-12, SK-LMS-1, 92.1, and U-87 with varieties of endothelial cells (red), i.e., PAE, HMEC-1, and TIME, to demonstrate tumor metastasis in vitro. The 3D rendered images show that the tumor cells (white arrowheads) migrated away from the primary mass (white arrows) and some cells even formed secondary colonies (green arrows). Tumor cells migrating along the highway of endothelial cells can be seen in the images taken from the frozen section (arrows, Figure 2f).
To show the detailed interactions of tumor and endothelial cells in our 3D platforms, we recorded a 72-hour time-lapse movie (Additional file 3: Movie 2). For the very first time, we observed how tumor and endothelial cells interacted with each other and how the early stages of tumor induced angiogenesis were initiated, including the phenomenon that tumor induced vascularization of endothelial cells increased dramatically as the tumor grew. See Additional file 2: Movie 1, which shows a fully developed vascular network after 9 days in culture and Additional file 3: Movie 2, which shows the tumor induced vascularization over 72 hours in culture. In addition, the formation of endothelial cell exosomes or microvesicles were observed and the shedding rate of these cellular fragments can be measured over time. Such cellular debris are known to contain both genetic and protein materials that regulate cell proliferation associated with both normal and disease physiology [30], which offer additional targets to measure drug efficiency.
Advanced Phenotypic Fluorescent 3D Platforms with Three Types of Cells
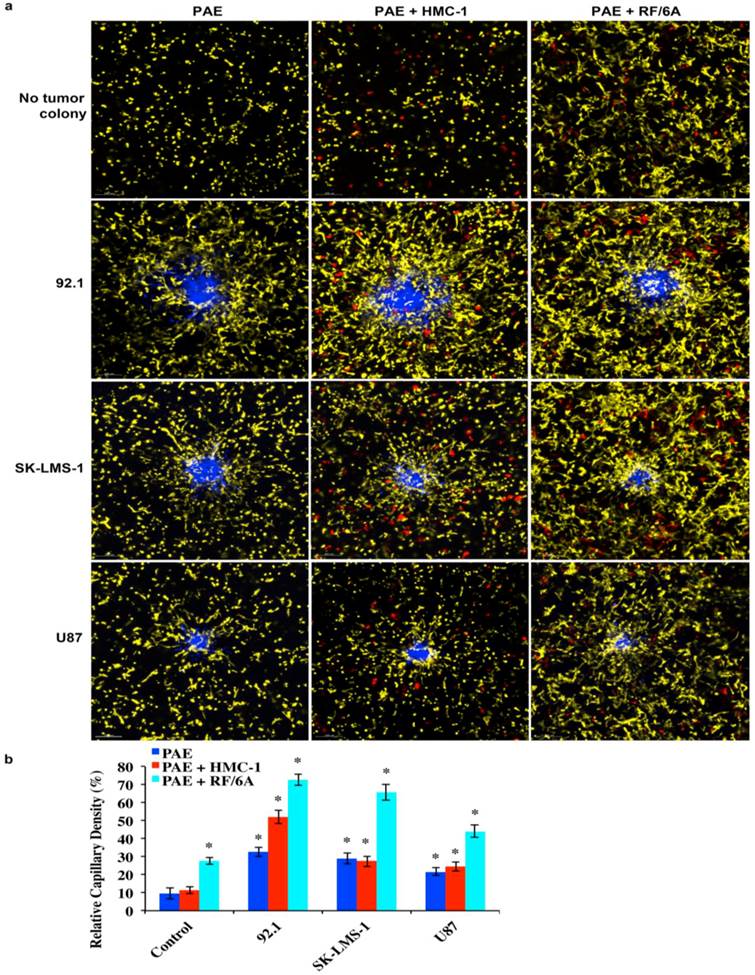
Because tumors grow in a 3D microenvironment and interact with multiple types of cells encompassed in an ECM [9, 10, 12, 31, 32], by incorporating a third type of cell population, we took our 3D platforms to a more complex level (advanced 3D platforms). The setup is shown in Figure 1b and the resulting images and quantitative data are shown in Figures 4a and 4b. Tumor cell (blue) induced neovacularization (yellow) was selectively enhanced depending on the neoplastic cells and third component cells (red) utilized. HMC-1 augmented 92.1 induced angiogenesis but had very little effect on SK-LMS-1 or U87 activity. Conversely, RF-6A (an immortalized monkey pericyte cell line) enhanced tumor induced vessel formation indiscriminately for the cancer cell types evaluated. As previously reported, pericytes have been shown to favor tumor angiogenesis and growth and are resistant to anti-cancer drugs [33, 34]. The observed phenomenon that mast cells are involved in the angoiogenic promotion of melanoma supports the notion that these inflammatory cells release potent proangiogenic factors [35, 36]. However, some clinical evidence demonstrates that mast cell accumulation benefits cancer patients [37]. Accordingly, our 3D platforms would be suitable to explore the involvement of mast cells, as well as other cell types, with regard to tumor progression.
Quantification of Vascularization in Advanced 3D Platforms. (a): 3D-rendered images of vascularization (yellow) induced by tumor colonies (blue) with/without a third cell population (red). PAE, an immortalized porcine aortic endothelial cell line. 92.1, a human ocular melanoma cell line; SK-LMS-1, a human leiomyosarcoma cell line; and U87, a human glioblastoma cell line; HMC-1, an immortalized human mast cell line; and s, an immortalized monkey pericyte line. Scale bar = 200μm. (b): quantified capillary density. N=3, Mean ±STD, * p < 0.05.
Drug Sensitivity Screening and Advanced Phenotypic Fluorescent 3D Platforms
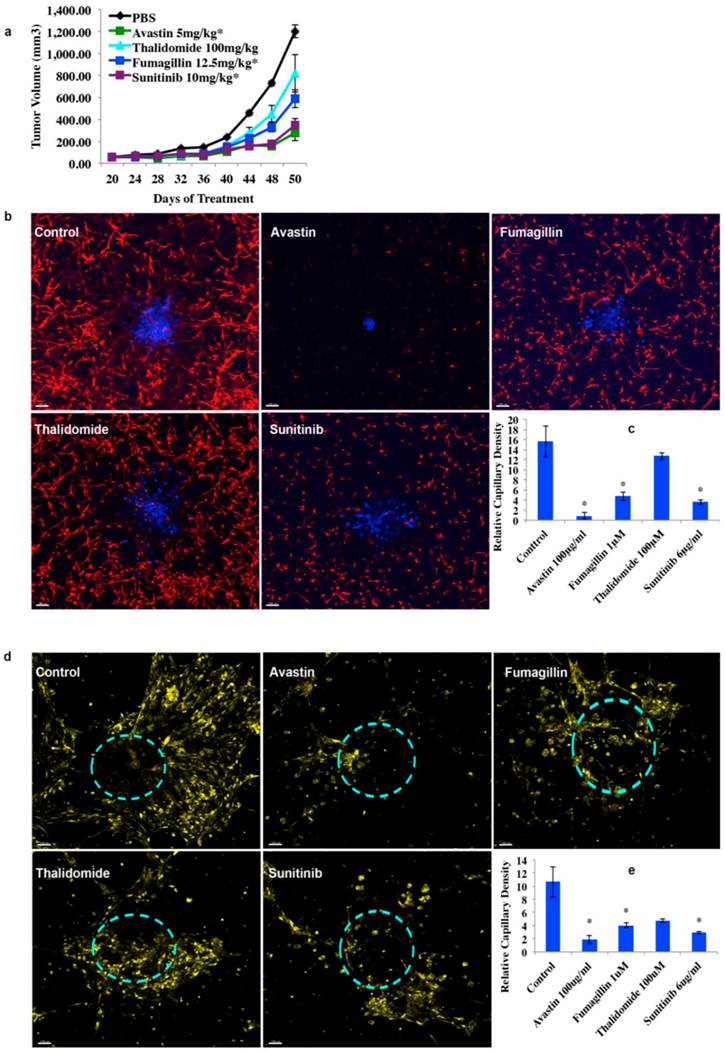
So far, we have shown that our 3D platforms closely mimic in vivo tumor features such as angiogenesis, metastasis, and interactions between different cell types and ECM. We have also demonstrated that our 3D platforms are more efficient and cost effective than mouse xenograft models regarding anticancer drug screening. We compared the sensitivities of four drugs on SK-LMS-1 (Figure 5) and A549 (data not shown) using our 3D platforms. Drug sensitivity results were obtained in 9 days using the basic 3D platforms (Figure 5b) and in only 5 days using the advanced 3D platforms (Figure 5d), but it took 50 days to obtain equivalent results from the mouse xenograft models (Figure 5a). The Figure 5d setup is shown in Figure 1b. The quantified capillary density data from the 3D rendered images in Figures 5b and 5d appear in Figures 5c and 5e, respectively. As shown, the drug sensitivity results between the in vivo (Figure 5a) and in vitro (Figures 5c and 5e) models are similar. Thus, our 3D platforms were much more efficient, as well as less costly, than animal studies in obtaining equivalent drug sensitivity results. It is noteworthy to mention that major variances in angiogenesis were observed among different anatomical xeno-biopsies from the same xenograft, see Additional file 1: Supplementary Figure 2. Clearly, peritumoral biopsies of SK-LMS-1 (Supplementary Figure 2a) and 92.1 (Supplementary Figure 2c) dramatically stimulated neovascularization as compared to non-necrotic intratumoral biopsies (Supplementary Figures 2b and d), respectively. This is consistent with the prior reports demonstrating that peritumoral sampling, rather than an intratumoral site, represents a more accurate depiction of the tumor's progress and prognosis characteristics [38].
Validation of the 3D Platforms as Ideal Surrogates for the Nude Mouse Tumor Xenograft Models due to their Efficiency and Effectiveness of Drug Sensitivity Screening. (a): inhibition of the nude mouse xenograft (SK-LMS-1) growth using the following four anti-angiogenic drugs: Avastin 5mg/kg, fumagillin 100mg/kg, and thalidomide 12.5mg/kg were injected introperitoneally (IP) three times a week, except for 10mg/kg of Sunitinib was administrated orally five times a week. The vehicle is PBS with 0.1% dimethyl sulfoxide (DMSO). The treatment was started after a palpable tumor mass was formed (~100mm3). Each treatment group was composed of 10 mice. The vehicle control was combination of IP and oral vehicle groups. SK-LMS-1 is a human leiomyosarcoma cell line. Mean ±STD, * p < 0.05. (b): inhibition of tumor (blue) induced vascularization (HMEC-1, red) using the same drugs referenced in (a) in the basic 3D platforms after 9 days in culture but with the following concentrations: Avastin (100μg/ml), fumagillin (1μM), Sunitinib (6μg/ml), and thalidomide (100μg/ml) in EBM2 medium with 1/10 of a bullet supplement and 0.1% DMSO. HMEC-1 is an immortalized human microvascular endothelial cell line. Scale = 100μm. (c): quantified capillary density of the tumor induced vascularization shown in (b). N=3, Mean ±STD, * p < 0.05. (d): inhibition of peritumoral xenograft biopsy (SK-LMS-1, a dotted-circle in light blue) induced vascularization (yellow) using the same drugs and concentrations described in (b) in the advanced 3D platforms after 5 days in culture. Scale = 100μm. (e): quantified capillary density of the tumor induced vascularization shown in (d). N=3, Mean ±STD, * p < 0.05.
Discussion
We have developed phenotypic fluorescent 3D platforms to simulate the in vivo microenvironment of tumors for high-throughput drug screening and personalized chemotherapy. These platforms should be the models of choice for the future because they do not have the correlative inadequacies of 2D cell assays and, unlike mouse xenograft models, are efficient and cost effective. Using multi-colored fluorescent cell lines, individual cellular components can be identified, tracked, and monitored live by a fluorescent microscope, and then isolated by a flow cytometer or a laser capture micro-dissection microscope for further protein/gene expression studies. Our novel 3D platforms are more convenient and controllable to better understand the processes of tumor induced angiogenesis and metastasis. More importantly, the 3D platforms are potentially ideal for personalized chemotherapy. Additionally, these platforms may be adapted for research and clinical applications for a variety of angiogenesis dominated diseases [1].
Ideal in vitro Angiogenic and Metastatic Platforms Based on A High-Resolution Phenotypic Profiling of a Multi-Cell Type Complex
Although 2D co-cultures are able to demonstrate tumor induced angiogenesis, their results did not correlate well with the angiogenic status of the tumor in vivo and proved vastly inferior to our 3D platforms. Likewise, other established 3D in vitro models also were insufficient [39]. Tumor growth depends on the development of an effective nutritional and waste removal system, as well as a supportive infrastructure [20, 40, 41]. Therefore, in vitro models that best mimic this complex interaction will generate more reliable data. Our 3D platforms are the first attempt to incorporate the collective aspects of the tumor microenvironment that utilize discriminatory colorimetric cell tracking and have proven to effectively parallel in vivo response profiles for a given tumor.
Tumor angiogenesis includes two events: the launching of the angiogenic switch phase and the vascularization phase [42]. Although some modified animal models have been used to identify the genes involved in tumor angiogenesis [43] and serve significant roles in defining angiogenic switch regulators, they are very time consuming and costly. Furthermore, it is not clear whether these modified animal models would affect the ultimate outcome regarding gene regulation [20, 44]. Researchers have long been interested in developing an in vitro 3D model wherein angiogenic switch regulators can be controlled and studied more conveniently.
Using our 3D platforms, one can selectively control the component makeup of the tumor microenvironment and assess how the different cell effectors interact with each other around the tumor colonies in real time. It is well known that an avascular microscopic cancer in excess of 2mm is blood vessel dependent for further growth to occur [20] and that the development of these new blood vessels (angiogenesis) are the recognized targets of chemotherapy [22]. Endothelial cells and a tumor colony were the first effectors used in our 3D platforms to simulate the in vivo tumor microenvironment. We observed that the tumor induced vascularization of the endothelial cells increased as the tumor grew. Because angiogenic and non-angiogenic tumor colonies are easily identified, the 3D platforms will enable researchers to compare gene/protein expression profiles in tumor colonies and the surrounding cells to ultimately identify the angiogenic switch and vascular regulators. Although tumor metastasis in vivo is an extremely inefficient process [45], it is still the primary cause of a lethal outcome in cancer patients [20, 27]. In the absence of efficient in vitro and in vivo metastatic models, investigators have had difficulty identifying and tracking early cellular events involved in cancer progression to a distal anatomical loci [28]. Consequently, it has been a major challenge to discover anti-metastatic drugs, especially considering the complexity of metastasis and the lack of research funding [46]. We have now demonstrated that at least one aspect of the metastatic process, angiotrophism, can be effectively studied in our 3D platforms to evaluate cancer cell migration along perivascular highways to distal sites from the primary tumor loci. Hence, this technology is able to assess anti-metastatic compounds and, in conjunction with flow cytometry or laser capture micro-dissection microscopy, to identify previously unknown molecular mechanisms responsible for tumor metastasis. Because these angiogenic switch regulators and metastatic inducers can be translated into meaningful clinical diagnostic biomarkers, the management of cancer treatment will be much more effective in devising intervention strategies to block dormant tumors from becoming symptomatic and progressing down the metastatic pathway.
A Surrogate for Animal Studies for Efficient and Effective Anti-angiogenic and Anti-metastatic Drug Screening on a High-throughput Scale
Our 3D platforms have been proven to recapitulate the angiogenic and metastatic status of a tumor in vivo. We have shown that drug sensitivity analysis can be performed with either tumor spheroids or xeno-biopsies in our 3D platforms, demonstrating a “Proof-of-Principle” for segueing this in vitro technology into the clinical setting. When assessing the anti-angiogenic effects of Avastin, sunitinib, thalidomide, and fumagillin in our 3D platforms, we demonstrated the drug response hierarchy matched the responses observed in the nude mouse xenograft model for a given tumor. When evaluating highly metastatic human tumor cell lines, such as melanoma [47], leiomyosarcoma [48], and rat pheochromocytoma [49], we observed that the individual cancer cells traveled along endothelial vascular highways (perivascular migration or angiotrophism) and migrated far beyond the cellular branch projections of the primary tumor colonies. In some cases, these migrated tumor cells formed secondary colonies. This phenomenon has been previously observed in pathological specimens of human glioma or glioblastoma [50] and melanoma [47]. Hence, our assays also offer a means to screen drugs with anti-metastatic potential. Since our 3D platforms employ a 96-well microtiter plate, it is readily adaptable to high-throughput drug screening with automated operations, thereby substantially reducing drug development costs.
A Robust Platform for Personalized Cancer Chemotherapies Based on A High-Throughput and Time-Sensitive Drug Sensitivity Screening Scale
It is of paramount importance that our 3D platforms have made personalized chemotherapy a reality. The drug sensitivity data acquired in only 5 days from this technology correlated exceedingly well with the observed results from nude mouse xenograft models. We have also demonstrated anatomical differences of xeno-biopsies regarding angiogenic potentials. These observations validate the existence of tumor heterogeneity and the importance of scrutinizing tissue selection for use in angiogenic analyses. Considering the successful achievements of our 3D platforms, these approaches can be easily segued to clinical practice for an adaptive treatment strategy by matching the most appropriate and effective drug regimen for a particular cancer patient. Furthermore, they are particularly useful for continuously monitoring the effectiveness of chemotherapeutics. Such an assessment will allow physicians to appropriately modify anti-angiogenesis based chemotherapies, thereby circumventing the possibility of drug resistance and will assist in developing a more effective treatment plan.
In summary, our novel phenotypic fluorescent 3D platforms recapitulate tumor in vivo angiogenic and metastatic features. These in vitro models are ideal for studying basic tumor biology and for efficiently and economically screening anti-angiogenic and anti-metastatic drugs on a high-throughput scale. More importantly, these platforms will eventually enable clinicians to provide personalized chemotherapy regimens for their patients. We also anticipate that the 3D platforms can be used to identify target pathways and logical avenues to pursue for management of vascular diseases other than cancer.
Supplementary Material
Additional File 1Supplementary Figure 1 and 2.
Movie 1.
Movie 2.
Acknowledgements
We thank Dr. Yvonne Gonzalez, NCI-Frederick, Inc., for performing the nude mouse xenograft experiments with SK-LMS-1 and A549 cells and in collecting the data; Dr. Charles Heller, NCI, for injecting the transfected tumor cells into nude mice for the xeno-biopsies; Dr. Dragon Maric, NINDS, and Ms. Barbara J. Taylor for enriching the transfected cells at NCI; Mr. Scott A. McGinnis for recording the 3D time-lapse movie at Nikon Instruments Inc., and Dr. David Burk for instructions on using the Imaris 7.4.0 to quantify the capillary density in the 3D z-stack images in the Cell Biology and Cell Imaging Core, Pennington Biomedical Research Center, Louisiana State University.
Author Contributions
C. F. and F. C. designed the experiments; C. F. and I. A. performed all of the in vitro experiments; D. S. provided some consultations; and C. F., I. A. and F. C. wrote the manuscript.
Competing Interests
The authors declare no competing financial interests.
References
1. Albini A, Tosetti F, Li VW, Noonan DM, Li WW. Cancer prevention by targeting angiogenesis. Nat Rev Clin Oncol. 2012;9:498-509 doi:10.1038/nrclinonc.2012.120
2. Kunz-Schughart LA, Freyer JP, Hofstaedter F, Ebner R. The use of 3-D cultures for high-throughput screening: the multicellular spheroid model. J Biomol Screen. 2004;9:273-85 doi:10.1177/1087057104265040
3. Horning JL, Sahoo SK, Vijayaraghavalu S, Dimitrijevic S, Vasir JK, Jain TK. et al. 3-D tumor model for in vitro evaluation of anticancer drugs. Mol Pharm. 2008;5:849-62 doi:10.1021/mp800047v
4. Pampaloni F, Reynaud EG, Stelzer EH. The third dimension bridges the gap between cell culture and live tissue. Nat Rev Mol Cell Biol. 2007;8:839-45 doi:nrm2236 [pii]. 10.1038/nrm2236
5. Abbott A. Cell culture: biology's new dimension. Nature. 2003;424:870-2 doi:10.1038/424870a. 424870a [pii]
6. Smalley KS, Lioni M, Herlyn M. Life isn't flat: taking cancer biology to the next dimension. In Vitro Cell Dev Biol Anim. 2006;42:242-7 doi:0604027 [pii]. 10.1290/0604027.1
7. Mitra M, Mohanty C, Harilal A, Maheswari UK, Sahoo SK, Krishnakumar S. A novel in vitro three-dimensional retinoblastoma model for evaluating chemotherapeutic drugs. Mol Vis. 2012;18:1361-78
8. Friedrich J, Seidel C, Ebner R, Kunz-Schughart LA. Spheroid-based drug screen: considerations and practical approach. Nat Protoc. 2009;4:309-24 doi:nprot.2008.226 [pii]. 10.1038/nprot.2008.226
9. Folkman J. The role of angiogenesis in tumor growth. Semin Cancer Biol. 1992;3:65-71
10. Folkman J, Shing Y. Angiogenesis. J Biol Chem. 1992;267:10931-4
11. Bissell MJ, Radisky D. Putting tumours in context. Nat Rev Cancer. 2001;1:46-54 doi:10.1038/35094059
12. Coussens LM, Werb Z. Inflammation and cancer. Nature. 2002;420:860-7 doi:10.1038/nature01322
13. Xu F, Burk D, Gao Z, Yin J, Zhang X, Weng J. et al. Angiogenic deficiency and adipose tissue dysfunction are associated with macrophage malfunction in SIRT1-/- mice. Endocrinology. 2012;153:1706-16 doi:10.1210/en.2011-1667
14. Petersen OW, Ronnov-Jessen L, Howlett AR, Bissell MJ. Interaction with basement membrane serves to rapidly distinguish growth and differentiation pattern of normal and malignant human breast epithelial cells. Proceedings of the National Academy of Sciences of the United States of America. 1992;89:9064-8
15. Lang SH, Sharrard RM, Stark M, Villette JM, Maitland NJ. Prostate epithelial cell lines form spheroids with evidence of glandular differentiation in three-dimensional Matrigel cultures. British journal of cancer. 2001;85:590-9 doi:10.1054/bjoc.2001.1967
16. Bianco C, Strizzi L, Ebert A, Chang C, Rehman A, Normanno N. et al. Role of human cripto-1 in tumor angiogenesis. J Natl Cancer Inst. 2005;97:132-41 doi:97/2/132 [pii]. 10.1093/jnci/dji011
17. Hamburger AW, Salmon SE. Primary bioassay of human tumor stem cells. Science. 1977;197:461-3
18. Brown TA, Fetter RD, Tkachuk AN, Clayton DA. Approaches toward super-resolution fluorescence imaging of mitochondrial proteins using PALM. Methods. 2010;51:458-63 doi:10.1016/j.ymeth.2010.01.001
19. Gimbrone MA Jr, Leapman SB, Cotran RS, Folkman J. Tumor dormancy in vivo by prevention of neovascularization. J Exp Med. 1972;136:261-76
20. Folkman J, Kalluri R. Cancer without disease. Nature. 2004;427:787. doi:10.1038/427787a. 427787a [pii]
21. Folkman J. What is the evidence that tumors are angiogenesis dependent? J Natl Cancer Inst. 1990;82:4-6
22. Folkman J. Tumor angiogenesis: therapeutic implications. N Engl J Med. 1971;285:1182-6
23. Folkman J. Antiangiogenesis in cancer therapy--endostatin and its mechanisms of action. Exp Cell Res. 2006;312:594-607 doi:S0014-4827(05)00545-8 [pii]. 10.1016/j.yexcr.2005.11.015
24. Folkman J, Haudenschild CC, Zetter BR. Long-term culture of capillary endothelial cells. Proc Natl Acad Sci U S A. 1979;76:5217-21
25. Folkman J, Haudenschild C. Angiogenesis by capillary endothelial cells in culture. Trans Ophthalmol Soc U K. 1980;100:346-53
26. Cukierman E, Pankov R, Stevens DR, Yamada KM. Taking cell-matrix adhesions to the third dimension. Science. 2001;294:1708-12 doi:10.1126/science.1064829
27. Sporn MB. The war on cancer. Lancet. 1996;347:1377-81
28. Sleeman JP, Nazarenko I, Thiele W. Do all roads lead to Rome? Routes to metastasis development. International journal of cancer Journal international du cancer. 2011;128:2511-26 doi:10.1002/ijc.26027
29. Quatresooz P, Pierard-Franchimont C, Noel F, Pierard GE. Thigmotropism of malignant melanoma cells. Dermatol Res Pract. 2012;2012:362784. doi:10.1155/2012/362784
30. Lee Y, El Andaloussi S, Wood MJ. Exosomes and microvesicles: extracellular vesicles for genetic information transfer and gene therapy. Human molecular genetics. 2012 doi:10.1093/hmg/dds317
31. Ribatti D, Crivellato E. Immune cells and angiogenesis. J Cell Mol Med. 2009;13(9A):2822-33 doi: 10.1111/j.1582-4934.2009.00810.x
32. Ribatti D, Crivellato E, Molica S. Mast cells and angiogenesis in haematological malignancies. Leuk Res. 2009;33:876-9 doi: 10.1016/j.leukres.2009.02.028
33. Bergers G, Song S, Meyer-Morse N, Bergsland E, Hanahan D. Benefits of targeting both pericytes and endothelial cells in the tumor vasculature with kinase inhibitors. The Journal of clinical investigation. 2003;111:1287-95 doi:10.1172/JCI17929
34. Allt G, Lawrenson JG. Pericytes: cell biology and pathology. Cells Tissues Organs. 2001;169:1-11
35. Kessler DA, Langer RS, Pless NA, Folkman J. Mast cells and tumor angiogenesis. Int J Cancer. 1976;18:703-9
36. Ribatti D, Crivellato E. Mast cells, angiogenesis, and tumour growth. Biochimica et biophysica acta. 2012;1822:2-8 doi:10.1016/j.bbadis.2010.11.010
37. Chan JK, Magistris A, Loizzi V, Lin F, Rutgers J, Osann K. et al. Mast cell density, angiogenesis, blood clotting, and prognosis in women with advanced ovarian cancer. Gynecol Oncol. 2005;99:20-5 doi:10.1016/j.ygyno.2005.05.042
38. Wang XL, Fang JP, Tang RY, Chen XM. Different significance between intratumoral and peritumoral lymphatic vessel density in gastric cancer: a retrospective study of 123 cases. BMC Cancer. 2010;10:299. doi:10.1186/1471-2407-10-299
39. Sutherland RM. Cell and environment interactions in tumor microregions: the multicell spheroid model. Science. 1988;240:177-84
40. Furuya M, Yonemitsu Y, Aoki I. III. Angiogenesis: complexity of tumor vasculature and microenvironment. Curr Pharm Des. 2009;15:1854-67
41. Folkman J. Angiogenesis: an organizing principle for drug discovery? Nat Rev Drug Discov. 2007;6:273-86 doi:nrd2115 [pii]. 10.1038/nrd2115
42. Hanahan D, Folkman J. Patterns and emerging mechanisms of the angiogenic switch during tumorigenesis. Cell. 1996;86:353-64 doi:S0092-8674(00)80108-7 [pii]
43. Hanahan D, Christofori G, Naik P, Arbeit J. Transgenic mouse models of tumour angiogenesis: the angiogenic switch, its molecular controls, and prospects for preclinical therapeutic models. Eur J Cancer. 1996;32A:2386-93
44. Knight ZA, Shokat KM. Chemical genetics: where genetics and pharmacology meet. Cell. 2007;128:425-30 doi:S0092-8674(07)00119-5 [pii]. 10.1016/j.cell.2007.01.021
45. Chambers AF, Naumov GN, Vantyghem SA, Tuck AB. Molecular biology of breast cancer metastasis. Clinical implications of experimental studies on metastatic inefficiency. Breast Cancer Res. 2000;2:400-7
46. Sleeman J, Steeg PS. Cancer metastasis as a therapeutic target. Eur J Cancer. 2010;46:1177-80 doi:S0959-8049(10)00166-8 [pii]. 10.1016/j.ejca.2010.02.039
47. Barnhill RL, Lugassy C. Angiotropic malignant melanoma and extravascular migratory metastasis: description of 36 cases with emphasis on a new mechanism of tumour spread. Pathology. 2004;36:485-90 doi:10.1080/00313020412331282708. 2PL240DM9Y1X13JG [pii]
48. Elhammady MS, Manzano GR, Lebwohl N, Levi AD. Leiomyosarcoma metastases to the spine. Case series and review of the literature. J Neurosurg Spine. 2007;6:178-83 doi:10.3171/spi.2007.6.2.178
49. Favier J, Plouin PF, Corvol P, Gasc JM. Angiogenesis and vascular architecture in pheochromocytomas: distinctive traits in malignant tumors. The American journal of pathology. 2002;161:1235-46 doi:10.1016/S0002-9440(10)64400-8
50. Samoto K, Ikezaki K, Ono M, Shono T, Kohno K, Kuwano M. et al. Expression of vascular endothelial growth factor and its possible relation with neovascularization in human brain tumors. Cancer research. 1995;55:1189-93
Author biography
Dr. Changge Fang is a highly proficient translational biologist who has extensive knowledge and experience in cellular and molecular biology, oncology, and immunology. She is skilled in performing high-throughput screening using cell-based assays and genomic approaches to identify potential oncogenic targets. Dr. Fang has extensive experience in evaluating and validating potential anti-angiogenic and anti-metastatic candidates, including peptides, antibodies, and small molecules, utilizing various in vitro cell-based assays as well as in vivo animal models. At the Angiogenesis Core Facility (ACF), National Cancer Institute (NCI), National Institutes of Health, and with the assistance of Dr. Frank Cuttitta, the former Director of the ACF, Dr. Fang designed and developed the 3D platforms (U.S. Pat. Appl. No. 12/802,666) for high-throughput drug screening and personalized chemotherapy. Dr. Fang obtained her Ph.D. in Physiology & Molecular Biology and a Masters in Physiology in Animal Sciences from the China Agricultural University in Beijing. From 2004 to 2012, Dr. Fang completed her postdoctoral training at various institutions at NIH, including NCI. On May 23, 2013, Dr. Fang was invited to the National Institute of Standards and Technology to give a lecture on “Three-Dimensional High-throughput Chemosensitivity Screening: The Model of Choice for Personalized Chemotherapy and Drug Development." In August 2012, Dr. Fang founded Advanced Personalized Diagnostics (APD), a life sciences company, to utilize the 3D platforms to provide chemosensitivity screening services to medical providers to develop a customized and evidence-based treatment plan for cancer patients, as well as drug development services to pharmaceutical companies and medical research facilities.
![]() Corresponding author: changgefangcom; Tel. 1(202)-277-1956; Fax. 1(703) 960-3216.
Corresponding author: changgefangcom; Tel. 1(202)-277-1956; Fax. 1(703) 960-3216.