Impact Factor ISSN: 1837-9664

 Global reach, higher impact
Global reach, higher impactJ Cancer 2013; 4(8):614-625. doi:10.7150/jca.7080 This issue Cite
Research Paper
CRM1 Inhibition Sensitizes Drug Resistant Human Myeloma Cells to Topoisomerase II and Proteasome Inhibitors both In Vitro and Ex Vivo
Joel G. Turner1, Jana Dawson1, Michael F. Emmons2, Christopher L. Cubitt3, Michael Kauffman4, Sharon Shacham4, Lori A. Hazlehurst2, Daniel M. Sullivan1 ![]()
1. Department of Blood and Marrow Transplantation and Chemical Biology and Molecular Medicine Program, H. Lee Moffitt Cancer Center and Research Institute, Tampa, Florida 33612
2. Molecular Oncology Department, H. Lee Moffitt Cancer Center and Research Institute, Tampa, Florida 33612
3. Translational Research Core Laboratory, H. Lee Moffitt Cancer Center and Research Institute, Tampa, Florida 33612
4. Karyopharm Therapeutics, Boston, MA 01760.
Received 2013-7-3; Accepted 2013-8-7; Published 2013-9-10
Abstract
Multiple myeloma (MM) remains an incurable disease despite improved treatments, including lenalidomide/pomalidomide and bortezomib/carfilzomib based therapies and high-dose chemotherapy with autologous stem cell rescue. New drug targets are needed to further improve treatment outcomes. Nuclear export of macromolecules is misregulated in many cancers, including in hematological malignancies such as MM. CRM1 (chromosome maintenance protein-1) is a ubiquitous protein that exports large proteins (>40 kDa) from the nucleus to the cytoplasm. We found that small-molecule Selective Inhibitors of Nuclear Export (SINE) prevent CRM1-mediated export of p53 and topoisomerase IIα (topo IIα). SINE's CRM1-inhibiting activity was verified by nuclear-cytoplasmic fractionation and immunocytochemical staining of the CRM1 cargoes p53 and topo IIα in MM cells. We found that SINE molecules reduced cell viability and induced apoptosis when used as both single agents in the sub-micromolar range and when combined with doxorubicin, bortezomib, or carfilzomib but not lenalidomide, melphalan, or dexamethasone. In addition, CRM1 inhibition sensitized MM cell lines and patient myeloma cells to doxorubicin, bortezomib, and carfilzomib but did not affect peripheral blood mononuclear or non-myeloma bone marrow mononuclear cells as shown by cell viability and apoptosis assay. Drug resistance induced by co-culture of myeloma cells with bone marrow stroma cells was circumvented by the addition of SINE molecules. These results support the continued development of SINE for patients with MM.
Keywords: CRM1, topoisomerase IIα, p53, multiple myeloma, nuclear export.
Introduction
The intracellular location of a protein is crucial to its normal functioning in a cell. Cancer cells utilize the processes of nuclear-cytoplasmic transport through the nuclear pore complex to effectively evade anti-cancer mechanisms [1-3]. Examples of nuclear proteins that are exported into the cytoplasm in cancer include the drug targets topoisomerase (topo) IIα [3] and BCR-ABL [4] and tumor-suppressor proteins such as retinoblastoma [5], APC [6], p53 [7], p21 [8], and p27 [9]. In addition, CRM1-mediated export is increased in various cancers (reviewed in Turner et al [2]). The therapeutic potential of various CRM1 inhibitors has begun to be addressed in the laboratory. Compounds under investigation include ratjadone compounds [3, 10-13], KOS-2464 [14], FOXO export inhibitors [15], valtrate [16], acetoxychavicol acetate [17], and most recently CBS9106 [18] and small-molecule selective inhibitors of nuclear export (SINE) [19]. Recent publications have indicated that SINE compounds may be effective against various malignancies, including leukemia [19-23], kidney cancer [24], mantle cell lymphoma [25], melanoma [26], and multiple myeloma (MM) [22].
In this study, we investigated whether SINE molecules would also be effective against MM in combination with drugs used to treat myeloma such as doxorubicin, bortezomib, carfilzomib, lenalidomide, dexamethasone, and melphalan. Using both human MM cell lines and patient bone marrow samples, we found that SINE molecules were effective both as single agents and when combined with the chemotherapeutic drugs doxorubicin, bortezomib, or carfilzomib but not lenalidomide, melphalan, or dexamethasone. These data support the ongoing development of these small-molecule CRM1 antagonists in patients with MM.
Materials and Methods
For human samples, informed consent approved by the University of South Florida Institutional Review Board was obtained from all patients, in accordance with the Declaration of Helsinki.
Cell lines
Human myeloma cell lines RPMI 8226 (8226) and NCI-H929 (H929), as well as HS-5 bone marrow stromal cells, were newly obtained from the American Type Culture Collection (Manassas, VA). Additional cancer and normal cell lines assayed included the normal fibroblasts WI-38 (American Type Culture Collection) and Flow2000 (Flow Laboratories), peripheral blood mononuclear cells (PBMCs) from normal donors (Florida Blood Services), and HL-60 human acute myeloid leukemia (AML) cells (American Type Culture Collection).
SINE CRM1 antagonist molecules
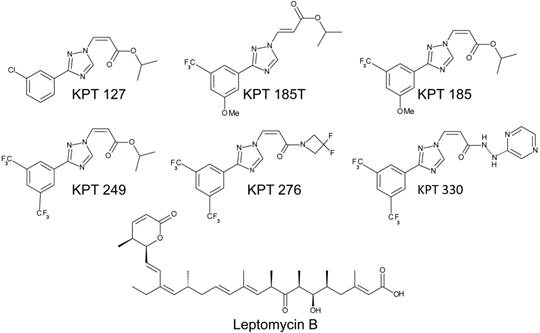
Studies were performed with specific SINE molecules developed by Karyopharm Therapeutics. These molecules included KPT127, KPT185, KPT249, KPT276, and KPT330 (Fig. 1). In addition, a trans-isomer of KPT185 (KPT185T) was used in all experiments as an inactive control molecule. KPT185T has shown ~100-fold less CRM1-inhibiting activity than KPT185 (the active, cis-isomer; unpublished results, Karyopharm Therapeutics). Leptomycin B (LMB) (ENZO Life Sciences), a classic CRM1 inhibitor [27], was used in all experiments as a positive control for CRM1-inhibiting activity. Both the SINE and LMB CRM1 inhibitors used in this study function by covalent modification of the active-site cysteine 528 [2, 27]. Stock solutions of SINE molecules (10 mM) were made in DMSO, and LMB (200 µM) was dissolved in absolute ethanol. Study drugs were stored in single-use aliquots at -80°C.
CRM1-targeted nuclear export inhibitors. Similar in mechanism to LMB, the SINE CRM1 inhibitors (KPT127, KPT185, KPT249, KPT 276, and KPT330) bind to the active site (CRM1 cysteine residue 528) and prevent transport receptor binding to the cargo protein. KPT185T is the inactive trans-isomer of KPT185.
Automated in vitro cell viability assay
The half-maximal inhibitory concentration (IC50; concentration of drug required for a 50% reduction in growth/viability) values and combination index (CI) values of the SINE molecules when used alone and in combination with other drugs were determined by a high-throughput CellTiter-Blue (Promega) cell viability assay as described previously [28]. Cell cultures grown at log-phase densities (2 x 105/mL) were used to determine IC50 values, and cells grown at plateau densities (3 x 106/mL) were used to determine synergistic activity. After additional reagents were added, including doxorubicin (Sigma), bortezomib (Fisher), carfilzomib (Selleck Chem), dexamethasone (Sigma), lenalidomide (Selleck Chem), and melphalan (Sigma), the plates were incubated for an additional 24 hours. All drugs were dissolved in DMSO except for melphalan, which was dissolved in an acid-alcohol solution (150 mM HCl (Sigma) in ethanol (Sigma)). Samples were assayed in replicates of four. Fluorescence data were used to calculate IC50 values using a sigmoidal equilibrium model regression with XLfit version 5.2 (ID Business Solutions Ltd.). For drug combination experiments, cell viability assays were performed as described above, and the results were analyzed for synergistic, additive, and antagonistic effects using the CI method developed by Chou and Talalay [29]. Cell lines assayed included human MM cell lines 8226 and H929, human AML cell line HL-60, and normal fibroblasts WI-38 and Flow2000.
Cell density and drug treatment
Our group has previously shown that MM cells grown at high density exhibit specific characteristics such as drug resistance and CRM1-dependent nuclear export of topo IIα [30-32]. Thus, human MM cells (H929) grown at 2 × 105 cells/mL were defined as low density (log phase), and cells grown at 3 × 106 cells/mL were defined as high density (plateau phase). Cell lines were placed at log- and plateau-density conditions and cultured for 20 hours with the SINE CRM1 inhibitors (300 nM) or with LMB (10 nM) with and without doxorubicin (1-2 µM), bortezomib (10 nM), or carfilzomib (20 nM). Optimal drug concentrations were determined by titration experiments for SINE compounds, doxorubicin, bortezomib, and carfilzomib. LMB concentrations were based on published IC50 data [14].
Immunofluorescent microscopy
Human H929 cells were treated with SINE KPT185T, KPT185, KPT249, and KPT330 at 100-300 nM or with LMB (10-100 nM) for 20 hours. Samples assayed for p53 or CRM1 were treated with 100 nM SINE or 10 nM LMB, and cells assayed for topo IIα were treated with 300 nM SINE or 100 nM LMB. Immunofluorescent microscopy was performed as previously described [3, 30]. Antibodies used were mouse monoclonal DO-1 (Santa Cruz) to detect p53, 454 polyclonal (produced in our lab) for topo IIα [33], and rabbit-polyclonal H-300 (Santa Cruz) for detection of CRM1. Antibodies were diluted 1:100 in a buffer containing 2% BSA-PBS, and slides were incubated for 1 hour at room temperature. After three washes with PBS, slides were incubated with secondary antibodies: anti-rabbit Alexa Fluor 488 (Invitrogen) or anti-mouse Alexa Fluor 594 (Invitrogen). Each secondary antibody was diluted 1:500 in 2% BSA-PBS and incubated for 40 minutes at room temperature. Slides were washed four times in PBS, air dried, and covered with cover glass and Vectashield mounting media containing anti-fade/4',6-diamidino-2-phenylindole (DAPI) (Vector Laboratories Inc). Immunofluorescence was observed with a Zeiss Automated Upright Fluorescent Microscope. Images were captured with a high-resolution CCD camera. To quantify topo IIα intracellular location, H929 cells treated with SINE were scored as nuclear or cytoplasmic when ≥90% of the fluorescence was in the respective cellular compartment; LMB was used as a positive control.
Western blot
Human H929 MM cells were incubated with SINE (100 nM) or LMB (10 nM) for 20 hours to determine their effect on CRM1 and topo IIα protein expression. Fifty micrograms of protein were loaded into each lane of an 8% SDS-PAGE gel (Bio-Rad) and transferred to PVDF membranes (Amersham) overnight (30 V at 4ºC) with the use of a Bio-Rad Mini-Transblot apparatus. Membranes were blocked for 1 hour at ambient temperature in a blocking buffer containing 0.1 M Tris-HCl (Sigma), 0.9% NaCl (Sigma), and 0.5% Tween 20 (Bio-Rad) (TBST) and 5% non-fat dry milk. CRM1 and topo IIα were identified by incubation with H-300 antibody or Ki-S1 antibody [34] (Millipore), respectively, at a 1:1000 dilution in blocking buffer for 1 hour at ambient temperature. Membranes were washed three times for 10 minutes in TBST and incubated for 1 hour with goat anti-rabbit polyclonal IgG antibody linked to a horseradish peroxidase antibody (Sigma) in blocking buffer at a 1:1000 dilution. Antibody binding was visualized by enhanced chemiluminescence (Amersham) on autoradiography film (Kodak). GAPDH was co-assayed with topo IIα and CRM1 as a protein loading control using the anti-GAPDH clone 6C5 (Millipore) at a 1:1000 dilution.
Cellular fractionation of p53 and topo IIα
Cellular fractionation experiments were performed to confirm whether SINE molecules prevented nuclear export of p53 and topo IIα. H929 cells were placed at plateau concentrations for assay of topo IIα and log-phase conditions for p53. SINE molecules were then added at 100 nM for 20 hours, with LMB at 10 nM used as control. Cells were collected, washed in PBS, and fractionated by a method adapted from Li et al [35]. To fractionate topo IIα, a high-salt lysis buffer containing 50 mM Tris, 0.1% Triton X-100 (Sigma), 400 mM NaCl, 10% glycerol (Sigma), and 5 mM EDTA (Sigma) was used. Separated fractions containing nuclei or precipitated cytosol were dissolved in 2% SDS buffer and assayed by SDS-PAGE Western blot analysis. Cytosolic fractions were probed with the anti-topo IIα Ki-S1 antibody. Nuclei were assayed for p53 protein (anti-p53 (DO-1)). In addition, blots were probed with anti-GAPDH clone 6C5 and anti-lamin (BD Biosciences) to verify cytosol and nuclear fraction separations, respectively.
Activated caspase 3 apoptosis assay and time course study
High-density (3 x 106) H929 MM cells were treated with 300 nM SINE and various concentrations of doxorubicin (0.1, 0.5, or 1 µM), bortezomib (2.5, 5, 10 nM), and carfilzomib (10, 20, 40 nM) for 20 hours and assayed for apoptosis (activated caspase 3). A time course experiment was performed in which H929 cells were co-incubated with 300 nM SINE and 1 µM doxorubicin for 4, 8, 16, 20, and 24 hours. In addition, H929 cells were incubated with 300 nM SINE and 10 nM bortezomib for 8, 16, 20, and 24 hours.
Cells were fixed and permeabilized, and apoptosis was measured by activated anti-caspase 3/Alexa Fluor 488 (Cell Signaling Technology) in accordance with the standard manufacturer's protocol. Percent apoptosis was analyzed by flow cytometry on a FACSCalibur bench-top analyzer with FlowJo analysis software (Becton-Dickinson).
Bone marrow aspirate processing
Bone marrow aspirates (20 mL) from patients were collected (sodium heparin tubes), and mononuclear cells were immediately isolated by centrifuging diluted marrow (1:1 with sterile PBS) over a Ficoll-Paque Plus (Amersham Biosciences) gradient at 400 x g for 35 minutes at ambient temperature. The interface was removed, cells were washed with cold PBS and counted, and 2 x 105 cells were plated on double cytoslides by centrifugation at 30 x g for 3 minutes. Slides were fixed with 4% paraformaldehyde for 10 minutes, washed in PBS, and stored at -80ºC. These slides were used to determine the percent plasma cell population. Plasma cells were identified by microscopic morphology of toluidine-stained cells. The isolated bone marrow mononuclear cells were plated at 4 x 106/mL in 200 µL RPMI (Fisher) containing 10% FBS in 96-well plates, treated with 300 nM SINE molecules with and without 2 µM doxorubicin, 10 nM bortezomib, or 20 nM carfilzomib, and incubated for 20 hours in a 5% CO2 humidified incubator. The following day, the cells were fixed and assayed for caspase activation and apoptosis.
Apoptosis assay of patient myeloma cells
After isolation and drug treatment (SINE molecules with and without doxorubicin, bortezomib, or carfilzomib) for 20 hours, bone marrow mononuclear cells were fluorescently labeled with antibodies against activated caspase 3 (ASP175-Alexa 488), CD138 (M115-Alexa 647, BD Bioscience), and light chain kappa (G20-193 V450, BD Bioscience) or lambda (JDC-12 V450, BD Bioscience). A BD Bioscience LSR II flow cytometer was used to gate MM cell populations, defined as cells that are both CD138 positive and light chain positive. Non-myeloma patient bone marrow cells were defined as cells that were both CD138 and light-chain double negative. The percentage of apoptotic cells from each sample was determined by activated caspase 3. Data analysis was performed using Flowjo version 9.4 software (Tree Star, Inc).
Co-culture experiments
Co-culture of HS-5 bone marrow stromal cells was used to simulate a drug-resistant bone marrow microenvironment, similar to that found in MM patients [36-38]. HS-5/GFP cells were plated (5 x 105 cells/mL) in 12-well plates for 24 hours before addition of H929 cells (5 x 105 cells/mL). In addition, a control group of H929 cells (5 x 105 cells/mL) grown in suspension without HS-5 cells were plated in 12-well plates. The cells were treated with 300 nM of the indicated CRM1 inhibitor and 2 µM doxorubicin or vehicle control (DMSO) for 20 hours. After drug treatment, cell death was assayed using FACS analysis to detect annexin V/APC (BD/Pharmingen)-positive cells in the GFP-negative quadrant. The experiment was repeated 3 times, with results shown as a representative figure.
Results
IC50 values for SINE molecules as single agents
Human log-phase myeloma and AML cell lines were incubated with SINE compounds for 48 hours (Table 1). IC50 data were produced by CT-Blue viability assays. As shown in Table 1, KPT127, KPT185, KPT249, KPT276, and KPT330 were found to be potent inhibitors of myeloma cells 8226 and H929 and AML cells HL-60, with IC50 values in the nanomolar to low micromolar range. Human H929 MM cells were up to 239-fold more sensitive to SINE than PBMCs and normal fibroblasts WI-38 and Flow2000, as shown by higher IC50 values in the micromolar range.
SINE IC50 values of normal, leukemic, and multiple myeloma log-phase cell lines with 48-hour drug exposure by CT-Blue assay.
| KPT127 (µM) | KPT185 (µM) | KPT249 (µM) | KPT276 (µM) | KPT330 (µM) | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Mean | SEM | n | Mean | SEM | n | Mean | SEM | n | Mean | SEM | n | Mean | SEM | n | |
| PBMCs | 13.6 | 0.99 | 5 | 13.1 | 1.0 | 6 | 18.3 | 1.7 | 6 | 74.6 | 15.6 | 4 | 16.9 | 1.76 | 4 |
| Flow20001 | 27.0 | 2.5 | 2 | 12.6 | 5.0 | 6 | 19.2 | 5.2 | 3 | 26.14 | 1 | 26.4 | 1 | ||
| Wi-381 | 16.8 | 4.3 | 2 | 11.1 | 4.0 | 5 | 13.4 | 5.1 | 3 | 3.52 | 0.55 | 3 | 10.5 | 1 | |
| HL-602 | 1.54 | 0.19 | 2 | 0.17 | 0.04 | 7 | 0.25 | 0.10 | 3 | 0.57 | 0.20 | 3 | 3.5 | 3.4 | 3 |
| H9293 | 0.86 | 0.05 | 2 | 0.12 | 0.02 | 9 | 0.17 | 0.03 | 5 | 0.32 | 0.04 | 5 | 0.115 | 0.005 | 2 |
| 82263 | 1.06 | 0.02 | 2 | 0.15 | 0.04 | 7 | 0.15 | 0.03 | 3 | 0.45 | 0.09 | 3 | 0.568 | 0.103 | 2 |
PBMCs, peripheral blood mononuclear cells. 1Normal fibroblasts. 2Leukemia cell line. 3Multiple myeloma cell line.
CRM1, p53, and topo IIα protein expression and intracellular localization
As shown by immunofluorescence microscopy, baseline CRM1 (green) nuclear localization and overall expression were high in untreated human MM cells and human MM cells treated with the inactive SINE trans-isomer KPT185T (Fig. 2A, first column). In addition, cells treated with the CRM1 inhibitor LMB had high levels of CRM1 protein. However, cells treated with the active SINE KPT185, KPT249, and KPT330 showed a decrease in CRM1 protein expression (Fig. 2A, first column). These data were confirmed by Western blot analysis (Fig. 2B). After 20 hours of treatment, protein levels of CRM1 were high in untreated controls and in KPT185T- and LMB-treated cells (Fig. 2B) but were significantly decreased in KPT185-, KPT249- and KPT330-treated cells (Fig. 2B). These data are in contrast to the effects seen on CRM1 mRNA, which is upregulated after treatment with all CRM1 inhibitors (Fig. 2B). CRM1 protein degradation was inhibited when cells were treated with the proteasome inhibitor bortezomib (Fig. 2B), indicating that SINE molecules may target CRM1 for proteosomal degradation.
CRM1, p53, and topo IIα localization in SINE-treated cells. A, Immunofluorescence microscopy. Human H929 MM cells were treated with SINE or leptomycin B for 20 hours. Samples assayed for p53 and CRM1 were treated with 100 nM SINE and 10 nM LMB. Samples assayed for topoisomerase (topo) IIα were treated with 300 nM SINE and 100 nM LMB. Intracellular localization and expression of p53, CRM1, and topo IIα were examined by immunofluorescence microscopy. Nuclei were counter-stained using DAPI (blue). Column 1: CRM1 (green) nuclear localization in low-density log-phase cells (2 x 105/mL) was increased in untreated controls, in KPT185T-treated cells, and in LMB-treated cells, but not in KPT185-, KPT249-, or KPT330-treated cells. Column 2: p53 (red) was exported from the nucleus in low-density log-phase (2 x 105/mL) untreated control, and KPT185T-treated cells; however, cells treated with KPT185, KPT249, KPT330, and LMB had increased nuclear accumulation of p53. Column 3: high-density (3 x 106/mL) untreated control cells exported topo IIα (green) to the cytoplasm (as did KPT185T treated cells) and had low levels of topo IIα in the nuclei (arrows) (blue/DAPI). KPT185, KPT249, KPT330, and LMB prevented nuclear export of topo IIα; therefore, the nuclei were green (topo IIα) (arrows). B, CRM1, p53, and topo IIα protein expression in SINE-treated cells. H929 cells were treated with 100 nM of each CRM1 inhibitor for 20 hours at high-density (3 x 106/mL) growth conditions. Whole cell lysates assayed for topo IIα protein demonstrated minimal change in total amount of topo IIα; however, cytoplasmic fractions showed that the nuclear export of topo IIα was inhibited by KPT185, KPT249, and LMB. GAPDH protein (loading control) showed that equal amounts of protein were loaded for topo IIα. H929 cells were also treated with 100 nM of each CRM1 inhibitor for 20 hours at log-phase growth conditions. Whole cell lysates assayed for p53 showed that total cellular p53 increased in cells treated with CRM1 inhibitors when compared to untreated and KPT185T-treated control samples. Nuclear fractions isolated from treated cells and assayed by Western blot demonstrated that nuclear p53 increased when cells were exposed to the active CRM1 inhibitors KPT185, KPT249, and LMB. KPT185 and KPT249, but not LMB or KPT185T, decreased CRM1 protein expression in whole cell lysates. However, when cells were treated with the proteasome inhibitor bortezomib, CRM1 protein levels did not decrease. Treated cells were also assayed for CRM1 mRNA levels. We found that all CRM1 inhibitors had significantly (P < 0.05) increased mRNA levels as compared to untreated and KPT185T-treated controls.
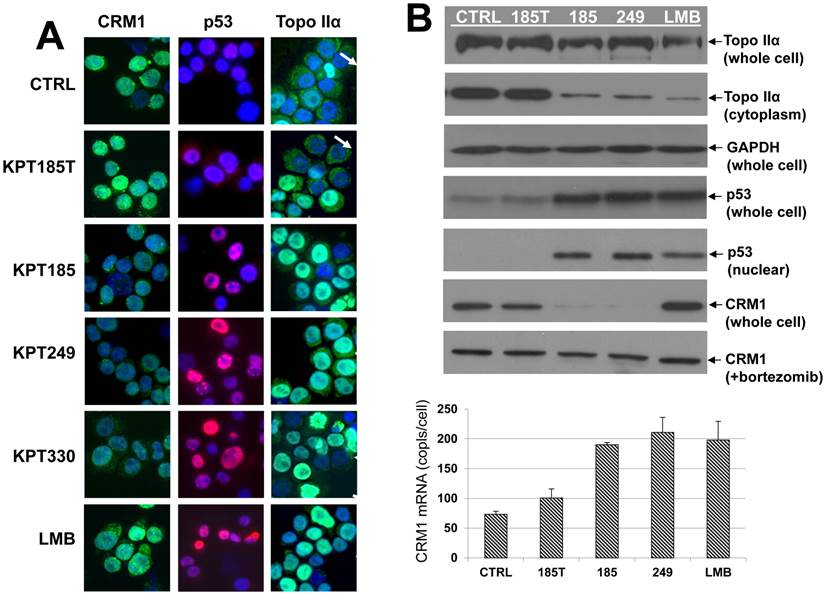
Based on relative fluorescence, overall p53 (red) protein levels were low in low-density control and KPT185T-treated cells (Fig. 2A, second column). However, cells treated with the CRM1 inhibitors KPT185, KPT249, KPT330, and LMB showed nuclear accumulation of high levels of p53 (Fig. 2A, second column). Nuclear fractions isolated from treated cells and analyzed by Western blot showed that CRM1 inhibition prevented nuclear export of p53 (Fig. 2B).
Topo IIα (green) was assayed by immunofluorescence microscopy in high-density cells (3 x 106/mL) after treatment with CRM1 inhibitors (Fig. 2A, third column). Untreated high-density cells expressed topo IIα (green) in both the nucleus and cytoplasm; however, KPT185, KPT249, and KPT330, as well as LMB, prevented nuclear export of topo IIα (Fig. 2A, third column). Whole cell lysates from SINE-treated MM cells were assayed for topo IIα protein (Fig. 2B) and demonstrated minimal change in the total amount of topo IIα. However, nuclear export of topo IIα was inhibited by KPT185, KPT249, and LMB and the corresponding cytoplasmic fraction was reduced (Fig. 2B). To quantify topo IIα intracellular localization, H929 cells treated with SINE were scored as nuclear or cytoplasmic when ≥90% of the fluorescence was in the respective cellular compartment. In untreated high-density controls, 52.5±4.9% of the cells had cytosolic topo IIα. KPT185- and KPT249-treated cells had 14.0±7.1% and 23.0±16.9%, respectively, cytosolic topo IIα. In LMB-treated controls, 9.5±10.6% of the cells had topo IIα in the cytoplasm. Therefore, CRM1 inhibitors prevented export of topo IIα from the nucleus to the cytoplasm.
SINE molecules synergize with doxorubicin, bortezomib, and carfilzomib
Human H929 MM cells were incubated at high-density conditions (3.0 x 106/mL) with SINE molecules, +/-doxorubicin, bortezomib, carfilzomib, lenalidomide, dexamethasone, and melphalan. When the percent viability versus drug concentration was plotted for each SINE molecule (Fig. 3), we found that the SINE molecules tested were strongly synergistic with doxorubicin, bortezomib, and carfilzomib in human H929 MM cells.
SINE molecules work synergistically with doxorubicin, bortezomib, and carfilzomib in H929 cells. SINE compound KPT185 was serially diluted, added to the wells containing H929 MM cells grown at plateau densities (2 x 106/mL), and incubated overnight at 37°C and 5% CO2. Doxorubicin (A), bortezomib (B), carfilzomib (C), lenalidomide (D), dexamethasone (E), and melphalan (F) were serially diluted and added the following day, and the plates were incubated an additional 24 hours. Samples were assayed for cell viability (CellTiter-Blue reagent) in replicates of 2-4 (see Table 2). Percent viability versus drug concentration was plotted. SINE compounds were found to be synergistic with doxorubicin, bortezomib, and carfilzomib, additive with melphalan, and non-synergistic/antagonistic for lenalidomide and dexamethasone in H929 cells.
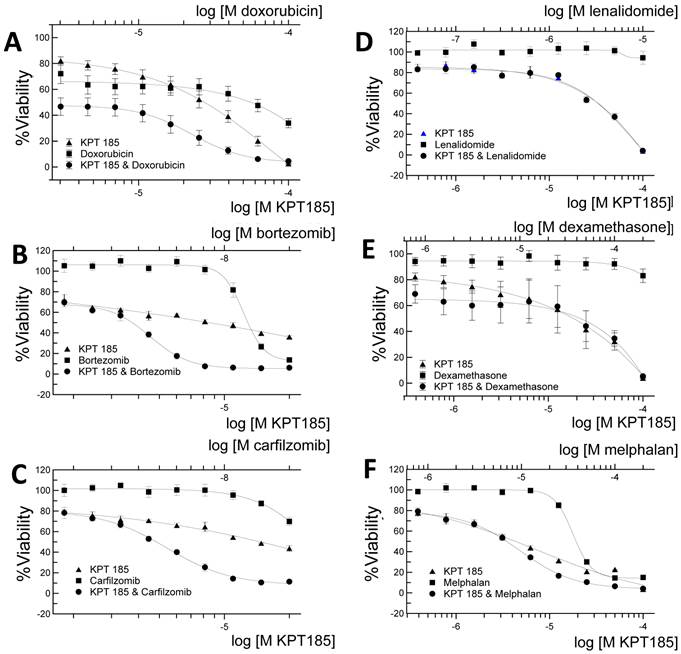
As shown in Table 2, SINE molecules KPT185, KPT249, and KPT330 worked synergistically with doxorubicin with combination indices of 0.377, 0.505, and 0.092, respectively. KPT185, KPT249, and KPT330 were synergistic with bortezomib (CI = 0.410, 0.888, and 0.502) and with carfilzomib (CI=0.322, 0.310, and 0.482). When co-incubated with melphalan, SINE produced a mild synergistic effect with KPT330 (CI=0.687) and KPT185 (CI=0.849) and an additive effect with KPT249 (CI=0.979). KPT185, KPT249, and KPT330 were antagonistic (CI >2.0, 1.263 and >2.0) when used in combination with dexamethasone. Lenalidomide was additive (CI=1.098) when used with KPT249 and antagonistic (CI>2.0) when used with KPT185 and KPT330.
Combination index values of high-density H929 cells pre-treated with SINE molecules for 24 hours followed by 24-hour incubation with additional myeloma chemotherapeutic drugs.
| SINE | Chemo drug | CI (mean) | CI (SEM) | n |
|---|---|---|---|---|
| KPT185 | Bortezomib | 0.410 | 0.019 | 3 |
| Carfilzomib | 0.322 | 0.136 | 3 | |
| Dexamethasone | >2.0 | * | 5 | |
| Doxorubicin | 0.377 | 0.060 | 4 | |
| Lenalidomide | >2.0 | * | 4 | |
| Melphalan | 0.849 | 0.104 | 4 | |
| KPT249 | Bortezomib | 0.888 | 0.166 | 3 |
| Carfilzomib | 0.310 | 0.013 | 2 | |
| Dexamethasone | 1.263 | 0.193 | 4 | |
| Doxorubicin | 0.505 | 0.055 | 9 | |
| Lenalidomide | 1.098 | 0.186 | 3 | |
| Melphalan | 0.979 | 0.164 | 3 | |
| KPT330 | Bortezomib | 0.502 | 0.040 | 2 |
| Carfilzomib | 0.482 | 0.043 | 2 | |
| Dexamethasone | >2.0 | * | 2 | |
| Doxorubicin | 0.092 | 0.025 | 3 | |
| Lenalidomide | >2.0 | * | 1 | |
| Melphalan | 0.687 | 0.254 | 3 |
Drug concentration ranges were as follows: SINE compounds KPT185, 249, and 330 were used at 4 nM to100 µM, doxorubicin at 4 nM to 100 µM, bortezomib at 40 pM to 10 µM, carfilzomib at 2 to 200 nM, and dexamethasone, melphalan and lenalidomide at 40 nM to 200 µM. *CI values and SEMs could not be calculated for several experiments due to the insufficient effects of single-agent lenalidomide or dexamethasone at the concentrations used. CI, combination index. The combination index method is based on that described by Chou and Talalay [29]. CI < 1, = 1, and >1 indicate synergism, additive effect, and antagonism, respectively.
Human myeloma cells are sensitized to the topo II inhibitor doxorubicin and proteasome inhibitors bortezomib and carfilzomib
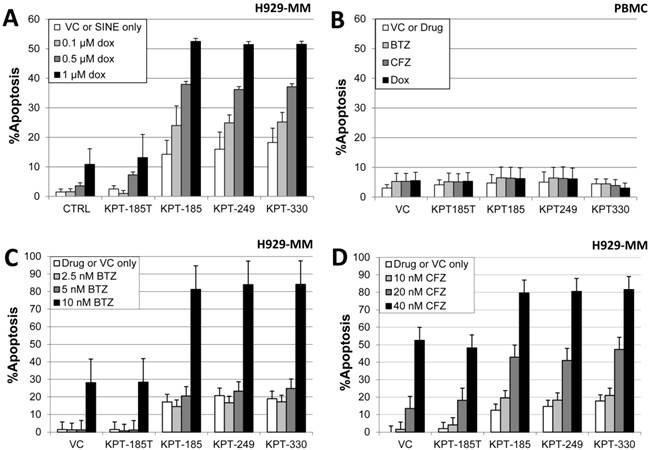
Since the activity of topo II inhibitors such as doxorubicin requires nuclear localization of topo IIα for their effect, forced nuclear retention of the target may augment or restore sensitivity to topo II inhibitors [3]. Therefore, high-density human MM H929 cells were co-treated with 300 nM SINE and doxorubicin (0.1, 0.5, and 1 µM) for 20 hours and assayed for apoptosis by activated caspase 3 (n=3). Fig. 4A demonstrates that high-density H929 cells were rendered more sensitive to the topo II inhibitor doxorubicin in a dose-dependent manner when co-incubated with KPT185, KPT249, and KPT330. The untreated and the inactive control KPT185T did not sensitize myeloma cells to doxorubicin. In a separate experiment, we found that human PBMCs from normal donors (n=3) were unaffected by treatment with SINE (300 nM) and were not sensitized to doxorubicin, bortezomib, or carfilzomib (Fig. 4B). In addition, high-density H929 myeloma cells co-incubated with SINE and 10 nM bortezomib were sensitized by KPT185, KPT249, and KPT330 (Fig. 4C). Co-incubation of SINE with the proteasome inhibitor carfilzomib also induced higher levels of apoptosis in H929 human myeloma cells (Fig. 4D). When compared to the inactive isomer KPT185T, cells treated with SINE and carfilzomib (20 and 40 nM) had significantly (P<0.05) higher levels of apoptosis after 20 hours of incubation (Fig. 4D).
Human myeloma cells are sensitized to doxorubicin, bortezomib, and carfilzomib. A, High-density (3.0 x 106 cells/mL) H929 cells were treated concurrently with 300 nM SINE and doxorubicin (dox; 0.1, 0.5, or 1 µM) for 20 hours and assayed for apoptosis by activated caspase 3. Apoptosis was induced in SINE/doxorubicin-treated cells in a dose-dependent manner (P < 0.021). B, Human peripheral blood mononuclear cells (PBMC) from normal donors (n=3) were unaffected by treatment with SINE (300 nM) and were not sensitized to doxorubicin, bortezomib (BTZ), or carfilzomib (CFZ). C, H929 MM cells co-incubated with SINE (100 nM) and bortezomib (2.5, 5, or 10 nM) for 20 hours were sensitized to 10 nM bortezomib (P < 0.0036). D, H929 cells co-incubated with SINE drugs +/- carfilzomib (10, 20, or 40 nM) demonstrated apoptosis (P < 0.05) in a dose-dependent manner.
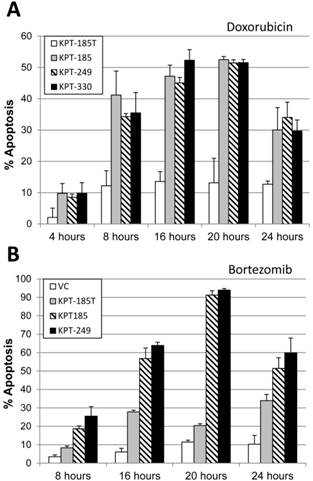
In a time course study, high-density H929 myeloma cells co-incubated with SINE and doxorubicin were sensitized by KPT185, KPT249, and KPT330 beginning at 8 hours after co-incubation with doxorubicin (Fig. 5A).This effect was found to plateau at 16-20 hours and decreased at 24 hours due to an increase in necrotic cell death as indicated by flow cytometry. Co-incubation of SINE with the proteasome inhibitor bortezomib also induced higher levels of apoptosis in H929 cells. When compared to the inactive isomer (KPT185T) or vehicle control-treated cells, co-incubation of bortezomib with KPT185 and KPT249 resulted in significantly (P<0.05) higher levels of apoptosis at 8, 16, 20, and 24 hours of incubation (Fig. 5B).
Human myeloma cells are sensitized to the topo IIα inhibitor doxorubicin and the proteasome inhibitor bortezomib. A, High-density (3.0 x 106 cells/mL) H929 myeloma cells were incubated concurrently with SINE (100 nM) and 1 µM doxorubicin over a time course of 4, 8, 16, 20, and 24 hours. MM cells demonstrated significant (P < 0.023) apoptosis beginning at 8 hours with co-treatment of SINE and doxorubicin. B, Human H929 MM cells co-incubated with SINE drugs ± bortezomib (10 nM) induced significantly (P < 0.05) higher levels of apoptosis than vehicle control and KPT185T at 16, 20, and 24 hours of incubation.
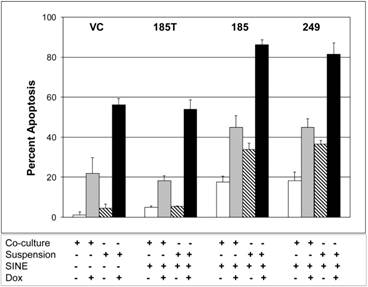
Stromal cell co-culture induces drug resistance
HS-5 stromal cells have been shown to support the proliferation of hematopoietic progenitor cells and promote drug resistance [36-38]. We found that MM cells co-cultured with HS-5 cells are relatively drug resistant to CRM1 inhibitors (P=0.00014) and doxorubicin (P=0.0002) when used as a single agents as compared to H929 cells grown in suspension (Fig. 6). Using flow cytometry analyses, we found that both KPT185 and KPT249 induced apoptosis when used as single agents and when used in combination with doxorubicin versus that shown in cells treated with the inactive trans-isomer (P<0.02) or DMSO vehicle control (P<0.0001) (Fig. 6). SINE compounds sensitized both suspension cells and co-cultured myeloma cells to doxorubicin.
Stromal cell co-culture induced drug resistance. H929 myeloma cells (5 x 105) co-cultured with HS-5 cells (5 x 105) are drug-resistant to CRM1 inhibitors (P=0.0001) and doxorubicin (P=0.0002) when used as a single agent compared with H929 cells grown in suspension. When compared to doxorubicin alone (columns 1 and 3 of the VC group (vehicle control)) (P<0.0001) or the inactive trans-isomer KPT-185T (P<0.02), both KPT185 and KPT249 sensitized H929 cells when used in combination with doxorubicin. SINE compounds sensitized both suspension cells and co-cultured myeloma cells to doxorubicin.
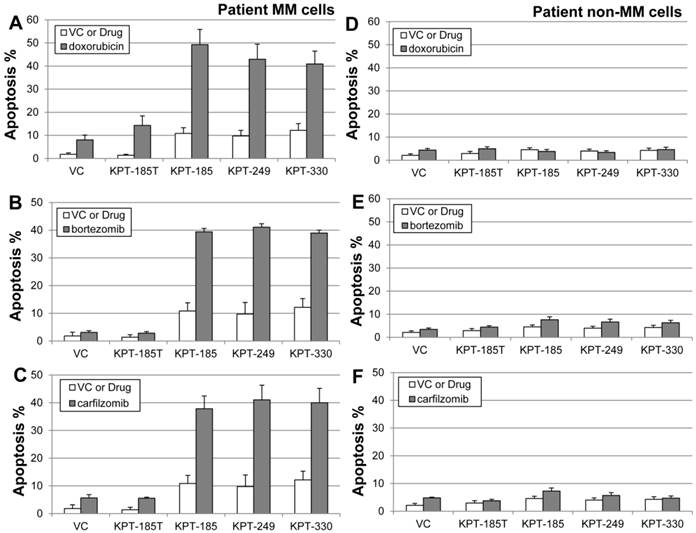
SINE KPT185, KPT249, and KPT330 sensitize patient myeloma cells to doxorubicin, bortezomib, and carfilzomib
To determine whether SINE would sensitize patient myeloma cells ex vivo, bone marrow mononuclear cells were isolated from both newly diagnosed and refractory/relapsed myeloma patients. Our results showed that SINE molecules highly sensitized patient MM cells (n=12) to doxorubicin (Fig. 7A), bortezomib (Fig. 7B), and carfilzomib (Fig. 7C). Both the drug-free controls and KPT185T did not sensitize cells. SINE-drug (doxorubicin, bortezomib, or carfilzomib) co-incubation synergistically induced apoptosis (activated caspase 3) in CD138/light-chain double-positive myeloma cell populations (Fig. 7A, 7B, and 7C) but not in non-myeloma CD138/light-chain double-negative cells (Fig. 7D, 7E, and 7F), indicating that SINE may specifically inhibit neoplastic cells in MM patients.
KPT185, KPT249, and KPT330 sensitize patient myeloma cells to doxorubicin, bortezomib, and carfilzomib. Bone marrow mononuclear cells were isolated and treated with SINE (300 nM) ± doxorubicin (2 µM), bortezomib (10 nM), carfilzomib (20 nM), or DMSO vehicle control (VC) for 20 hours. Treated cells were fluorescently labeled with antibodies against activated caspase 3, CD138, and light chain (kappa or lambda). Results for patient multiple myeloma cells (n=12 patient samples) (A-C) and patient non-myeloma cells (n=12) (D-F) are shown. Apoptosis was induced when SINE molecules were co-incubated with doxorubicin (P = 0.0004), bortezomib (P = 0.003), or carfilzomib (P = 0.002) in CD138/light-chain double-positive myeloma cell populations (A-C) but not in non-myeloma CD138/light-chain double-negative cells (P > 0.25) (D-F). This indicates that SINE may specifically inhibit neoplastic cells in MM patients.
Discussion
In this study, we examined the cytotoxic effects of SINE in MM cells. We showed by immunofluorescence microscopy and nuclear-cytoplasmic fractionation that these molecules were able to prevent nuclear export of the tumor suppressor protein p53 and the nuclear drug target topo IIα. Cell viability data demonstrated that SINE molecules were effective as single agents against human myeloma H929 and 8226 and HL-60 AML cells at nanomolar concentrations but were up to 239-fold less toxic to normal cells. SINE molecules, when used in combination with anti-MM agents (doxorubicin, bortezomib, and carfilzomib), were found to sensitize drug-resistant MM cells in both the high-density cell culture model and in co-culture with HS-5/GFP bone marrow stromal cells. Further studies are needed to determine the mechanism of resistance to CRM1 inhibitors in the bone marrow co-culture model.
Combining SINE molecules with doxorubicin, bortezomib, or carfilzomib induced apoptosis and decreased cell viability of myeloma cells synergistically. The synergistic effect of KPT185, KPT249, and KPT330 with myeloma drugs (doxorubicin, bortezomib, and carfilzomib) was dose dependent, and time-course studies performed in high-density drug-resistant MM cultures showed that apoptosis was induced after an 8-hour co-treatment with doxorubicin-SINE or a 16-hour co-treatment with bortezomib-SINE. Normal human PBMCs were unaffected by SINE treatment and were not sensitized to doxorubicin, bortezomib, or carfilzomib. Co-incubation of SINE with the proteasome inhibitor bortezomib was found to strongly induce apoptosis in H929 human myeloma cells. KPT185 and KPT249 used in combination with bortezomib had significantly (P<0.05) higher levels of apoptosis at 8, 16, 20 and 24 hours of incubation. In studies done on MM patient bone marrow mononuclear cells, SINE-doxorubicin co-incubation synergistically induced activated caspase 3 in CD138/light-chain double-positive myeloma cell populations but not in CD138/light-chain double-negative non-myeloma cells, indicating that SINE may specifically inhibit neoplastic cells in MM patients. We found that CRM1 protein levels are decreased by SINE but not by LMB. This is an unusual finding because both SINE and LMB inhibit CRM1 activity by covalent modification of the active site cysteine 528. It is possible that SINE molecules may signal proteosomal degradation of CRM1 but LMB does not. When SINE-treated cells were co-incubated with the proteasome inhibitor bortezomib, CRM1 protein levels were increased to the levels of untreated controls and LMB. Interestingly, CRM1 mRNA levels were increased by both SINE and LMB even with the concurrent decrease of CRM1 protein found in SINE-treated cells.
Recent publications on the effects of SINE compounds on AML have shown that SINE compounds have anti-proliferative effects on AML cell lines and primary AML samples [19, 20]. In addition, when used as single agents, SINE induced apoptosis and accumulation of p53 in the nucleus of AML cells [19]. We found parallel results in human MM cell lines and in patient MM bone marrow mononuclear cells with SINE as single agents; in addition, SINE compounds sensitized MM cells both in vitro and ex vivo to doxorubicin, bortezomib, and carfilzomib.
NF-κB is constitutively active in multiple myeloma, resulting in increased cell survival and proliferation. In a recent publication [23], CRM1 inhibitors have been shown to increase IκB (inhibitor of NF-κB) levels in CLL cells. This increase in IκB may result in inhibition of NF-κB and result in decreased viability of cancer cells. The addition of the proteasome inhibitor bortezomib will further inhibit IkB degradation, synergize with CRM1, and possibly result in increased cell death. Furthermore, increased nuclear p53 from the CRM1-bortezomib combination observed in this paper would also induce apoptosis of myeloma cells. Therefore, inhibiting the anti-apoptotic effect of NF-κB and addition of the pro-apoptotic effect of p53 may synergistically induce cell death in myeloma.
High-density drug-resistant MM cell lines and cells made resistant by stromal cell co-culture were sensitive to SINE treatment. KPT185, KPT249, and KPT330 induced apoptosis both as single agents and in combination with doxorubicin, bortezomib, or carfilzomib as compared to the inactive trans-isomer (P<0.02) or DMSO vehicle control (P<0.0001). SINE compounds were found to sensitize both suspension cells and co-cultured myeloma cells to doxorubicin.
Drug-resistant myeloma cells export topo IIα into the cytoplasm of the cell. Once topo IIα is trafficked from the nucleus into the cytoplasm, topo II inhibitors such as doxorubicin can no longer induce topo II/DNA cleavable complexes and cell death. In a previous study with the CRM1-specific inhibitor ratjadone C, our group found that CRM1 inhibition by specific chemical inhibitors (ratjadone/LMB) or by siRNA knockdown sensitized drug-resistant myeloma cells to doxorubicin and etoposide [3]. In the current study, we found that SINE molecules exhibit an anti-myeloma effect similar to ratjadone or siRNA knockdown of CRM1. SINE molecules have been shown to effectively inhibit CRM1-mediated nuclear export without the off-target cytotoxic effects found in LMB-based molecules [19]. KPT330, an orally bioavailable SINE molecule with potency similar to KPT185 and KPT249, is currently undergoing phase I studies in both solid and hematologic malignancies, including patients with MM. These data support such clinical studies, as well as studies investigating combinations of SINE with doxorubicin, liposomal doxorubicin, or bortezomib in MM.
Acknowledgements
We thank Rasa Hamilton (Moffitt Cancer Center) for editorial assistance. This work was supported in part by the Flow Cytometry Core Facility at the H. Lee Moffitt Cancer Center & Research Institute. We especially thank Jodi Kroeger for her expert assistance with flow cytometry. This work was also supported by the Bankhead-Coley Team Science Project Grant 2BT03-43424 and in part by Karyopharm Therapeutics (Boston, MA 01760). The work was also supported in part by the National Cancer Institute, part of the National Institutes of Health, through grant number 2 P30-CA76292-14.
Competing Interests
Authors and co-authors from the Moffitt Cancer Center have no conflict of interest to disclose. Co-authors from Karyopharm Therapeutics (Michael Kauffman and Sharon Shacham) provided the CRM1 inhibitors studied in this paper.
References
1. Turner JG, Sullivan DM. CRM1-mediated nuclear export of proteins and drug resistance in cancer. Curr Med Chem. 2008;15:2648-55
2. Turner JG, Dawson J, Sullivan DM. Nuclear export of proteins and drug resistance in cancer. Biochem Pharmacol. 2012;83:1021-32 doi:10.1016/j.bcp.2011.12.016
3. Turner JG, Marchion DC, Dawson JL, Emmons MF, Hazlehurst LA, Washausen P. et al. Human multiple myeloma cells are sensitized to topoisomerase II inhibitors by CRM1 inhibition. Cancer Res. 2009;69:6899-905 doi:10.1158/0008-5472.CAN-09-0484
4. Ren R. Mechanisms of BCR-ABL in the pathogenesis of chronic myelogenous leukaemia. Nature reviews Cancer. 2005;5:172-83 doi:10.1038/nrc1567
5. Jiao W, Datta J, Lin HM, Dundr M, Rane SG. Nucleocytoplasmic shuttling of the retinoblastoma tumor suppressor protein via Cdk phosphorylation-dependent nuclear export. J Biol Chem. 2006;281:38098-108
6. Rosin-Arbesfeld R, Cliffe A, Brabletz T, Bienz M. Nuclear export of the APC tumour suppressor controls beta-catenin function in transcription. Embo J. 2003;22:1101-13
7. O'Brate A, Giannakakou P. The importance of p53 location: nuclear or cytoplasmic zip code? Drug Resist Updat. 2003;6:313-22
8. Keeshan K, Cotter TG, McKenna SL. Bcr-Abl upregulates cytosolic p21WAF-1/CIP-1 by a phosphoinositide-3-kinase (PI3K)-independent pathway. Br J Haematol. 2003;123:34-44
9. Min YH, Cheong JW, Kim JY, Eom JI, Lee ST, Hahn JS. et al. Cytoplasmic mislocalization of p27Kip1 protein is associated with constitutive phosphorylation of Akt or protein kinase B and poor prognosis in acute myelogenous leukemia. Cancer Res. 2004;64:5225-31
10. Burzlaff A, Kalesse M, Kasper C, Scheper T. Multi parameter in vitro testing of ratjadone using flow cytometry. Appl Microbiol Biotechnol. 2003;62:174-9
11. Kalesse M, Christmann M, Bhatt U, Quitschalle M, Claus E, Saeed A. et al. The chemistry and biology of ratjadone. Chembiochem. 2001;2:709-14
12. Koster M, Lykke-Andersen S, Elnakady YA, Gerth K, Washausen P, Hofle G. et al. Ratjadones inhibit nuclear export by blocking CRM1/exportin 1. Exp Cell Res. 2003;286:321-31
13. Meissner T, Krause E, Vinkemeier U. Ratjadone and leptomycin B block CRM1-dependent nuclear export by identical mechanisms. FEBS Lett. 2004;576:27-30
14. Mutka SC, Yang WQ, Dong SD, Ward SL, Craig DA, Timmermans PB. et al. Identification of nuclear export inhibitors with potent anticancer activity in vivo. Cancer Res. 2009;69:510-7
15. Kau TR, Schroeder F, Ramaswamy S, Wojciechowski CL, Zhao JJ, Roberts TM. et al. A chemical genetic screen identifies inhibitors of regulated nuclear export of a Forkhead transcription factor in PTEN-deficient tumor cells. Cancer Cell. 2003;4:463-76
16. Murakami N, Ye Y, Kawanishi M, Aoki S, Kudo N, Yoshida M. et al. New Rev-transport inhibitor with anti-HIV activity from Valerianae Radix. Bioorganic & medicinal chemistry letters. 2002;12:2807-10
17. Tamura S, Shiomi A, Kaneko M, Ye Y, Yoshida M, Yoshikawa M. et al. New Rev-export inhibitor from Alpinia galanga and structure-activity relationship. Bioorganic & medicinal chemistry letters. 2009;19:2555-7 doi:10.1016/j.bmcl.2009.03.047
18. Sakakibara K, Saito N, Sato T, Suzuki A, Hasegawa Y, Friedman JM. et al. CBS9106 is a novel reversible oral CRM1 inhibitor with CRM1 degrading activity. Blood. 2011;118:3922-31 doi:10.1182/blood-2011-01-333138
19. Ranganathan P, Yu X, Na C, Santhanam R, Shacham S, Kauffman M. et al. Pre-clinical activity of a novel CRM1 inhibitor in acute myeloid leukemia. Blood. 2012 doi:10.1182/blood-2012-04-423160
20. Etchin J, Sun Q, Kentsis A, Farmer A, Zhang ZC, Sanda T. et al. Anti-leukemic activity of nuclear export inhibitors that spare normal hematopoietic cells. Leukemia. 2012 doi:10.1038/leu.2012.219
21. Etchin J, Sanda T, Mansour MR, Kentsis A, Montero J, Le BT. et al. KPT-330 inhibitor of CRM1 (XPO1)-mediated nuclear export has selective anti-leukaemic activity in preclinical models of T-cell acute lymphoblastic leukaemia and acute myeloid leukaemia. Br J Haematol. 2013;161:117-27 doi:10.1111/bjh.12231
22. Tai YT, Landesman Y, Acharya C, Calle Y, Zhong MY, Cea M. et al. CRM1 inhibition induces tumor cell cytotoxicity and impairs osteoclastogenesis in multiple myeloma: molecular mechanisms and therapeutic implications. Leukemia. 2013 doi:10.1038/leu.2013.115
23. Lapalombella R, Sun Q, Williams K, Tangeman L, Jha S, Zhong Y. et al. Selective inhibitors of nuclear export show that CRM1/XPO1 is a target in chronic lymphocytic leukemia. Blood. 2012;120:4621-34 doi:10.1182/blood-2012-05-429506
24. Inoue H, Kauffman M, Shacham S, Landesman Y, Yang J, Evans CP. et al. CRM1 Blockade by Selective Inhibitors of Nuclear Export Attenuates Kidney Cancer Growth. J Urol. 2012 doi:10.1016/j.juro.2012.10.018
25. Zhang K, Wang M, Tamayo AT, Shacham S, Kauffman M, Lee J. et al. Novel selective inhibitors of nuclear export CRM1 antagonists for therapy in mantle cell lymphoma. Exp Hematol. 2013;41:67-78 doi:10.1016/j.exphem.2012.09.002
26. Salas FRA, Chung HW, Landesman Y, Senapedis W. et al. CRM1 and BRAF inhibition synergize and induce tumor regression in BRAF mutant melanoma. Mol Cancer Ther. 2013 doi:10.1158/1535-7163MCT-12-1171
27. Kudo N, Matsumori N, Taoka H, Fujiwara D, Schreiner EP, Wolff B. et al. Leptomycin B inactivates CRM1/exportin 1 by covalent modification at a cysteine residue in the central conserved region. Proc Natl Acad Sci U S A. 1999;96:9112-7
28. Gray J, Cubitt CL, Zhang S, Chiappori A. Combination of HDAC and topoisomerase inhibitors in small cell lung cancer. Cancer Biol Ther. 2012;13:614-22 doi:10.4161/cbt.19848
29. Chou TC, Talalay P. Quantitative analysis of dose-effect relationships: the combined effects of multiple drugs or enzyme inhibitors. Adv Enzyme Regul. 1984;22:27-55
30. Valkov NI, Gump JL, Engel R, Sullivan DM. Cell density-dependent VP-16 sensitivity of leukaemic cells is accompanied by the translocation of topoisomerase IIalpha from the nucleus to the cytoplasm. Br J Haematol. 2000;108:331-45
31. Engel R, Valkov NI, Gump JL, Hazlehurst L, Dalton WS, Sullivan DM. The cytoplasmic trafficking of DNA topoisomerase IIalpha correlates with etoposide resistance in human myeloma cells. Exp Cell Res. 2004;295:421-31
32. Turner JG, Engel R, Derderian JA, Jove R, Sullivan DM. Human topoisomerase IIalpha nuclear export is mediated by two CRM-1-dependent nuclear export signals. J Cell Sci. 2004;117:3061-71
33. Sullivan DM, Latham MD, Rowe TC, Ross WE. Purification and characterization of an altered topoisomerase II from a drug-resistant Chinese hamster ovary cell line. Biochemistry. 1989;28:5680-7
34. Boege F, Andersen A, Jensen S, Zeidler R, Kreipe H. Proliferation-associated nuclear antigen Ki-S1 is identical with topoisomerase II alpha. Delineation of a carboxy-terminal epitope with peptide antibodies. Am J Pathol. 1995;146:1302-8
35. Li Y, Bor YC, Misawa Y, Xue Y, Rekosh D, Hammarskjold ML. An intron with a constitutive transport element is retained in a Tap messenger RNA. Nature. 2006;443:234-7 doi:10.1038/nature05107
36. Roecklein BA, Torok-Storb B. Functionally distinct human marrow stromal cell lines immortalized by transduction with the human papilloma virus E6/E7 genes. Blood. 1995;85:997-1005
37. Emmons MF, Gebhard AW, Nair RR, Baz R, McLaughlin ML, Cress AE. et al. Acquisition of resistance toward HYD1 correlates with a reduction in cleaved alpha4 integrin expression and a compromised CAM-DR phenotype. Mol Cancer Ther. 2011;10:2257-66 doi:10.1158/1535-7163.MCT-11-0149
38. Nair RR, Emmons MF, Cress AE, Argilagos RF, Lam K, Kerr WT. et al. HYD1-induced increase in reactive oxygen species leads to autophagy and necrotic cell death in multiple myeloma cells. Mol Cancer Ther. 2009;8:2441-51 doi:10.1158/1535-7163.MCT-09-0113
Author contact
![]() Corresponding author: Daniel M. Sullivan, H. Lee Moffitt Cancer Center and Research Institute, 12902 Magnolia Drive, Tampa, FL 33612, USA; Tel: 813-745-3878; Fax: 813-745-1436; E-mail: dan.sullivanorg.
Corresponding author: Daniel M. Sullivan, H. Lee Moffitt Cancer Center and Research Institute, 12902 Magnolia Drive, Tampa, FL 33612, USA; Tel: 813-745-3878; Fax: 813-745-1436; E-mail: dan.sullivanorg.