Impact Factor ISSN: 1837-9664

 Global reach, higher impact
Global reach, higher impactJ Cancer 2014; 5(6):457-464. doi:10.7150/jca.9145 This issue Cite
Research Paper
Regulation of Desmocollin3 Expression by Promoter Hypermethylation is Associated with Advanced Esophageal Adenocarcinomas
Qinggang Wang1,2, DunFa Peng2, Shoumin Zhu2, Zheng Chen2,3, TianLing Hu2, Mohammed Soutto2, Rama Saad2,4, Shutian Zhang1 ![]() , Wael EI-Rifai2,5
, Wael EI-Rifai2,5 ![]()
1. Department of Gastroenterology, Beijing Friendship Hospital, Capital Medical University, Beijing China;
2. Department of Surgery, Vanderbilt-Ingram Cancer Center, Vanderbilt University Medical Center, Nashville, Tennessee, USA;
3. Department of General Surgery; The First Affiliated Hospital of Nanjing Medical University, Nanjing, China;
4. Department of Biology, The American University in Cairo, Cairo, Egypt;
5. Department of Cancer Biology, Vanderbilt University Medical Center, Nashville, Tennessee, USA.
Received 2014-3-17; Accepted 2014-4-14; Published 2014-5-14
Abstract
BACKGROUND: Desmocollin3 (DSC3) is a member of the cadherin superfamily of calcium-dependent cell adhesion molecules and plays an important role in tumor invasion and metastasis. In this study, we investigated the epigenetic mechanism that regulates DSC3 expression in esophageal adenocarcinomas (EACs). METHODS: Expression of DSC3 was analyzed by quantitative real-time polymerase chain reaction (qRT-PCR). The promoter DNA methylation level of DSC3 was examined using quantitative bisulfite pyrosequencing. RESULTS: The qRT-PCR analysis demonstrated significant down-regulation of the DSC3 mRNA levels in human EAC cell lines and tissue samples (P<.001). In addition, the EAC cell lines and tumor samples have aberrant promoter hypermethylation as compared to normal esophageal samples (P<.001). DSC3 promoter hypermethylation (>10% methylation level) was detected in 97.5% (39/40) of EAC samples whereas none of the normal tissue samples showed hypermethylation (P<.0001). There was a significant inverse correlation between promoter DNA methylation levels and mRNA expression folds for DSC3 (coefficient r=-0.685, P<.0001). Treatment of FLO-1 and SKGT4 EAC cells with 5-Aza-deoxytidine led to a significant reduction in the promoter DNA methylation levels with restoration of the DSC3 expression, suggesting that promoter DNA methylation is a key epigenetic mechanism regulating DSC3 expression. High DSC3 promoter DNA methylation levels were significantly correlated with advanced tumor stage (P<.001) and lymph node metastasis (P<.001). CONCLUSION: Taken together, our results demonstrate that epigenetic silencing of DSC3 is a frequent finding in EAC that is possibly associated with advanced stages.
Keywords: epigenetics, DSC3, esophageal, cancer, metastasis.
Introduction
The incidence rate of esophageal adenocarcinoma (EAC) has increased rapidly in the United States and other Western countries [1-3]. EAC is the most frequent human esophageal malignancy in the US. Risk factors of EAC include male gender, obesity, gastroesophageal reflux disease (GERD), smoking, and low fruit and vegetable intake [4-7]. Although Barrett's esophagus is the major risk factor for the development of EAC, the molecular underpinnings during EAC development and progression are still largely unknown.
Recent evidences demonstrated that hypermethylation of the gene promoter CpG island, alone or in coordination with other genetic or epigenetic mechanisms, is one of the major mechanisms in silencing tumor suppressor genes [8, 9] as well as others which are associated with key oncogenic functions such as angiogenesis, invasion, metastasis, and progression. Cell-cell adhesion plays a critical role in cancer invasion and metastasis. Loss of cell adhesion and alterations in the expression of cadherins are common features of tumor cells and markers for aggressive tumor growth and poor prognosis [10, 11]. Desmocollin3 (DSC3) is a member of the cadherin superfamily of calcium-dependent cell adhesion molecules and a principle component of desmosomes. DSC family members are uniquely expressed in epidermal tissue, with DSC3 mainly present in the basal and spinous layers [12]. One of the more intriguing functions of desmosomal proteins, as they relate to cancer, is their ability to inhibit cell motility [13, 14]. Down-regulation of DSC3 was reported in colorectal cancer and skin tumor [15, 16], possibly through epigenetic mechanisms and/or regulation by p53 [17]. In this study, we demonstrated that frequent loss of DSC3 in EAC cell lines and primary tumor tissues is mediated by aberrant cytosine methylation of promoter regions. Furthermore, we investigated the effect of demethylation on EAC cell lines and confirmed that demethylation treatment can restore DSC3 expression. Our results indicated that epigenetic silencing of DSC3 is a molecular event in esophageal adenocarcinomas that is progressive with advanced stages of the disease.
Materials and methods
Cell culture
DSC3 gene expression and DNA methylation were analyzed in 10 cell lines originating from esophagus, including normal esophageal squamous epithelium (HET1A and HEEC), Barrett's esophagus (BART, CPA, and CPB) and adenocarcinomas (OE33, FLO-1, OE19, SKGT4, and JHU-Eso-Ad1). These cell lines were obtained from the American Type Culture Collection (ATCC, Manassas, VA, USA), Sigma Aldrich (St. Louis, MO, USA), ScienCell Research Laboratories (Carlsbad, CA, USA), Dr. Rhonda Souza (University of Texas Southwestern), Dr. David Beer (University of Michigan), and Dr. Jim Eshleman (John Hopkins University). The esophageal adenocarcinoma cell lines OE33, OE19, FLO-1, SKGT4, and JHU were cultured in Dulbecco's modified Eagle's medium (DMEM) media. The immortalized Barrett's esophagus cell lines were cultured with epithelial cell medium 2 (ScienCell). All the cell lines were supplemented with 10% fetal bovine serum and antibiotics (100 u/ml penicillin and 100 µg/ml streptomycin (Invitrogen, Carlsbad, CA, USA) on primaria plates and flasks (BD Biosciences, Bedford, MA, USA) in a 37°C incubator with 5% CO2.
Esophageal tumor specimens
All frozen tissue samples were obtained from the National Cancer Institute Cooperative Human Tissue Network and the archives of pathology at Vanderbilt University (Nashville, TN, USA). The use of de-identified archival tissue samples was approved by the Vanderbilt institutional review board. Histopathological diagnosis of the EACs was verified based on hematoxylin and eosin stained sections according to the Vienna classification of gastrointestinal epithelial neoplasia [18]. The patient ages ranged from 42-78 years (median at 62.4 years). The samples included 46 EAC and 30 normal esophageal squamous mucosa tissue samples. The adenocarcinomas ranged from well-differentiated to poorly-differentiated, stages I-IV, with a mix of intestinal- and diffuse-type tumors.
Quantitative real-time RT-PCR (qRT-PCR) analyses of DSC3 expression
Total RNAs from all the cell lines and frozen primary tissue samples were isolated by using the RNeasy Mini Kit (Qiagen, Valencia, CA, USA), and single stranded cDNA was subsequently synthesized using the iScript cDNA synthesis kit (Bio-Rad, Hercules, CA, USA). The primers were designed using the online software, Primer 3 (http://frodo.wi.mit.edu/cgi-bin/primer3/primer3_www.cgi). The forward and reverse primers were designed to span two different exons. DSC3 [forward] 5'-ATCTTCAGTCGTGCTGGTGA-3' and DSC3 [reverse] 5'-ACTTGACCGGATGAGGTCTG-3'. The qRT-PCR was performed on a CFX Connect real-time system (Bio-Rad) using Bio-Rad iQ SYBR Green Supermix with the threshold cycle number determined by the use of Bio-Rad's CFX manager 3.0 software. Reactions were performed in triplicate, and the threshold numbers were averaged. The results of the DSC3 gene were normalized to HPRT1, which had minimal variation in all normal and tumor samples tested [19]. Expression fold was calculated according to the formula 2(Rt-Et)/2(Rn-En), where Rt is the threshold cycle number for the reference gene observed in the tumor. Et is the threshold cycle number for the experimental gene observed in the tumor. Rn is the threshold cycle number for the for the reference gene observed in the normal samples. En is the threshold cycle number for the experimental gene observed in the normal samples. For normalization of tumor data and calculation of fold changes, the Rn and En values were calculated as an average of 30 normal samples.
DNA sodium bisulfate modification and pyrosequencing analysis
DNA from cell lines and the frozen primary tissue samples were purified using a DNeasy tissue kit (Qiagen). The bisulfite modification of the DNA was performed using an EZ DNA Methylation-Gold Kit (ZYMO Research, Orange, CA, USA), according to the manufacturer's protocol. A 20 ng aliquot of modified DNA was subjected to polymerase chain reaction (PCR) amplification of the specific promoter region containing a CpG island, by using of a primer set designed using PSQ assay design software (Qiagen), where one of the primers was biotin labeled. The primer set does not include CpG site that enables us to amplify both methylated and unmethylated sequences of the DSC3 gene. The primers were designed [Forward] AGGGGAGTGGGAGAATTGGT, [Reverse] CCTACCCCAAAACAACTTCACTTCT. Platinum PCR SuperMix High Fidelity (Invitrogen) was used to prepare the PCR solution. PCR products were checked by gel electrophoresis to confirm the size of the products and rule out the formation of primer dimers. Quantitative pyrosequencing analyses were done using Biotage PyroMark MD system (Qiagen) following the protocol provided by the manufacturer. The results were analyzed by Pyro Q-CpG 1.0.9 software (Qiagen). Based on control normal samples and internal quality controls provided in the software analysis, we used a 10% DNA methylation level as a cutoff for identification of DNA hypermethylation [20, 21].
5-Aza-2'-Deoxycytidine and Trichostatin-A Treatment
For validation of the role of epigenetics in transcriptional regulation of DSC3 in vitro, esophageal cancer cell lines FLO-1 and SKGT4 were used. Cells were maintained in Dulbecco's modified Eagle's medium (DMEM), supplemented with 10% fetal bovine serum (FBS) and antibiotics (Invitrogen) as described above. Cells were seeded at low density for 24 hours and then treated with 5 μM 5-Aza-2'-deoxycytidine (5-Aza-2') (Sigma-Aldrich) for 72 hours and/or 200 nM Trichostatin-A (TSA) (Wako, Osaka, Japan) for 24 hours. Total RNA and DNA were isolated and purified by an RNeasy kit and DNeasy tissue kit (Qiagen), as described above. The DNA methylation levels of DSC3 CpG nucleotides of the promoter were determined by pyrosequencing before and after treatments. The DSC3 mRNA expression levels were determined by qRT-PCR, as described above.
Statistical analysis
The student t test was used to compare DNA methylation and mRNA expression between normal and tumor samples. The correlations between the DNA methylation level and mRNA expression fold were determined by Spearman Rank Correlation. All P-values were based on two-sided tests and differences were considered statistically significant when P-value≤.05.
Results
Down-regulation of DSC3 gene expression and promoter hypermethylation of DSC3 in esophageal adenocarcinoma cell lines
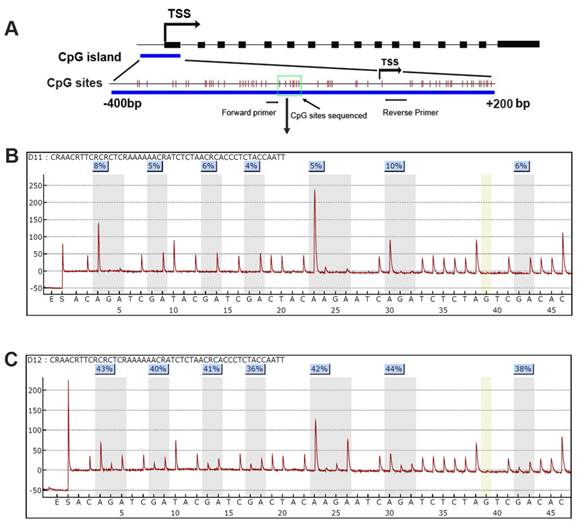
The DSC3 gene has 16 exons with a CpG island located around its transcription start site (TSS, Figure 1). To study DSC3 gene expression and DNA methylation in esophageal adenocarcinoma, we have collected a panel of 10 cell lines originated from esophagus, including normal esophageal squamous epithelium (HET1A and HEEC), Barrett's esophagus (BART, CPA, and CPB) and adenocarcinomas (OE33, FLO-1, OE19, SKGT4, and JHU-Eso-Ad1). We first analyzed the expression of DSC3 in esophageal cancer cell lines by real-time PCR. We detected significant reduction in DSC3 mRNA expression in esophageal adenocarcinoma cell lines as compared to non-neoplastic esophageal cells (Figure 2A). The pyrosequencing analysis demonstrated significantly higher DNA methylation levels in 4 of 5 EAC cell lines as compared to the non-neoplastic cells (Figure 2B).
DSC3 promoter CpG island and Pyrosequencing. A) A schematic chart shows DSC3 genomic structure. DSC3 has 16 exons as shown in black boxes. A CpG island with dense CpG sites is present from -400 to +200 bp relative to the transcription start site (TSS); each vertical bar represents one CpG site. Forward and reverse primers for Pyrosequencing assay PCR locate in the gap of CpG site and the 7 CpG sites sequenced is shown within green box. B) and C) Representative Pyrosequencing profile of the 7 CpG sites in a matched normal (B) and tumor (C) sample pair, showing DNA methylation level for each individual CpG site.
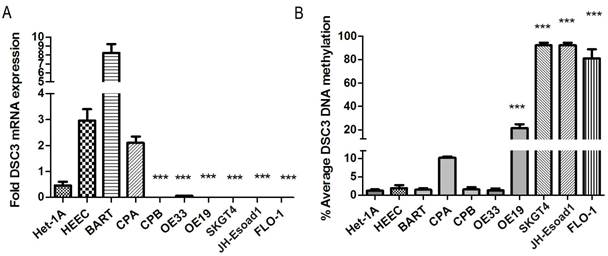
DSC3 mRNA expression and DNA methylation level in non-neoplastic esophageal cell lines and EAC cell lines. A) The expression of DSC3 in EAC cell lines were significantly down-regulated compared with non-neoplastic esophageal cell lines. B) DSC3 promoter DNA methylation levels in EAC cell lines were significantly higher than in non-neoplastic esophageal cell lines. *** P<.001, compared with the average value of normal samples.
Frequent Promoter DNA hypermethylation of DSC3 correlates with down-regulation of DSC3 mRNA expression in primary esophageal cancers
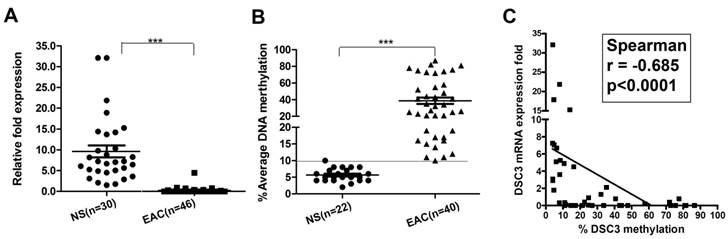
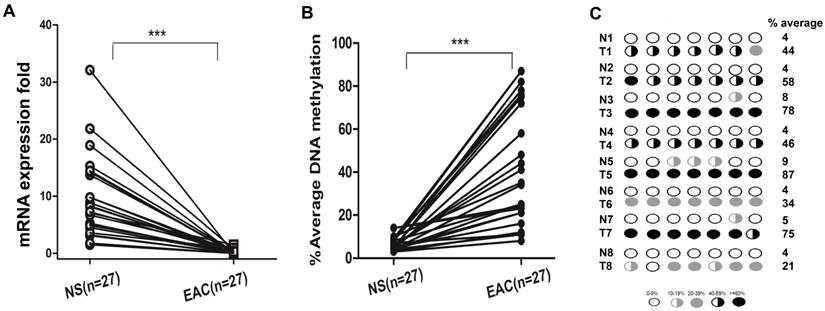
We next analyzed DSC3 mRNA expression and promoter methylation status in normal and EAC human samples. We found that in EAC tissues, DSC3 gene expression was significantly lower than in normal tissues (0.24 vs 9.36, P<.001; Figure 3A) with increased promoter DNA methylation levels (% average 38.6 vs 5.86, P<.001; Figure 3B). Based on our results in normal samples and our previous studies, we used 10% average DNA methylation level as a cutoff threshold for hypermethylation. While 97.5% (39/40) EAC samples displayed hypermethylation, none of the 22 (0%) normal samples showed hypermethylation (Table 1). Spearman's rank correlation analysis demonstrated a significant inverse correlation between DSC3 promoter DNA methylation and mRNA expression fold (coefficient r=-0.685, P<.0001; Figure 3C). We further confirmed the above results by analyzing DNA methylation in 25 tumor samples and their matching adjacent histologically normal tissues from the same patients (P<.001, Figure 4A and 4B). A representative DNA methylation status of an individual CpG site in 8 matched normal and tumor samples is illustrated in Figure 4C. These results suggest that the DSC3 promoter hypermethylation is a key molecular factor involved in suppression of its mRNA expression in EACs.
Frequency of DSC3 hypermethylation in normal and EAC samples.
| N | DSC3 DNA methylation Level | P value | ||
|---|---|---|---|---|
| ≤ 10% | > 10% | |||
| Normal | 22 | 22 | 0 | <.0001 |
| EAC | 40 | 1 | 39 | |
DSC3 mRNA expression and DNA methylation level in human normal and EAC tissues. A) DSC3 mRNA expression was significantly higher in normal tissues, while down-regulated in tumor tissues (relative fold was 9.36 vs 0.24; P<.001). B) DSC3 promoter methylation level was low in normal samples but aberrant hypermethylation was found in EAC tissues (% average DNA methylation level was 5.86 vs 38.6 in normal and tumor respectively; P<.001). A line on 10% level displays the cutoff threshold for hypermethylation. C) The Spearman rank correlation analysis between DNA methylation level and mRNA expression fold of DSC3. Significant inverse correlation was found (r=-0.685, P<.0001).
DSC3 mRNA expression and promoter DNA methylation status in matched human normal tissues and EAC tissues. A) DSC3 gene expression was significantly down-regulated in tumors. B) Tumor tissues had aberrant hypermethylation levels when compared with matched normal tissues. C) Methylation levels of 8 representative matching normal and tumor samples.
Demethylation of DSC3 promoter restores DSC3 mRNA expression in EAC cell lines
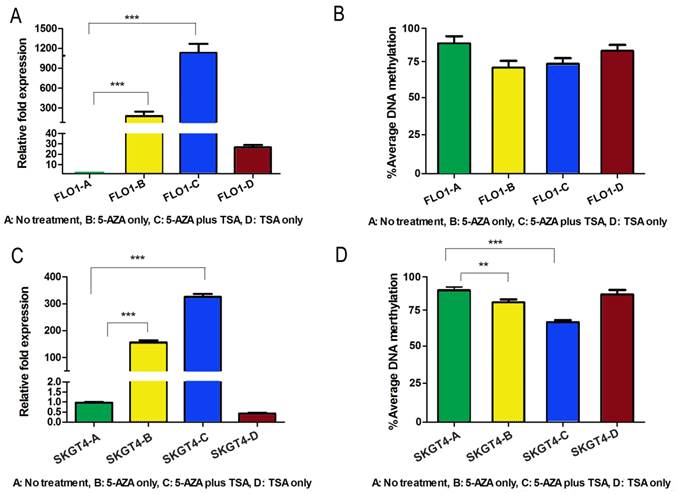
Because both DNA methylation and histone de-acetylation are epigenetic events that regulate gene expression at the promoter level, we checked whether interference with the activities of DNA methyltransferases, using 5-Aza-2', and/or histone deacetylases, using TSA, could restore the gene expression in esophageal adenocarcinoma cell lines. To confirm the epigenetic silencing of DSC3 gene expression, FLO-1 and SKGT4, which have DSC3 promoter hypermethylation and loss of DSC3 mRNA expression, were treated with 5-Aza-2' alone or in combination with TSA. The results show that treatment with 5-Aza-2' alone, and not TSA alone, led to significant restoration of DSC3 mRNA expression in both FLO-1 and SKGT4 cell lines (P<.001; Figure 5A and 5C). The expression of DSC3 was enhanced upon combination of 5-Aza-2' with TSA (P<.001; Figure 5A and 5C). These results suggest that promoter hypermethylation is the primary epigenetic event whereas histone de-acetylation has an added contribution to the regulation of DSC3 expression. In concordance with these results, the DSC3 promoter DNA methylation level decreased in FLO-1 (from 92.2% to 77.2%) and in SKGT4 (from 92% to 69%), respectively (Figure 5B and 5D).
Treatment with 5-Aza and TSA reverses DNA methylation and gene expression patterns of DSC3 in esophageal adenocarcinoma cell lines. A) Treatment restores gene expression in FLO-1 cell line. 5-Aza treatment alone led to significantly induction of relative mRNA expression from 1-180 fold. B) DSC3 promoter methylation level decreased in FLO-1 after treatments. C) Treatment restores gene expression in SKGT4 cell line. 5-Aza treatment alone led to significant induction of relative expression from 1-150 fold. D) DSC3 promoter methylation level significantly decreased in SKGT4 after treatments.
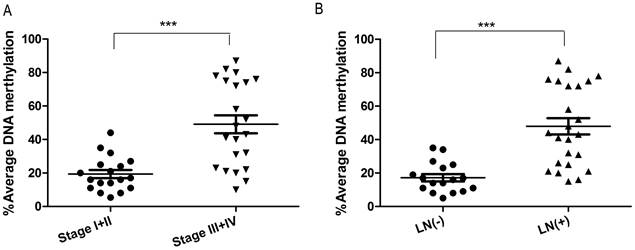
DNA methylation of DSC3 gene promoter region correlated with advanced tumor stages
To investigate whether DSC3 promoter methylation is associated with tumor biology, we evaluated DNA methylation levels and clinical pathological parameters. As shown in Figure 6, DSC3 DNA hypermethylation was significantly correlated with advanced tumor stage (P<.001) and lymph node metastasis (P<.001).
DSC3 methylation is associated with advanced tumor stage and lymph node metastasis. A) Increased DNA methylation of DSC3 promoter correlates with advanced tumor staging (P<.001). B) Increased DNA methylation of DSC3 promoter correlates with lymph node metastasis (P<.001).
Discussion
Esophageal adenocarcinomas are characterized by poor outcome, which is mainly attributed to the diagnosis of the disease at late stages with high occurrence of metastatic lesions and poor response to therapy. Although the molecular and genetic events underlying tumor metastasis are still not well understood, intense investigation into this process has led to the notion that genes and signaling pathways involved in cell adhesion and migration are critical in tumor invasion and metastasis [22]. Desmocollins are transmembrane glycoproteins and members of the cadherin superfamily of calcium-dependent cell-cell adhesion molecules. There are three DSC isoforms (DSC1-3); each is a product of a separate gene. The genomic organization of DSCs is similar to that of classic cadherins [23]. In this study, we analyzed the mRNA expression of DSC3 in esophageal adenocarcinoma cell lines and non-cancer esophageal epithelial cell lines. We found that the expression of DSC3 was significantly down-regulated or silenced in cancer cell lines as compared with non-cancer epithelial cell lines. We also confirmed that in human EAC tissues, DSC3 mRNA expression was significantly decreased compared with human normal esophageal tissues. Our results support earlier findings in other tumor subtypes, which have found down-regulation of DSC3 in breast cancer and colorectal cancer [17, 24]. To explore the mechanism responsible for the gene silencing in EACs, bisulfite methylation pyrosequencing was applied to quantify the DSC3 promoter methylation levels in the cell lines and normal and cancer tissues. The results demonstrated hypermethylation of DSC3 in almost all EAC samples with a strong inverse correlation between the DNA methylation and gene expression levels (r=-0.685). These data suggest that epigenetic promoter methylation is a possible regulatory mechanism of DSC3 gene expression in EAC. In fact, our results using 5-Aza and TSA confirmed the epigenetic regulation of DSC3 and suggested that DNA methylation plays a greater role in regulating the expression of DSC3 than histone de-acetylation. These results add to previous findings of DSC3 methylation in colorectal [25] and lung cancers [26-28]. Of note, in our assay, we detected DSC3 hypermethylation (> 10%) in all EACs except one (97.5%, 39/40) whereas none of the 22 (0%) normal esophageal epithelial samples showed DSC3 hypermethylation. These results suggest that DSC3 could serve as biomarker for Barrett's tumorigenesis and early detection of EAC; however, large cohort studies are needed to validate these primary observations.
Previous studies have observed that loss or reduction of desmosomes contribute to the development and/or the progression of various human epithelial cancers [29, 30]. Analysis of clinical-pathological data indicated that DSC3 prompter hypermethylation was highly correlated with advanced tumor stage and lymph node metastasis (P<.001). This finding is consistent with the known function of DSC3, which is a member of the cadherin superfamily of calcium-dependent cell-cell adhesion molecules. Similarly, loss of expression of DSC3 induced the progression of oral carcinomas and correlated with lymph node metastasis and cell proliferation [31]. Taken together, our results suggest that DSC3 could be a novel tumor suppressor gene with inhibitory functions in invasion and metastasis in EAC.
In conclusion, this is the first study reporting DNA hypermethylation and silencing of DSC3 in EACs. Further studies examining the functions of DSC3 in EAC are needed to uncover the mechanisms by which desmosomal signaling regulates cell migration, proliferation and other biological processes in EAC.
Abbreviations
NS, normal esophagus, BE, Barrett's esophagus; EAC, esophageal adenocarcinoma; GERD, gastroesophageal reflux disease; DSC3, Desmocollin3
Acknowledgements
This study was supported by grants from the National Institute of Health; R01CA106176, Vanderbilt SPORE in Gastrointestinal Cancer (P50 CA95103), Vanderbilt Ingram Cancer Center (P30 CA68485) and the Vanderbilt Digestive Disease Research Center (DK058404); and Department of Veterans Affairs. The contents of this work are solely the responsibility of the authors and do not necessarily represent the official views of the National Cancer Institute, Department of Veterans Affairs, or Vanderbilt University.
Competing Interests
All the authors declared no conflict of interest for the purpose of this study.
References
1. Hur C, Miller M, Kong CY, Dowling EC, Nattinger KJ, Dunn M. et al. Trends in esophageal adenocarcinoma incidence and mortality. Cancer. 2013;119:1149-58 doi:10.1002/cncr.27834
2. Pennathur A, Gibson MK, Jobe BA, Luketich JD. Oesophageal carcinoma. Lancet. 2013;381:400-12 doi:10.1016/S0140-6736(12)60643-6
3. Pohl H, Welch HG. The role of overdiagnosis and reclassification in the marked increase of esophageal adenocarcinoma incidence. Journal of the National Cancer Institute. 2005;97:142-6 doi:10.1093/jnci/dji024
4. Carr JS, Zafar SF, Saba N, Khuri FR, El-Rayes BF. Risk factors for rising incidence of esophageal and gastric cardia adenocarcinoma. Journal of gastrointestinal cancer. 2013;44:143-51 doi:10.1007/s12029-013-9480-z
5. Lassen A, Hallas J, de Muckadell OB. Esophagitis: incidence and risk of esophageal adenocarcinoma--a population-based cohort study. Am J Gastroenterol. 2006;101:1193-9
6. Pohl H, Wrobel K, Bojarski C, Voderholzer W, Sonnenberg A, Rosch T. et al. Risk factors in the development of esophageal adenocarcinoma. Am J Gastroenterol. 2013;108:200-7 doi:10.1038/ajg.2012.387
7. Reid BJ, Li X, Galipeau PC, Vaughan TL. Barrett's oesophagus and oesophageal adenocarcinoma: time for a new synthesis. Nature reviews. 2010;10:87-101 doi:10.1038/nrc2773
8. Esteller M, Corn PG, Baylin SB, Herman JG. A gene hypermethylation profile of human cancer. Cancer Res. 2001;61:3225-9
9. Herman JG, Baylin SB. Gene silencing in cancer in association with promoter hypermethylation. N Engl J Med. 2003;349:2042-54
10. Brooke MA, Nitoiu D, Kelsell DP. Cell-cell connectivity: desmosomes and disease. J Pathol. 2012;226:158-71 doi:10.1002/path.3027
11. Le Bras GF, Taubenslag KJ, Andl CD. The regulation of cell-cell adhesion during epithelial-mesenchymal transition, motility and tumor progression. Cell adhesion & migration. 2012;6:365-73 doi:10.4161/cam.21326
12. Delva E, Tucker DK, Kowalczyk AP. The desmosome. Cold Spring Harbor perspectives in biology. 2009;1:a002543. doi:10.1101/cshperspect.a002543
13. Runswick SK, O'Hare MJ, Jones L, Streuli CH, Garrod DR. Desmosomal adhesion regulates epithelial morphogenesis and cell positioning. Nat Cell Biol. 2001;3:823-30 doi:10.1038/ncb0901-823
14. Tselepis C, Chidgey M, North A, Garrod D. Desmosomal adhesion inhibits invasive behavior. Proc Natl Acad Sci U S A. 1998;95:8064-9
15. Chen J, O'Shea C, Fitzpatrick JE, Koster MI, Koch PJ. Loss of Desmocollin 3 in skin tumor development and progression. Mol Carcinog. 2012;51:535-45 doi:10.1002/mc.20818
16. Knosel T, Chen Y, Hotovy S, Settmacher U, Altendorf-Hofmann A, Petersen I. Loss of desmocollin 1-3 and homeobox genes PITX1 and CDX2 are associated with tumor progression and survival in colorectal carcinoma. Int J Colorectal Dis. 2012;27:1391-9 doi:10.1007/s00384-012-1460-4
17. Cui T, Chen Y, Yang L, Knosel T, Zoller K, Huber O. et al. DSC3 expression is regulated by p53, and methylation of DSC3 DNA is a prognostic marker in human colorectal cancer. Br J Cancer. 2011;104:1013-9 doi:10.1038/bjc.2011.28
18. Soutto M, Peng D, Razvi M, Ruemmele P, Hartmann A, Roessner A. et al. Epigenetic and genetic silencing of CHFR in esophageal adenocarcinomas. Cancer. 2010;116:4033-42 doi:10.1002/cncr.25151
19. El-Rifai W, Moskaluk CA, Abdrabbo MK, Harper J, Yoshida C, Riggins GJ. et al. Gastric cancers overexpress S100A calcium-binding proteins. Cancer Res. 2002;62:6823-6
20. Peng D, Hu TL, Jiang A, Washington MK, Moskaluk CA, Schneider-Stock R. et al. Location-specific epigenetic regulation of the metallothionein 3 gene in esophageal adenocarcinomas. PLoS One. 2011;6:e22009. doi:10.1371/journal.pone.0022009 PONE-D-11-06954 [pii]
21. Peng DF, Razvi M, Chen H, Washington K, Roessner A, Schneider-Stock R. et al. DNA hypermethylation regulates the expression of members of the Mu-class glutathione S-transferases and glutathione peroxidases in Barrett's adenocarcinoma. Gut. 2009;58:5-15 doi:10.1136/gut.2007.146290
22. Canel M, Serrels A, Frame MC, Brunton VG. E-cadherin-integrin crosstalk in cancer invasion and metastasis. J Cell Sci. 2013;126:393-401 doi:10.1242/jcs.100115
23. Dusek RL, Godsel LM, Green KJ. Discriminating roles of desmosomal cadherins: beyond desmosomal adhesion. J Dermatol Sci. 2007;45:7-21 doi:10.1016/j.jdermsci.2006.10.006
24. Oshiro MM, Kim CJ, Wozniak RJ, Junk DJ, Munoz-Rodriguez JL, Burr JA. et al. Epigenetic silencing of DSC3 is a common event in human breast cancer. Breast Cancer Res. 2005;7:R669-80 doi:10.1186/bcr1273
25. Oster B, Thorsen K, Lamy P, Wojdacz TK, Hansen LL, Birkenkamp-Demtroder K. et al. Identification and validation of highly frequent CpG island hypermethylation in colorectal adenomas and carcinomas. International journal of cancer. 2011;129:2855-66 doi:10.1002/ijc.25951
26. Cui T, Chen Y, Yang L, Knosel T, Huber O, Pacyna-Gengelbach M. et al. The p53 target gene desmocollin 3 acts as a novel tumor suppressor through inhibiting EGFR/ERK pathway in human lung cancer. Carcinogenesis. 2012;33:2326-33 doi:10.1093/carcin/bgs273
27. Cui T, Chen Y, Yang L, Mireskandari M, Knosel T, Zhang Q. et al. Diagnostic and prognostic impact of desmocollins in human lung cancer. Journal of clinical pathology. 2012;65:1100-6 doi:10.1136/jclinpath-2011-200630
28. Yang L, Chen Y, Cui T, Knosel T, Zhang Q, Albring KF. et al. Desmoplakin acts as a tumor suppressor by inhibition of the Wnt/beta-catenin signaling pathway in human lung cancer. Carcinogenesis. 2012;33:1863-70 doi:10.1093/carcin/bgs226
29. Dubash AD, Green KJ. Desmosomes. Curr Biol. 2011;21:R529-31 doi:10.1016/j.cub.2011.04.035
30. Dusek RL, Attardi LD. Desmosomes: new perpetrators in tumour suppression. Nature reviews. 2011;11:317-23 doi:10.1038/nrc3051
31. Wang L, Liu T, Wang Y, Cao L, Nishioka M, Aguirre RL. et al. Altered expression of desmocollin 3, desmoglein 3, and beta-catenin in oral squamous cell carcinoma: correlation with lymph node metastasis and cell proliferation. Virchows Arch. 2007;451:959-66 doi:10.1007/s00428-007-0485-5
Author contact
![]() Corresponding authors: Wael El-Rifai, MD, PhD, Department of Surgery, Cancer Biology, Vanderbilt University Medical Center, 760 Preston Research Building, 2220 Pierce Ave, Nashville TN, 37232. Email: wael.el-rifaiedu. Shutian Zhang, MD, PhD, Department of Gastroenterology, Beijing, Friendship Hospital, Beijing Digestive Disease Center, Capital Medical University, 95 Yongan Road, Beijing, China, 100050. Email: zhangstedu.cn.
Corresponding authors: Wael El-Rifai, MD, PhD, Department of Surgery, Cancer Biology, Vanderbilt University Medical Center, 760 Preston Research Building, 2220 Pierce Ave, Nashville TN, 37232. Email: wael.el-rifaiedu. Shutian Zhang, MD, PhD, Department of Gastroenterology, Beijing, Friendship Hospital, Beijing Digestive Disease Center, Capital Medical University, 95 Yongan Road, Beijing, China, 100050. Email: zhangstedu.cn.