Impact Factor ISSN: 1837-9664

 Global reach, higher impact
Global reach, higher impactJ Cancer 2014; 5(6):480-490. doi:10.7150/jca.8731 This issue Cite
Research Paper
Geldanamycin and Its Derivatives Inhibit the Growth of Myeloma Cells and Reduce the Expression of the MET Receptor
Artur Jurczyszyn1, Anna Zebzda2, Jacek Czepiel3, William Perucki4, Stanisława Bazan-Socha5, Dorota Cibor3, Danuta Owczarek3, Marcin Majka2 ![]()
1. Department of Hematology, Jagiellonian University Medical College, Krakow, Poland
2. Department of Transplantology, Jagiellonian University Medical College, Krakow, Poland
3. Department of Gastroenterology, Hepatology and Infectious Diseases, Jagiellonian University Medical College, Krakow, Poland
4. Students' Scientific Society, Jagiellonian University Medical College, Krakow, Poland
5. Second Department of Internal Medicine, Jagiellonian University Medical College, Krakow, Poland
Received 2014-1-31; Accepted 2014-3-19; Published 2014-5-31
Abstract
Introduction. Geldanamycin (GA) is an ansamycin antibiotic that exhibits potent anti-neoplastic properties. The aim of this study was to assess the impact of GA and its derivatives on the growth and invasiveness of myeloma cell lines and CD138+ cells derived from the bone marrow of patients with multiple myeloma.
Materials and methods. We evaluated cell proliferation, survival, apoptosis, cell cycle of myeloma cells, and the expression of cell surface proteins after incubation with geldanamycin or its derivatives.
Results. GA and its analogs have an effect on myeloma cells by inhibiting their growth in a time and dose-dependent manner. Myeloma cell lines demonstrated decreased proliferation after incubation with 10 nM of GA or 100 nM GA analogs. The first significant effects of GA on U266 cells was observed after 24 hours. After 24 hours, U266 cells incubated with 100 nM GA were in both early and late stages of apoptosis; 17AEP and 17DMAG caused apoptosis of similar intensity to GA. It has been observed that GA and its derivatives cause caspase-3 activation. Analysis of the activity of AKT and MAP 42/44 kinases was performed by incubating U266 cells for 24 and 48 hours in100 nM of GA and its derivatives. After 24 hours incubation, no significant changes in protein expression were observed, while after 48 hours, the strongest changes were seen in AKT protein expression after incubation with GA and 17AEP-GA. In studies of the cell cycle, it was found that 100 nM 17AEP-GA and 17-DMAP-GA cause cell cycle abnormalities. We observed a nearly two-fold increase in U266 cells in the G1 phase and a simultaneous decrease in the percentage of cells in the G2/M phase, indicating that cells were halted in the G1 phase. In the case of the INA6 cells, proliferation was halted in both the G1 and G2/M phases.
Conclusions. GA and the analogues that we tested can inhibit myeloma cell growth by induction of apoptosis and blockage of cell cycle progression, and have an effect on the down-regulation of the MET receptor. The GA derivatives tested, despite their modifications still retain strong anticancer properties. Specifically, two analogues of GA, 17AEP-GA and 17DMAG due to their properties can be more effective and safer chemotherapeutic agents than 17AAG, which is currently used and described in literature.
Keywords: multiple myeloma, geldanamycin, 17DMAG, 17AEP-GA, 17DMAP-GA
Introduction
Multiple myeloma (MM) is a malignant proliferation of plasma cells producing and secreting monoclonal immunoglobulins or light chains thereof. Among all malignant tumors, the incidence of MM is 1-2%, which represents approximately 15% of hematopoietic tumors [1]. The most characteristic symptoms include the following: clearly marked multiple osteolytic foci in the bones accompanied by pain and pathological fractures, renal failure, anemia, and susceptibility to infection. The introduction of new drugs in the past decade to treat MM has doubled median survival of patients, which is currently 6-7 years. Ongoing research into new therapeutic agents raises hopes of a greater increase in survival. Current research is targeted not only at drugs which effect the myeloma cells themselves but also at other factors that affect the interactions of these cells with the bone marrow (BM) stroma [2,3].
Heat shock protein 90 (HSP90) comprises about 1-2% of all cellular proteins. This protein plays an important role in maintaining homeostasis in both physiological and stress conditions. HSP90 is a conserved protein responsible for the correct folding of proteins and stabilization of their structure. It is secreted in response to stress (infectious agents, temperature, heavy metals, acidification) and is involved in intracellular signaling [4]. HSP90 cooperates with other proteins by forming complexes with them to maintain the active form of the target proteins and direct them to a degradation pathway if they are abnormally conformed [4]. Cancer cells appear to be especially dependent on HSP90 activity. In neoplastic cells abnormal HSP90 complexes are present which stabilize mutated forms of proteins, via strong binding to carrier proteins, thus avoiding degradation [4]. HSP90 are produced in excess and mutate assuming an unstable form. HSP90 target proteins regulate a number of processes determining the growth and spread of cancer cells, specifically factors: stimulating angiogenesis (factor-1 induced by hypoxia HIF1α, MET, Src, vascular endothelial growth factor VEGF), cell cycle control (CDK4, CDK6, cyclin D), inhibition of apoptosis (AKT, insulin-like growth factor 1 receptor IGF-1R), survival and invasiveness (matrix metalloproteinase-2 MMP-2 and urokinase-2 ) [5]. The role of HSP90 in cancer cells is to maintain homeostasis, despite unfavorable conditions (hypoxia, malnutrition) and intensive production of abnormal proteins [6]. It is therefore hypothesized that the level of chaperone proteins in cancer cells may correlate with the degree of their malignancy. Increased activity of HSP90 was noted in a variety of tumors [7, 8], hence the use of inhibitors of HSP90 as a trial in the treatment of proliferative diseases [4, 9].
Geldanamycin (GA), belongs to the ansamycin antibiotic subclass, and was first isolated in 1970 in actinomycete culture of Streptomyces hygroscopicus. In 1993, Whitesell et al found that GA specifically inhibits the formation of complexes of HSP90-v-SRC, suggesting HSP90 has an important role in the process of malignant transformation [10]. Further studies have shown that GA also downregulates a number of other proteins involved in tumorogenesis, such as mutant p53, mutant B-RAF, AKT, BCR-ABL, and ERBB2 [11]. GA's anti-tumor effect results from several mechanisms including: inhibition of the proliferative effect of proteins such as RAF-1, a reduction in expression of the mutant or overproduced oncogenic proteins, inhibition of proteins determining neoplastic cell survival such as AKT, blocking the cell cycle, inhibition of angiogenesis, and the reduction of invasiveness and the ability to metastasize [12].
The aim of this study was to assess the impact of GA and its derivatives on the growth and invasiveness of myeloma cell lines and CD138+ cells derived from the BM of patients with MM.
Materials and methods
Patients
BM samples were collected from 20 patients with MM, including 12 men and 8 women, with age ranging from 45 to 78 years (average 61.5 years), and with marrow plasmacytosis from 15% to 65% (average 35.6%). Patients were treated at the Hematological Clinic of the University Hospital in Krakow. Additionally, samples were obtained from 10 volunteers, including 5 men and 5 women, with age ranging from 48 to 68 years (average 60.5 years). Diagnosis of MM was based on histological increase of myeloid cells on BM biopsy and monoclonal hyperglobulinemia. BM was collected by aspiration from the posterior iliac crest into a sterile Vacutainer tube (Becton Dickinson) with anticoagulant (EDTA or heparin). Informed consent was obtained from all the participants, and the study was approved by the local ethics committee.
Isolation and culture of primary myeloma cells
Primary myeloma cells were isolated from fresh BM aspirates of MM patients. BM mononuclear cells were separated by centrifuging the total aspirate on a Ficoll gradient (1000 x g, 30 min), followed by incubating (15 min, 4°C) with anti-CD138+-coupled magnetic beads, and then isolating the cells using the MiniMACS system (Miltenyi, Biotec, Auburn, CA, USA). Isolated cells were cultured in the presence of 10% fetal bovine serum (FBS) and 2 ng/ml IL-6. Purity of isolated cells was verified by flow cytometry (FACS Canto, Becton Dickinson, Germany) using monoclonal anti-phycoerythrin (PE) labeled CD138+ (BD PharMingen, San Diego, USA).
Isolation and culture of normal cells
The study used mesenchymal stem cells (MSC) derived from the BM of healthy donors (BM-MSC) and patients with MM (MMBM-MSC). BM-MSC were isolated from BM of healthy donors or patients with MM using the protocol described by Friedenstein et al. [13], using the fraction of BM mononuclear cells, obtained by centrifugation of the total BM on Ficoll gradient (1000 x g, 30 min), and then selecting MSC by adhesion to a plastic surface. Mononuclear cells were seeded on plastic bottles (Sarstedt AG & Co., Germany) at a concentration of 4 x 105/cm2. Culture of BM-MSC and MMBM-MSC was carried out on Dulbecco's modified eagle's medium (DMEM) (Sigma-Aldrich) and supplemented with 10% FBS, for incubation of MSC (Stem Cell Technologies) supplemented with an antibiotic (PAA Laboratories GmbH) at 37°C in an atmosphere of 5% CO2 and 95% humidity. After reaching 90% density, the cells were passaged using 0.05% trypsin (trypsin-EDTA, PAA Laboratories GmbH) and were seeded at a concentration of 1 x 104/cm2 into new culture flasks.
Breeding myeloma cell lines
Myeloma cell lines were cultured using RPMI 1640 medium (Gibco BRL, Grand Island, NY, USA) and supplemented with FBS (PAA Laboratories, Pasching, Austria), 2 mM L-glutamine and the following antibiotics: 100 IU/ml penicillin and 10 ug/ml streptomycin (Gibco BRL, Grand Island, NY, USA) at 37 ° C in an atmosphere of 5% CO2 and 95% humidity. Cell lines were passaged two times a week. Names and myeloma line culture conditions are shown in Table 1.
Names and culture conditions of myeloma cell lines
| Line | %FBS | IL-6 (ng/ml) |
|---|---|---|
| U226 | 15 | - |
| RPMI | 20 | - |
| INA6 | 10 | 2 |
| CAG | 10 | - |
Geldanamycin
Geldanamycin, 17-Allylamino-17-demethoxygeldanamycin (17AAG), 17-(Dimethylaminoethylamino)-17-demethoxygeldanamycin (17DMAG), 17-[2-(Pyrrolidin-1-yl)ethyl] aminno-17-demethoxygeldanamycin(17AEP-GA), and 17-(Dimethylaminopropylamino)-17-demethoxygeldanamycin (17DMAP-GA) were purchased at Invivogen (San Diego, CA, USA). GA and 17AAG were dissolved in DMSO (Merck, Darmstadt, Germany) while 17DMAG, 17AEP-GA, and 17DMAP-GA were dissolved in water.
Measuring the proliferation/survival of myeloma cells using MTS assay
U266, RPMI, and CAG cell lines were plated in complete RPMI medium to 96 well culture plates (1x103/well), followed by addition of GA at concentrations of 1, 10, 100, 500, and 1000 nM, at 24, 48, 72, and 96 hours after inhibitor exposure. Evaluation of cell survival was performed using a colorimetric MTS assay (Cell Titer 96 Aqueous One Solution Cell Proliferation Assay, Promega, Madison, WI, USA). Tetrazolium salt (MTS), as a result of redox activity of mitochondria, is reduced to colored formazan. The resulting colored product, reflecting the number of metabolically active cells was assessed using a EL800 reader (BIO -TEK Instruments, USA) at a wave length of 490 nm. The results are shown as a percent of control, where the GA untreated cells were used as control. There were three independent experiments.
Detection of apoptosis
Apoptosis was assessed using annexin V binding assay with simultaneous use of propidium iodide (PI). Annexin V binds to phosphatidyl serine, which is present on the surface of apoptotic cells. Propidium iodide enters the cells due to loss of membrane integrity and thus stains necrotic cells or cells in the late stages of apoptosis. Cells were washed with cold Phosphate Buffered Saline (PBS). Cell pellets were resuspended in 100μl of binding buffer (Becton Dickinson Pharmingen, San Diego, CA, USA), then 5 ml of phycoerythrin-labeled annexin V and 10 ml of PI were added (Pharmingen Becton Dickinson). After 15 minutes of incubation at room temperature, 400 ml of binding buffer was added and the solution was analyzed using a FacsCanto flow cytometer (Becton Dickinson, San Jose, CA, USA). Analysis of the results was performed using the Facs Diva (Becton Dickinson).
Cell Cycle Analysis
An analysis was performed of the cell cycle after 48 hours of incubation with GA and its derivatives. Distribution of the number of cells in the different cell cycle phases were assessed by measuring DNA content using fluorescent labeled propidium iodide. Samples for flow cytometry analysis were prepared by fusing the cells in 75% aqueous ethanol solution and incubating for at least 12 hours at 40°C. The samples were then digested with RNase [50 ug/ml] (ICN Polfa, Rzeszów, Poland) for 30 min at 37°C, and then incubated with PI [50 ug/ml] (Sigma), and diluted in a sodium citrate solution (Sigma) for 30 min at room temperature. The analysis was performed using a Facs Canto flow cytometer (Becton Dickinson). Analysis of the results was performed using the Facs Diva program.
Evaluation of Protein Expression
Western Blot was used to determine the expression of protein in conjunction with immunohistochemical staining and histological preparations.
Determination of protein concentration
Cells were lysed on ice for 10 minutes using M-PER buffer (Pierce, Rockford, IL, USA) supplemented with phosphatase and proteinase inhibitors as follows: 10 mg/ml aprotinin, 1 mg/ml pepstatin A, 10 mg/ml leupeptin, 1 M NaF, 10 mM sodium pyruvate, 500 mM EDTA, and 10 mg/ml PMSF (Sigma). Concentration measurement was performed by the Bradford method using Bio-Rad reagent protein assay (Bio-Rad, Hercules, CA, USA) and an EL800 reader. The protein concentration in the samples was determined on the basis of a calibration curve on which albumin was used as the standard.
Evaluation of protein expression by Western blot
Isolated protein was denatured in the presence of Bond-Braker (Pierce) and loading buffer (NuPage LDS Sample Buffer, Invitrogen) for 10 minutes at 70°C. Prepared samples containing 10 mg of protein were loaded and separated on a polyacrylamide gel with 10% SDS (SDS-PAGE) at a voltage of 150 V for 100 min. The next step was to transfer the sample to PVDF nitrocellulose membrane (BioRad) at 100 V for 60 min. We used Precision Protein Standard (BioRad) as a size marker. The presence of proteins including MET, AKT, MAP, and HSP90 was detected using rabbit monoclonal anti-MET (Santa Cruz Biotech, Santa Cruz, CA, USA), anti-AKT, anti-MAP and anti-HSP90 (Cell Signaling, Danvers, MA, USA) antibodies. As secondary antibodies, anti-rabbit IgG was used as well as related anti-mouse Horse Radish Peroxidase (HRP) (Santa Cruz Biotech). The number of separated products was controlled using rabbit anti-GAPDH (Santa Cruz Biotech). The phosphorylation of p44/42 MAPK kinase (Tyr204/Thr202) and AKT (Ser473) was studied using a mouse monoclonal antibody against phosphorylated p44/42 MAPK, and rabbit antibodies against the AKT kinase (Cell Signaling). Mouse anti-IgG and rabbit anti-HRP-linked antibodies were used as secondary antibodies. The number of separated products was controlled using rabbit anti-GAPDH (Santa Cruz Biotech). After the standard washing conditions, vinyl membrane (PVDF) were coated with a chemiluminescent substrate for HRP (ECL reagent, Amersham Life Sci., Littre Chalfont, UK) and exposed to photographic film (Hyper Film ECL, Amersham Life Sci.).
RNA isolation and reverse transcription reaction
RNA was isolated using RNeasy Mini Kit (Qiagen, Valencia, CA, USA). The concentration and purity of the RNA obtained was evaluated by measuring the absorbance at a wavelength of 260nm and 280nm using a DU 640B spectrophotometer (Beckman Coulter, Fullerton, CA, USA). RNA was transcribed into cDNA using MMLV reverse transcriptase (Promega) and random primers (Promega).
PCR reaction in real time
Evaluation of gene expression was performed by quantitative PCR in real time (qRT-PCR) based on specific TaqMan probes (Applied Biosystems, Foster City, CA, USA) using an ABI PRISM 7300 Sequence Detection System (Applied Biosystems). The reaction mixture contained the ingredients set forth in Table 2. A list of probes used in the experiments is included as Table 3. Figure 1 is a diagram of the qRT PCR reaction.
The qRT PCR reaction
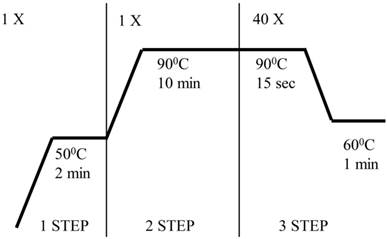
Composition of ingredients used in the qRT-PCR reaction
| TaqMan PCR Master Mix | 25μl |
|---|---|
| cDNA | 100ng |
| 20x probe | 2.5μl |
| Water | To final volume of 50μl |
Probes used for qRT PCR
| TaqMan Probe | Number (Applied Biosystems) |
|---|---|
| MET | Hs01565589_ml |
| HSP90 | Hs00743767_ml |
| GAPDH | Hs99999905_ml |
To calculate results, we used a method of analyzing the relative gene expression - ΔΔCt, which is based on comparison of Ct of the tested and reference gene.
Statistical Analysis
Statistical analysis of results was performed using Microsoft Excel and GraphPad Prism 4.02. Experiments were carried out at least 3 times. Quantitative results are presented in graphs or tables, and are expressed as mean with an error indicating the standard deviation or standard error. The captions under the graphs show the number of times the experiments were carried out and the size of the sample in a single experiment. The statistical significance of differences between groups was tested using Student t test or one-way analysis of variance at a significance level of 0.05.
Results
GA and its derivatives inhibit the growth of myeloma cell line
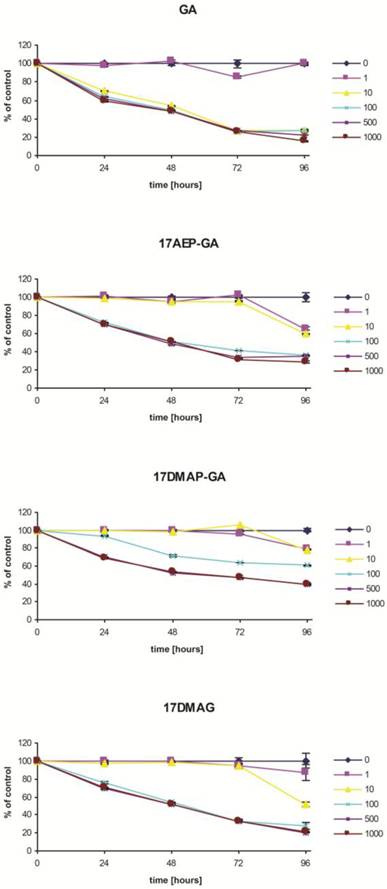
Blocking of HSP90 can affect many cellular processes, including growth. To determine the effect of GA on the growth of the U266, RPMI, INA6, and CAG myeloma cell lines, MTS assay was performed. It was found that GA its and analogs impair the myeloma cells by inhibiting their growth in a time and dose dependent manner (Fig. 2). It was also observed that the effect of GA and its different analogs varies depending on the myeloma line (Fig. 3). Myeloma cell lines showed inhibition of proliferation after administration of 10 nM of GA and 100 nM of GA analogs. We also examined whether the effect of GA and analogues on myeloma cells is dependent on length of exposure. It was observed that inhibition of proliferation was greater with longer exposure to the inhibitor. The first significant effects of GA on U266 cells was observed after 24 hours incubation with a 10 nM dose of GA or a 100nM dose of GA analogs. The inhibition of growth of U266 proceeded with the length of incubation with the inhibitor. Similar effects of GA and its analogs was observed in the other myeloma cell lines (RPMI and CAG). We chose the U266 line and a 100nM dose of GA analogs for further experimentation.
Effects of GA and analogues on MM line U266 at 24, 48, 72, and 96 hours incubation. p<0.001 compared to the control for GA and its analogues and at all time points for concentrations of 100, 500, and 100 nM.
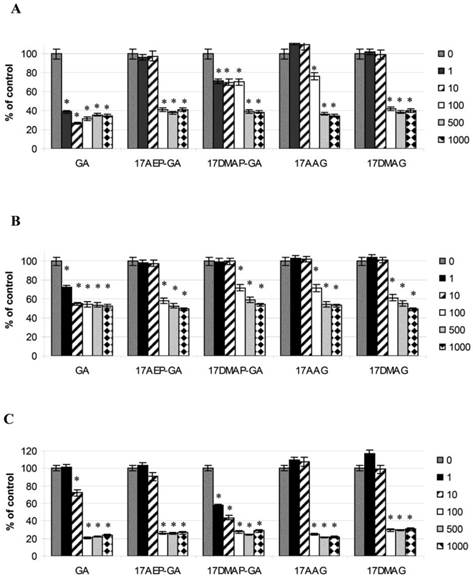
Effects of GA and its analogues on the RPMI (A), U266 (B), and CAG (C) MM cell lines after 96 hours of incubation.* p<0.005.
GA and its derivatives induce apoptosis of myeloma cell line
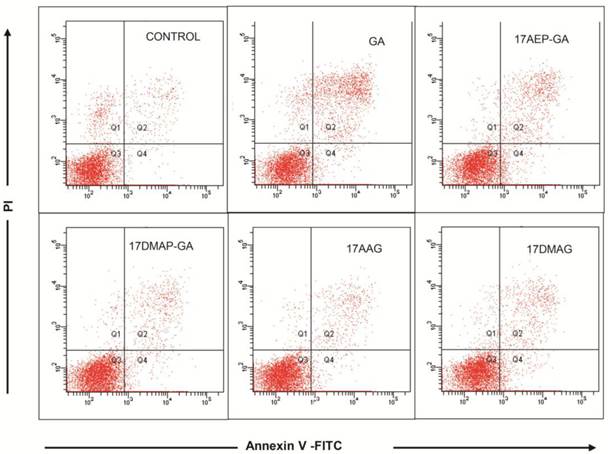
Inhibition of cell growth may be due to induction of apoptosis and cell cycle arrest, among other mechanisms. Using annexin V and propidium iodide staining, we examined whether GA induces apoptosis of myeloma cells. 24 hour incubation of U266 cells with 100 nM GA resulted in the emergence of cell populations at both early (annexin V-binding cells, negative for propidium iodide incorporation) and late stages of apoptosis (annexin V-binding cells, positive for propidium iodide ) (Fig. 4).
Effects of GA and analogues on apoptosis of myeloma cells. The effect of 24 hours of incubation with 100 nM GA and its analogues on apoptosis of the U266 MM line. Exemplary graphs selected from three independent experiments.
48 hours exposure to 100nM of GA and its analogs caused increased apoptosis, thus increasing the percentage of annexin V-binding cells. In order to compare the sensitivity of the myeloma cell lines both dependent and independent of IL-6, analogous experiments were carried out on the U266 and INA6 cell lines. The results are shown in Table 4.
Effect of GA on apoptosis of myeloma cells-binding of annexin V. The increase in annexin V binding cells relative to the control after 24 and 48 hours exposure to a 100 nM dose of GA and its derivatives. Results are from three independent experiments.
| U266 | INA6 | |||
|---|---|---|---|---|
| 24 hours | 48 hours | 24 hours | 48 hours | |
| GA | + | ++ | + | +++ |
| 17AEP-GA | + | ++ | + | +++ |
| 17DMAP-GA | + | + | + | ++ |
| 17AAG | + | + | + | ++ |
| 17DMAG | + | ++ | + | +++ |
The following convention was used for this experiment:
+ Up to 30% apoptotic cells++ Up to 50% apoptotic cells+++Greater than 50% apoptotic cells
17AEP-GA and 17DMAG cause a similar level of apoptosis when compared to GA. The U266 line proved to be less sensitive to GA analogues than INA6 dependent IL6. Both 17DMAP -GA and 17AAG showed weak proapoptotic activity in the U266 and INA6 cell lines.
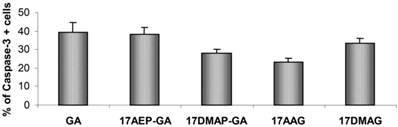
The process of programmed cell death is carried out by activation of proteolytic enzymes - caspases, which irreversibly digest numerous cytoplasmic and nuclear proteins. In our experiments we examined whether GA and its derivatives result in activation of caspase-3. It has been previously observed that GA and its derivatives cause caspase-3 activation (Fig. 5). The highest percentage of cells displaying examined features of apoptosis, was observed after 48 hours incubation with GA, 17AEP-GA, and 17DMAG.
Effects of GA and its derivatives on the activity of Caspase-3 in U266 cells. Increase in the number of cells with activated caspase -3 relative to the control after 48 hours exposure to a 100 nM dose of GA and its derivatives. Results are from two independent experiments.
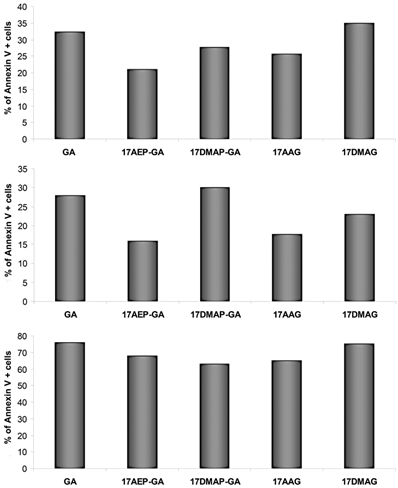
GA and its derivatives induce apoptosis of myeloma cells harvested from patients
CD138+ cells isolated from the BM of patients with MM were analyzed for level of apoptosis. 3 independent experiments were performed. The results of the experiments show the greatest percentage of cells displaying apoptotic features after 48 hours of incubation with GA and 17DMAG (Fig. 6).
Effect of GA on apoptosis of myeloma cells-binding of annexin V. The increase in annexin V binding cells relative to control after 48 hours exposure to 100 nM dose of GA and its derivatives. Results are from analysis of three independent patients.
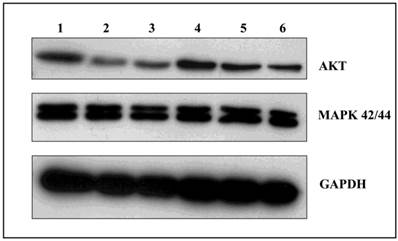
GA and its derivatives decrease the expression of AKT protein
Among the many cellular proteins, regulatory proteins play a very important role in the regulation of the controlled process of death. Three independent experiments were conducted examining the levels of AKT kinase, one of the key proteins that control the process of apoptosis, and MAP 42/44 which has an influence on gene expression, cellular division, cellular differentiation, cellular movement, and apoptosis. Analysis of the activity of AKT kinase and MAP 42/44 were made by incubating U266 cells for 24 and 48 hours with 100 nM of GA and its derivatives. After 24 hours incubation, there were no significant changes in protein expression (data not shown), while after 48 hours the strongest effects was seen in expression of the AKT protein when incubated with GA or 17AEP-GA (Fig. 7). The strongest effects were observed after incubation with GA, 17AEP-GA, and 17DMAG.
Effects of GA and its derivatives on the expression of AKT and MAP. 1-control, 2-GA, 3-17AEP-GA, 4-17DMAP-GA, 5-17AAG, 6-17DMAG.GAPDH-control constitutive protein.
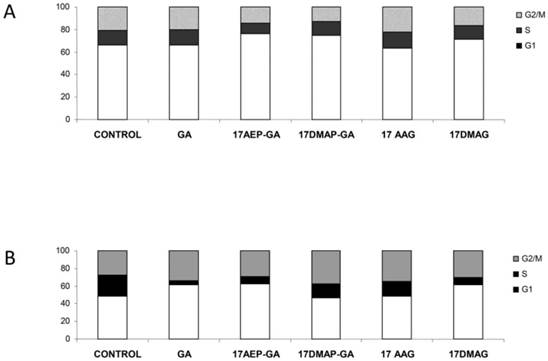
GA and its derivatives cause cell cycle inhibition in myeloma cells
Inhibition of cell growth may also be due to inhibition of the cell cycle. It was found that 100 nM 17AE -GA and 17-DMAP-GA causes abnormalities in the cell cycle. The U266 cells were observed in G1 phase lock, which is manifested by a nearly two-fold increase in the percentage of cells in this phase of the cycle and the simultaneous lowering of the percentage of cells in G2/M. In the case of the INA6 line, inhibition of the cell cycle occurred in the G1 and G2/M phases. Incubation with GA resulted in an increase in the percentage of cells in G1 phase by about 15%, similar to the percentage of cells in the G2/M phase, whereas the number of cells in S phase decreased. A similar mechanism was observed in the case of GA derivatives, the effect with 17AEP-GA and 17DMAG showing the strongest effects. (Fig. 8).
Effects of GA and its derivatives on the cell cycle of MM. Panel A- U266 line, panel B-INA6 line (selected representative experiments)
HSP90 expression in the cells of MM is higher than in normal cells
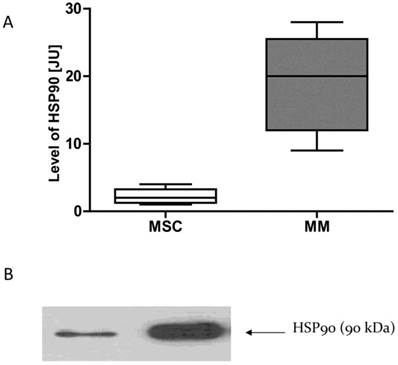
Given that normal cells and neoplastic cells exhibit different sensitivity to the GA and its derivatives, we examined whether this phenomenon may be due to differences in the expression of HSP90. mRNA and proteins were isolated from the MSC cells and MM cells obtained from MM patients. Real-time PCR was used to test the expression of mRNA for HSP90. The amount of HSP90 protein in normal cells and cancer cells were compared using the Western Blot technique. It has been observed that cancer cells have higher expression of HSP90 as compared to normal cells at both the mRNA and protein levels (Fig. 9).
HSP90 expression in normal and malignant cellsA-HSP90 mRNA expression, B-HSP90 protein expression. * p<0.05 compared with the level of HSP90 in normal cells. In the case of normal cells, four samples are included (n=4), while in the case of myeloma cells, 7 samples are included (n=7).
GA and its derivatives reduce MET receptor expression in myeloma cells
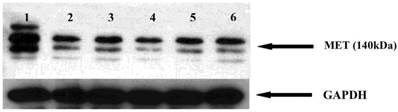
The MET receptor interacts with HSP90. We therefore examined whether HSP90 inhibitors affect the MET receptor expression in myeloma cells. After 48 hours incubation with GA and analogues, the level of MET protein was analyzed by Western blot. It was found that geldanamycin decreases the expression of the MET receptor in the U266 (Fig. 10).
Effects of GA and its derivatives on MET protein expression in myeloma cells. 1-control, 2-GA, 3-17DMAP-GA, 4-17DMAG, 5-17AAG,6-17AEP-GA.
Discussions
GA inhibits the growth of myeloma cells
The anti-tumor effect of GA is primarily due to inhibition of the activity of proteins that promote cell proliferation, such as RAF-1. Additional factors include: reduction in expression of mutant or excessively produced oncogenic proteins, inhibition of the activity of regulatory cell survival proteins, such as AKT, inhibition of the cell cycle, inhibition of angiogenesis and reduction of invasiveness metastatic ability of neoplastic cells [14-18]. In recent years, studies have also demonstrated positive effects of GA when used with other compounds. Parthenolide enhanced geldanamycin-induced changes in apoptosis-related protein levels in ovarian carcinoma cell lines [19]. Wu et al., suggested GA enhances Fisetin induced cytotoxicity via induction of apoptosis in human colonic cancer cells [20]. The present study examined the impact of GA on the proliferation of MM cells, cell apoptosis, cell cycle, and the expression of AKT kinase and the oncogene MET. Previous reports have indicated different sensitivities to GA activity in vitro in a variety of cancers. For glioma cell lines, IC50 (50 % growth inhibition) was estimated at 0.4-3 nM, breast cancer lines reported IC50 at 2-20 nM, small cell lung cancer lines at 50-100 nM, ovarian cancer lines at 2000 nM, and in the case of T-cell leukemia lines between 10-700 nM [21-24]. The biological effect that GA has covers a wide range of intracellular pathways controlled by HSP90. Tumor cells exhibit diverse genetic variation, which results in binding to a different protein profile, and this may affect the susceptibility of these cells to GA [25]. In our study we found that GA inhibits the proliferation of myeloma cells. It has been shown that the tested myeloma cell lines show varying sensitivity to GA. The selected concentration of 100 nM for U266 line does not deviate from the range of dose values determined for GA for other cell lines.
Genetic variability of cancer cells may not only lead to varying degrees of sensitivity to the GA, but also result in different biological effects [25]. It was shown that GA can induce cell cycle arrest at different stages [14], as well as induce apoptosis or cell differentiation [26]. We demonstrated that 24 hour incubation with GA causes earlier signs of apoptosis such as membrane dissymmetry in MM cells. Intracellular proteolytic enzymes, caspases, mobilize apoptosis. It has been found that GA activates caspase-3 in MM cells. This activation of caspase-3 leads to apoptotic death, as evidenced in leukemia cells.GA, through its interactions with HSP90 and ubiquitin, initiates the degradation of many cellular proteins, such as AKT, RAF-1, or CDK-4 [27]. As in the case of the osteosarcoma or neuroblastoma cell lines [16,28], we demonstrated that GA significantly decreases the expression AKT protein in the MM U266 cell line. AKT kinase is a protein that controls the process of programmed cell death. AKT kinase activation leads to the induction of both anti-apoptotic factors such as NF-kB and inhibition of proapoptotic factors such as BAD [29]. Reducing the expression of AKT disrupts the balance of signals that control cell survival and consequently can lead to apoptosis. Thus, it seems that one reason for GA-dependent apoptosis of myeloma cells, may be a reduction in the expression of AKT protein.
Another manifestation of GA activity is cell cycle arrest, wherein the locking mechanism depends on the type of tumor, and very often even the type of cell line. A G2/M phase lock is frequently observed, causing accumulation of cells in this phase due to the inability to complete mitosis [30, 31]. G1 phase locking is less common [32]. Srethapakdi et al. showed that in breast cancer cell lines, the occurrence of locking in the G1 phase, caused by inhibition of HSP90 proteins, correlate with the presence of functional RB protein [32]. In our study we noted that inhibition of the cell cycle occurred in MM cells, but the mechanism of this phenomenon seems to be different for different cells. In the case of the U266 cell line, accumulation was observed in the G1 phase, whereas in the case of the INA6 cell line, we also noted lock in the G2/M phase. It is possible that this phenomenon is due to genetic differences between the cells.
The MET receptor is one of the target proteins of HSP90 [27]. Webb et al demonstrated that GA reduces the MET receptor expression in tumor cells, thereby causing inhibition of HGF-dependent urokinase-type plasminogen activator [18]. Plasmin activation disorders can lead to impairment of the maturation process of the extracellular matrix, and thereby limit the ability of tumor cells to metastasize. Lowering the MET receptor expression may also affect the inhibition of tumor cell migration as demonstrated in the example of small cell lung cancer [23]. In our study, 100 nM of GA resulted in a reduction of MET protein expression in U266 cells.
GA derivatives have similar effects on MM cells as GA
Although encouraging results have been obtained in vitro, animal models of GA show GA to be toxic, particularly to hepatocytes [33]. One reason for this may be the presence of a methoxy group at the 17 carbon of the benzoquinone ring. As this group does not interact with the protein HSP90, many modifications may be made within the substituent [34]. 17AAG, one of the first GA analogues, has proved to be significantly less toxic while maintaining its antineoplastic traits and is now undergoing many clinical trials [35]. Shulte et al. have shown that GA and 17AAG not only have similar biological properties, but they also have comparable antitumor activity [35]. Enthusiasm for potential clinical applications of 17AAG have been weakened because the compound is insoluble in aqueous solutions. Another intensively studied GA analogue is 17DMAG, which appears to have all the advantages of 17AAG, and the additional benefit of excellent solubility in water. Smith et al have demonstrated that 17DMAG is on average 3 times more potent than 17AAG in the inhibition and proliferation of various tumor cell types, including melanoma, bladder cancer, colon cancer, pancreatic cancer, lung cancer, prostate cancer, and breast cancer [36]. In our experiments, we compared GA and its derivatives 17AAG and 17DMAG, already in phase I and phase II clinical trials, as well as 17AEP-GA and 17DMAP-GA [37]. We examined whether GA derivatives, despite their modifications, retain their antitumor properties in MM cells. It was found that the GA derivatives we tested do in fact inhibit the growth of MM cells and their effect increases with the length of exposure. As in the case of GA, analogs initiate apoptosis, resulting in the activation of caspase-3, among other proteins. We also observed a decrease in the expression of the anti-apoptotic protein AKT. Moreover, GA derivatives caused blockade of the cell cycle. This effect was confirmed by examining the expression of MET protein derivatives. On the basis of the above observations, we concluded that the mechanism of action of GA and its derivatives is similar. In the case of U266 cells, significant differences were demonstrated in the susceptibility of these cells to GA and its derivatives. On the basis of the MTS assay, it was concluded that 17DMAG, 17AEP -GA, and 17DMAP-GA have similar anticancer properties as GA.
Although it was found that GA derivatives bind to HSP90 with differing affinities, a correlation between this fact and the ability to inhibit tumor cell growth has not been established. It is possible that the differing effects of the tested inhibitors result from their ability to penetrate into the cell [34]. The differences in the inhibition of MM cell growth, depending on the inhibitor used, were also reflected in the intensity of phenomena such as apoptosis and cell cycle blockade. There have also been differences in the action of inhibitors on protein expression of AKT and MET. Based on our studies in vitro, it can be concluded that 17AEP-GA and 17DMAG are effective inhibitors of HSP90 with properties comparable to GA. 17AAG, in contrast, appears to be the least effective inhibitor of the growth of MM cells.
Studies of apoptosis were also performed on cells derived from patients with myeloma, and after 48 hours of incubation, the largest percentage of apoptotic cells was observed after application of GA and 17DMAG.
To eliminate adverse effects on normal cells, we also compared the level of target proteins for GA and analogs in normal and malignant cells derived from patients with MM. The difference in HSP90 protein and mRNA levels confirmed that normal and malignant cells respond differently to GA.
In conclusion GA and its tested analogues inhibit the growth of MM cells via induction of apoptosis, inhibition of cell cycle progression, and reduction of the expression of the MET receptor. The GA analogues tested, despite their modifications, maintain strong anticancer properties. It is possible that two analogues of GA, 17AEP-GA and 17DMAG, due to their properties may prove to be more effective and safer chemotherapeutic agents than the currently used 17AAG, as reported in literature.
Abbreviations
MM: multiple myeloma; HSP90: heat shock protein 90; HIF1α: factor-1 induced by hypoxia; VEGF: vascular endothelial growth factor; IGF-1R: insulin-like growth factor 1 receptor; MMP-2: matrix metalloproteinase-2; GA: geldanamycin; BM: bone marrow; FBS: fetal bovine serum; PE: phycoerythrin; MSC: mesenchymal stem cells; BM-MSC: mesenchymal stem cells from healthy donors; MMBM -MSC: mesenchymal stem cells from patients with multiple myeloma; DMEM: Dulbecco's modified eagle's medium; 17AAG: 17-Allylamino-17-demethoxygeldanamycin; 17DMAG: 17-(Dimethylaminoethylamino)-17-demethoxygeldanamycin; 17AEP-GA: 17-[2-(Pyrrolidin-1-yl)ethyl] aminno-17-demethoxygeldanamycin; 17DMAP-GA: 17-(Dimethylaminopropylamino)-17-demethoxygeldanamycin; PI: propidium iodide; PBS: phosphate buffered saline; HRP: horse radish peroxidase; qRT-PCR: quantitative polymerase chain reaction in real time.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Rajkumar CV. Treatment of myeloma cure vs control. Mayo Clin Proc. 2008;83:1142-1145
2. Gahrton G. New therapeutic targets in multiple myeloma. Lancet. 2004;364:1648-1649
3. Turner JG, Dawson J, Emmons MF. et al. CRM1 inhibition sensitizes drug resistant human myeloma cells to topoisomerase II and proteasome inhibitors both in vitro and ex vivo. J Canc. 2013;4:614-625
4. Goetz MP, Toft DO, Ames MM. et al. The Hsp90 chaperone complex as a novel target for cancer therapy. Ann Oncol. 2003;14:1169-1176
5. Hanahan D, Weinberg RA. The hallmarks of cancer. Cell. 2000;100:57-70
6. Mosser DD, Morimoto RI. Molecular chaperones and the stress of oncogenesis. Oncogene. 2004;23:2907-2918
7. Hermisson M, Strik H, Rieger J. et al. Expression and functional activity of heat shock proteins in human glioblastoma multiforme. Neurology. 2000;54:1357-1365
8. Yano M, Naito Z, Yokoyama M. et al. Expression of hsp90 and cyclin D1 in human breast cancer. Cancer Lett. 1999;137:45-51
9. Lesko E, Gozdzik J, Kijowski J. et al. HSP90 antagonist, geldanamycin, inhibits proliferation, induces apoptosis and blocks migration of rhabdomyosarcoma cells in vitro and seeding into bone marrow in vivo. Anticancer Drugs. 2007;18:1173-1181
10. Whitesell L, Mimnaugh EG, De Costa B. et al. Inhibition of heat shock protein HSP90-pp60v-src heteroprotein complex formation by benzoquinone ansamycins: essential role for stress proteins in oncogenic transformation. Proc Natl Acad Sci USA. 1994;91:8324-8328
11. Neckers L. Hsp90 inhibitors as novel cancer chemotherapeutic agents. Trends Mol Med. 2002;8:S55-61
12. Powers MV, Workman P. Targeting of multiple signaling pathways by heat shock protein 90 molecular chaperone inhibitors. Endocr Relat Cancer. 2006;13:S125-135
13. Friedenstein AJ, Gorskaja JF, Kulagina NN. Fibroblast precursors in normal and irradiated mouse hematopoietic organs. Exp Hematol. 1976;4:267-274
14. Bedin M, Gaben AM, Saucier C. et al. Geldanamycin, an inhibitor of the chaperone activity of HSP90, induces MAPK-independent cell cycle arrest. Int J Cancer. 2004;109:643-652
15. Blagosklonny MV. Hsp-90-associated oncoproteins: multiple targets of geldanamycin and its analogs. Leukemia. 2002;16:455-462
16. Kim S, Kang J, Hu W. et al. Geldanamycin decreases Raf-1 and Akt levels and induces apoptosis in neuroblastomas. Int J Cancer. 2003;103:352-359
17. Sanderson S, Valenti M, Gowan S. et al. Benzoquinone ansamycin heat shock protein 90 inhibitors modulate multiple functions required for tumor angiogenesis. Mol Cancer Ther. 2006;5:522-532
18. Webb CP, Hose CD, Koochekpour S. et al. The geldanamycins are potent inhibitors of the hepatocyte growth factor/scatter factor-met-urokinase plasminogen activator-plasmin proteolytic network. Cancer Res. 2000;60:342-349
19. Lee CS, Kim YJ, Lee SA. et al. Combined effect of Hsp90 inhibitor Geldanamycin and Parthenolide via Reactive Oxygen Species-mediated apoptotic process on epithelial ovarian cancer cells. Basic Clin Pharmacol Toxicol. 2012;111:173-181
20. Wu MS, Lien GS, Shen SC. et al. HSP90 inhibitors, Geldanamycin and Radicicol, enhance Fisetin-induced cytotoxicity via induction of apoptosis in human colonic cancer cells. Evid Based Complement Alternat Med. 2013:987612. doi: 10.1155/2013/987612
21. Hartmann F, Horak EM, Cho C. et al. Effects of the tyrosine-kinase inhibitor geldanamycin on ligand-induced Her-2/neu activation, receptor expression and proliferation of Her-2-positive malignant cell lines. Int J Cancer. 1997;70:221-229
22. Jones DT, Addison E, North JM. et al. Geldanamycin and herbimycin A induce apoptotic killing of B chronic lymphocytic leukemia cells and augment the cells' sensitivity to cytotoxic drugs. Blood. 2004;103:1855-1861
23. Maulik G, Kijima T, Ma PC. et al. Modulation of the c-Met/hepatocyte growth factor pathway in small cell lung cancer. Clin Cancer Res. 2002;8:620-627
24. Yang J, Yang JM, Iannone M. et al. Disruption of the EF-2 kinase/Hsp90 protein complex: a possible mechanism to inhibit glioblastoma by geldanamycin. Cancer Res. 2001;61:4010-4016
25. Maloney A, Clarke PA, Workman P. Genes and proteins governing the cellular sensitivity to HSP90 inhibitors: a mechanistic perspective. Curr Cancer Drug Targets. 2003;3:331-341
26. Nimmanapalli R, O'Bryan E, Bhalla K. Geldanamycin and its analogue 17-allylamino- 17-demethoxygeldanamycin lowers Bcr-Abl levels and induces apoptosis and differentiation of Bcr-Abl-positive human leukemic blasts. Cancer Res. 2001;61:1799-1804
27. Dai C, Whitesell L. HSP90: a rising star on the horizon of anticancer targets. Future Oncol. 2005;1:529-540
28. Bagatell R, Beliakoff J, David CL. et al. Hsp90 inhibitors deplete key anti-apoptotic proteins in pediatric solid tumor cells and demonstrate synergistic anticancer activity with cisplatin. Int J Cancer. 2005;113:179-188
29. Cheng JQ, Lindsley CW, Cheng GZ. et al. The Akt/PKB pathway: molecular target for cancer drug discovery. Oncogene. 2005;24:7482-7492
30. Kefford R, Millward M, Hersey P. Phase 2 Trial of Tanespimycin (KOS-953), a Heat shock protein-90 (Hsp90) inhibitor in patients with metastatic melanoma. J Clin Oncol. ASCO. 2007;25:8558
31. Pennacchietti S, Michieli P, Galluzzo M. et al. Hypoxia promotes invasive growth by transcriptional activation of the met protooncogene. Cancer Cell. 2003;3:347-361
32. Srethapakdi M, Liu F, Tavorath R. et al. Inhibition of Hsp90 function by ansamycins causes retinoblastoma gene product-dependent G1 arrest. Cancer Res. 2000;60:3940-3946
33. Supko JG, Hickman RL, Grever MR. et al. Preclinical pharmacologic evaluation of geldanamycin as an antitumor agent. Cancer Chemother Pharmacol. 1995;36:305-315
34. Tian ZQ, Liu Y, Zhang D. et al. Synthesis and biological activities of novel 17-aminogeldanamycin derivatives. Bioorg Med Chem. 2004;12:5317-5329
35. Schulte TW, Neckers LM. The benzoquinone ansamycin 17-allylamino-17-demethoxygeldanamycin binds to HSP90 and shares important biologic activities with geldanamycin. Cancer Chemother Pharmacol. 1998;42:273-279
36. Smith DF, Whitesell L, Nair SC. et al. Progesterone receptor structure and function altered by geldanamycin, an hsp90-binding agent. Mol Cell Biol. 1995;15:6804-6812
37. Fukuyo Y, Hunt CR, Horikoshi N. Geldanamycin and its anti-cancer activities. Cancer Letters. 2010;290:24-23
Author contact
![]() Corresponding author: Marcin Majka PhD, Professor, Department of Transplantology, Jagiellonian University, Medical College, Wielicka St 265, Krakow, Poland. mmajkakrakow.pl; tel/fax 126582011
Corresponding author: Marcin Majka PhD, Professor, Department of Transplantology, Jagiellonian University, Medical College, Wielicka St 265, Krakow, Poland. mmajkakrakow.pl; tel/fax 126582011