Impact Factor ISSN: 1837-9664

 Global reach, higher impact
Global reach, higher impactJ Cancer 2014; 5(7):510-517. doi:10.7150/jca.9215 This issue Cite
Research Paper
Glutathione Peroxidase 7 Suppresses Bile Salt-Induced Expression of Pro-Inflammatory Cytokines in Barrett's Carcinogenesis
Dun-Fa Peng1, Tian-Ling Hu1,3, Mohammed Soutto1,3, Abbes Belkhiri1, Wael El-Rifai1,2,3 ![]()
1. Department of Surgery, Vanderbilt University Medical Center, Nashville, TN, USA;
2. Department of Cancer Biology, Vanderbilt University Medical Center, Nashville, TN, USA;
3. Tennessee Valley Healthcare System, Department of Veterans Affairs, Nashville, TN, USA.
Received 2014-3-25; Accepted 2014-5-1; Published 2014-6-10
Abstract
Esophageal adenocarcinoma (EAC) is the most frequent malignancy in the esophagus in the US and its incidence has been rising rapidly in the past few decades. Chronic gastroesophageal reflux disease (GERD), where the esophageal epithelium is abnormally exposed to acid and bile salts, is a pro-inflammatory condition that is the main risk factor for the development of Barrett's esophagus (BE) and its progression to EAC. Glutathione peroxidase 7 (GPX7) is frequently silenced through DNA hypermethylation during Barrett's tumorigenesis. In this study, we investigated the role of GPX7 in regulating the bile salts-induced inflammatory signaling in Barrett's carcinogenesis. Using quantitative real-time PCR (qRT-PCR), we demonstrated a significant induction in the expression levels of pro-inflammatory cytokines (TNF-α, IL-1β, IL-6, and IL-8) and chemokines (CXCL-1 and CXCL-2) in esophageal cells after exposure to acidic (pH4) or neutral (pH7) bile salts. Western blot analysis showed that exposure to acidic and neutral bile salts increased p-NF-κB-p65 (S536) protein levels independent of ROS. Reconstitution of GPX7 expression in EAC cells abolished the increase of p-p65 (S536) protein levels and mRNA expression of cytokines and chemokines upon treatment with acidic and neutral bile salts. Examination of human primary EAC tissues by qRT-PCR demonstrated significant overexpression of cytokines (TNF-α, IL-1β and IL-8) in EAC samples, as compared to normal samples, with significant inverse correlation with GPX7 expression level. Taken together, the loss of GPX7 expression promotes bile salt-induced activation of pro-inflammatory cytokines and chemokines; important contributors to GERD-associated Barrett's carcinogenesis.
Keywords: glutathione, inflammation, cytokine, reflux, GERD, bile salts, Barrett's, cancer, esophagus, NF-κB.
Introduction
Esophageal adenocarcinoma (EAC) is the most frequent malignancy in the esophagus in the US. The incidence rate for EAC has increased 4-10% per year among men since 1976, more rapidly than for any other type of cancer [1]. The current paradigm suggests that EAC develops through chronic gastro-esophageal reflux disease (GERD), Barrett's esophagus (BE), dysplasia, and adenocarcinoma sequence [2-4]. GERD is a condition whereby esophageal epithelium of the lower esophagus is abnormally exposed to a mixture of acid and bile salts [3, 5]. GERD is associated with mucosal injury and inflammation and leads to the development of BE which consists of metaplastic glandular intestinal cells, the main risk factor for the development of EAC [6-8]. The risk of developing EAC is 30 times higher for chronic GERD patients and up to 125 times higher for GERD patients with BE than for the general population [4, 6]. In this regard, EAC is considered an example of inflammation-associated cancer [2].
Chronic inflammation has been consistently linked with human carcinogenesis in several cancer types [9, 10]. Nuclear factor-kappa B (NF-κB) is one of the few key regulatory signaling molecules; the aberrant activation of NF-κB is associated with inflammation and cancer in mouse models and human disease [11-14]. Activation of NF-κB plays an important role in integrating multiple stress stimuli and regulating immune responses [14, 15]. A variety of stimuli such as infections, inflammatory cytokines, UV-irradiation and oxidative stress, can activate NF-κB signaling. Bile acids, in particular, deoxycholic acid has been shown to activate NF-κB pathway [16, 17]. Upon activation of NF-κB by phosphorylation, it translocates to the nucleus where it regulates the transcription of several pro-inflammatory cytokines such as TNF-α, IL-1β, IL-6, and IL-8 and chemokines such as CXCL-1 and CXCL-2 [14, 18].
The glutathione peroxidases (GPXs) are composed of a major antioxidant enzyme family with 8 members, GPX1-GPX8 [19]. We have previously shown that GPX7 has very limited or none of the glutathione peroxidase enzyme activity but can neutralize H2O2 in vitro independent of glutathione and protect normal esophageal epithelia from acidic bile salts-induced oxidative stress, oxidative DNA damage and double strand breaks [20]. GPX7 can alleviate oxidative stress generated from polyunsaturated fatty acid metabolism [21] and may act as an oxidative stress sensor that regulates GRP78 chaperone activity to reduce oxidative stress [22]. A recent discovery showed that GPX7 deficiency in mice leads to systemic oxidative stress, increased tumor incidence and shortened life span [22], consistent with our recent findings showing that GPX7 possesses tumor suppressor functions in EAC [23]. Loss of expression and dysfunction of GPX7 are frequent in EAC and its precancerous lesions [20, 23, 24]. In the present study, we show that GPX7 has a potential role in modulating the expression of bile salts-induced pro-inflammatory cytokines associated with Barrett's carcinogenesis.
Materials and methods
Cell lines
The immortalized human normal esophageal squamous cell line (HET1A) and the esophageal adenocarcinoma cell lines (FLO-1 and OE33) were obtained from American Type Culture Collection (ATCC, Manassas, VA) and were cultured in Dulbecco's modified Eagle's medium (DMEM), supplemented with 10% fetal bovine serum and antibiotics (Invitrogen, Carlsbad, CA). Immortalized Barrett's esophagus cell line (BAR-T, a kind gift from Dr. Rhonda Souza) was cultured with epithelial cell medium 2 (ScienCell, Carlsbad, CA), supplemented with 5% fetal bovine serum and antibiotics on primaria plates and flasks (BD Biosciences, Bedford, MA). All cell lines were grown at 37°C in 5% carbon dioxide.
Chemicals
A bile salts cocktail consisting of an equal molar mixture of sodium salts of glycocholic acid, taurocholic acid, glycodeoxycholic acid, glycochenodeoxycholic acid, and deoxycholic acid was prepared in reflection to the mixture of bile acids in distal esophagus during gastro-esophageal reflux disease, as previously reported [25]. In all experiments, we used 100 µM of the bile salts cocktail (20 μM of each of the above bile salts) final concentration in either pH4 or pH7 medium.
Tissue samples
55 frozen tissue samples (30 EAC, 25 normal esophagus samples) were collected. All tissue samples were de-identified and obtained from the archives of pathology at Vanderbilt University (Nashville, TN) and from the National Cancer Institute Cooperative Human Tissue Network. The use of specimens from the tissue repository was approved by the Vanderbilt Institutional Review Board. All EAC originated from the lower esophagus or gastro-esophageal junction corresponding to AEG type 1, as previously described [26].
Quantitative real-time RT-PCR analysis of gene expression
Total RNA was isolated using the RNeasy mini kit (Qiagen, Valencia, CA). Single-stranded complementary DNA was subsequently synthesized from RNA using the iScript cDNA synthesis kit (Bio-Rad, Hercules, CA). Quantitative real-time RT-PCR (qRT-PCR) was performed using a CFX Connect real-time system (Bio-Rad) with the threshold cycle number determined by the use of Bio-Rad's CFX manager 3.0 software. The sequence of primers is provided in Table 1. For primary tissues, the mRNA expression result was first normalized to the average value of HPRT1 of the same sample, and then compared to the value of the matched normal or the average normal if the matched normal sample was not available. For in vitro assays, the mRNA expression result was first normalized to the average value of HPRT1 of the same sample then compared to the value of the untreated control cells [24, 27].
Primers used in quantitative real time RT-PCR.
| Gene name | RefSeq No. | Forward primer | Reserve primer | Product size (bp) |
|---|---|---|---|---|
| GPX7 | NM_015696 | AACTGGTGTCGCTGGAGAAG | AAACTGGTTGCAGGGGAAG | 161 |
| TNF | NM_000594 | TCTTCTCCTTCCTGATCGTG | GCCAGAGGGCTGATTAGAGA | 118 |
| IL-6 | NM_000600 | CCCTGAGAAAGGAGACATGTAA | TCTTTTTCAGCCATCTTTGGA | 100 |
| IL-8 | NM_000584 | TCCTGATTTCTGCAGCTCTGT | AAATTTGGGGTGGAAAGGTT | 107 |
| IL-1β | NM_000576 | CAGCCAATCTTCATTGCTCA | GGCCATCAGCTTCAAAGAAC | 121 |
| CXCL1 | NM_001511 | GAAAGCTTGCCTCAATCCTG | CACCAGTGAGCTTCCTCCTC | 107 |
| CXCL2 | NM_002089.3 | CTCAAGAATGGGCAGAAAGC | CTTCAGGAACAGCCACCAAT | 134 |
| HPRT1 | NM_000194 | TTGGAAAGGGTGTTTATTCCTCA | TCCAGCAGGTCAGCAAAGAA | 146 |
Construction of GPX7 expression plasmids
A full length of GPX7 coding sequence with Flag tag was amplified from normal cDNA by PCR using Platinum PCR SuperMix High Fidelity (Invitrogen) and was cloned into PcDNA 3.1 and pACCMV.pLpA plasmids, as previously described [20]. For stable transfection, cells were transfected with PcDNA-GPX7 or PcDNA 3.1 control plasmids. Cells were selected in G418 (600 µg/ml) for two weeks. For transient expression of GPX7, cells were infected with 5 MOI (multiplicity of infection) of GPX7 expression or control adenoviruses. 48h after infection cells were used for experiments.
Western blotting analysis
Western blot analysis was performed using standard protocol. Protein concentration of lysates with proteinase and phosphatase inhibitors was measured by a Bio-Rad protein assay using a FLUO Star OPTIMA microplate reader (BMG). Equal amounts of proteins were loaded onto 12.5% SDS-PAGE and transferred to nitrocellulose membrane. Anti-GPX7 antibody was purchased from Proteintech Group (Chicago, IL). Anti-p65 and anti-p-p65 (S536) antibodies were obtained from Cell Signaling (Danvers, MA). The quantification of the protein bands intensity was performed using ImageJ software (NIH) and was normalized to the corresponding band density of the β-actin of the same samples.
Statistical analysis
Data are expressed as the mean ± SD for parametric data. Unpaired Student t test was performed for two independent variables. Spearman's correlation analysis was used to analyze the correlation between GPX7 expression and TNF-α, IL-1β, or IL-8 expression in esophageal tissues. All statistical analyses were done using GraphPad Prism 4 software. For all analyses, P≤.05 is considered statistically significant.
Results
Acidic and neutral bile salts induce the expression of pro-inflammatory cytokines and chemokines in esophageal cells
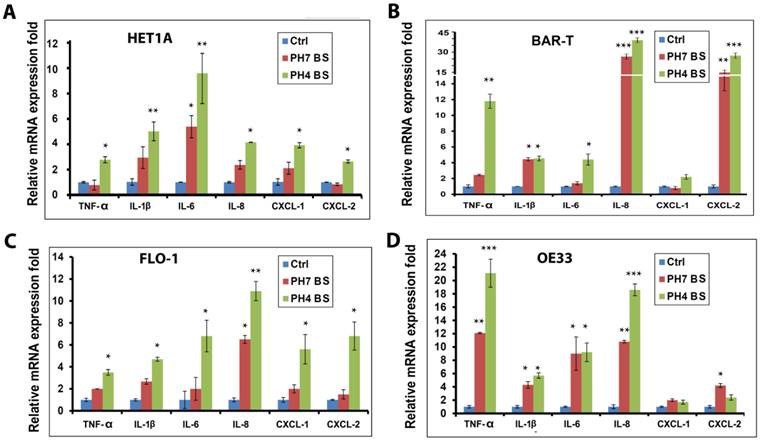
The bile salts and gastric acid components of GERD induce mucosal damage and inflammation in the distal region of the esophagus. To mimic the pathophysiological condition in GERD, a bile salts cocktail was prepared using a mixture of 5 bile salts that mimic human conditions, as described in Materials and Methods section. To investigate if this bile salts cocktail can induce pro-inflammatory cytokines and chemokines, we treated normal esophageal squamous cells (HET1A), Barrett's esophageal cells (BAR-T), and esophageal adenocarcinoma cells (FLO-1 and OE33) with the cocktail at pH4 (mimicking GERD) or pH7 to exclude the acid effects. Interestingly, both treatment conditions induced a significant increase in mRNA expression levels of major cytokines (TNF-α, IL-1β, IL-6, and IL-8) and chemokines (CXCL-1 and CXCL-2) as compared to untreated control cells (Figure 1A-D), suggesting bile salts as the major pro-inflammatory component of GERD. Nonetheless, a relatively higher expression for most of the cytokines and chemokines was observed at the acidic conditions, suggesting an additive effect of acid (Figure 1A-D).
Acidic (pH4) and neutral (pH7) bile salts up-regulate cytokines and chemokines expression in esophageal cells. HET1A (A, normal esophageal squamous cells), BAR-T (B, Barrett's esophagus cells), FLO-1 (C, EAC cells), and OE33 (D, EAC cells) were treated with acidic (pH4) and neutral (pH7) bile salts for 10 min, qRT PCR was used to analyze mRNA expression of the cytokines (TNF-α, IL-1β, IL-6, and IL-8), and chemokines (CXCL-1 and CXCL-2). pH4 BS, acidic bile salts; pH7 BS, neutral bile salts; Ctrl, control untreated. *P<.05, **P<.01, ***P<.001.
Acidic and neutral bile salts mediate phosphorylation and activation of NF-κB in esophageal cells
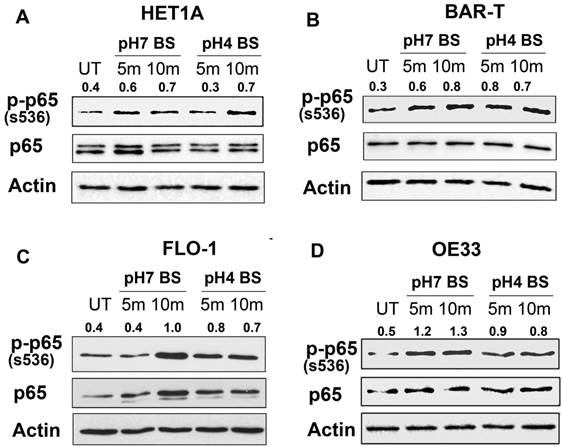
Because the pro-inflammatory cytokines and chemokines are known transcriptional targets of NF-κB, we next investigated whether bile salts could activate NF-κB signaling. Our Western blot analysis data indicated that treatment of esophageal cells with acidic or neutral bile salts induced a marked increase in p-p65 (S536) protein levels (Figure 2A-D). Interestingly, we did not observe significant differences in the levels of p-p65 (S536) protein between acidic and neutral bile salts treatment conditions (Figure 2A-D). This finding is in agreement with our observation of induction of cytokines and chemokines by bile salts. It further supports our conclusion that bile salts components, and not acidity, are the main mediators of the pro-inflammatory signaling in Barrett's cells.
Acidic (pH4) and neutral (pH7) bile salts induce activation of NF-κB in esophageal cells. HET1A (A), BAR-T (B), FLO-1 (C) and OE33 (D) cells were treated with acidic (pH4) and neutral (pH7) bile salts for 10 min. Cells were then harvested and Western blot analysis was used to determine the protein level of p-p65 (S536). The relative protein band density of p-p65 (S536) was normalized to β-actin.
Bile salts-induced activation of NF-κB signaling is largely independent of ROS
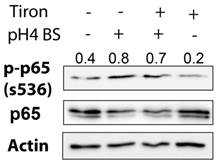
Previous studies have shown that activation of NF-κB could occur through reactive oxygen species (ROS) [28]. To examine if ROS mediates the activation of NF-κB by bile salts in esophageal cells, we utilized Tiron compound, a universal ROS scavenger [29]. Treatment of OE33 cells with Tiron (10 mM) alone significantly diminished the basal protein expression of p-p65 (S536) as compared to untreated control cells (Figure 3). However, pre-treatment of the cells with Tiron (1h prior to bile salts exposure) failed to block the induction of p-p65 (S536) protein expression by bile salts (Figure 3). These results indicated that ROS plays a minimal role, if any, in the activation of NF-κB pathway by bile salts.
Bile salts-induced activation of NF-κB is independent of ROS. OE33 cells were treated with acidic (pH4) bile salts (pH4 BS, 100 µM for 10 min), Tiron (10 mM for 1 h) or Tiron (10 mM for 1 h) followed by acidic bile salts (pH4 BS, 100 µM for 10 min). Western blot analysis was used to determine the p-p65 (S536) protein level. The relative protein band density of p-p65 (S536) was normalized to β-actin.
GPX7 expression suppresses the bile salts-induced activation of NF-κB and the up-regulation of the cytokines and chemokines
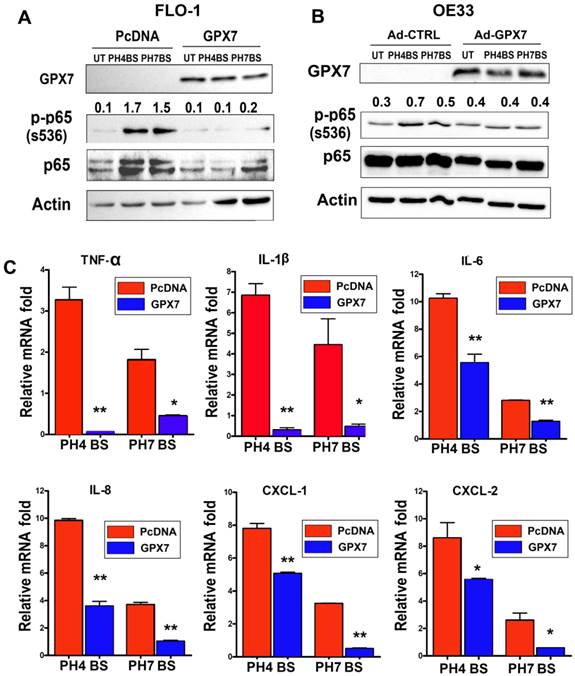
We have previously shown that GPX7 plays a tumor suppressor function in esophageal adenocarcinoma [23]. Aberrant activation of NF-κB has been associated with inflammation and tumor development and progression [11, 30, 31]. Therefore, we next examined if GPX7 could suppress the NF-κB activation induced by exposure to bile salts. As expected, treatment of FLO-1 cells with bile salts induced a marked increase in p-p65 (S536) protein level as compared to untreated control cells (Figure 4A). However, the reconstitution of GPX7 expression abrogated the bile salts-induced increase of p-p65 (S536) protein level in FLO-1 cells (Figure 4A). These results were confirmed in a different esophageal adenocarcinoma cell model (OE33) (Figure 4B). The qRT-PCR data demonstrated that expression of NF-κB target genes, cytokines (TNF-α, IL-1β, IL-6, and IL-8) and chemokines (CXCL-1and CXCL-2), were significantly suppressed in FLO-1 cells stably expressing GPX7 as compared to control cells (Figure 4 C).
GPX7 suppresses bile salts-induced activation of NF-κB and expression of cytokines and chemokines. FLO-1 cells (A) stably expressing GPX7 (GPX7) or control plasmid (PcDNA), and OE33 cells (B) transiently expressing GPX7 (Ad-GPX7) or control (Ad-CTRL) were treated with acidic (pH4) or neutral (pH7) bile salts for 10 minutes. Western blot analysis was used to determine the p-p65 (S536) protein level. The relative protein band density of p-p65 (S536) was normalized to β-actin. C) qRT-PCR analysis of gene expression of NF-κB target genes (TNF-α, IL-1β, IL-6, IL-8, CXCL1, and CXCL2) in FLO-1 cells stably expressing GPX7 (GPX7) or control plasmid (PcDNA) after exposure to acidic (pH4) or neutral (pH7) bile salts. *P<.05, **P<.01.
GPX7 expression is inversely correlated with expression of pro-inflammatory cytokines in human esophageal tissues
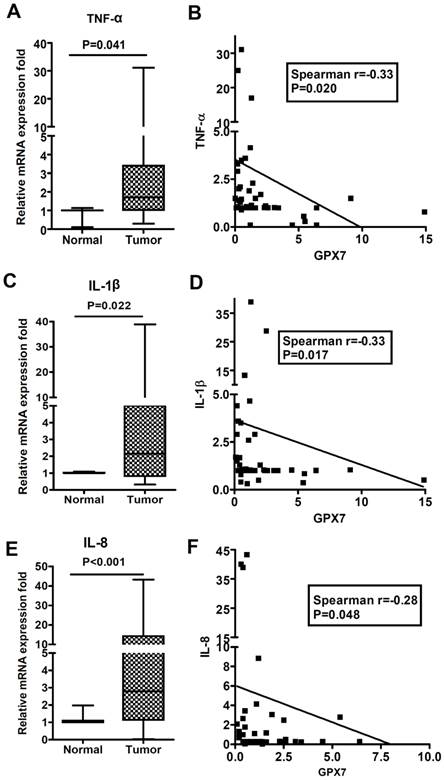
We have previously reported that GPX7 is frequently silenced in Barrett's carcinogenesis through location-specific promoter DNA hypermethylation [23, 24]. We next examined if the loss of GPX7 expression correlates with the major pro-inflammatory cytokines associated with NF-κB activation in EAC. Using qRT-PCR, we evaluated the gene expression of TNF-α, IL-1β and IL-8 in a panel of normal esophageal and EAC samples. We found that expression of these cytokines was significantly up-regulated in EAC as compared to normal esophageal samples (Figure 5A, C and E). Notably, GPX7 expression was significantly inversely correlated with TNF-α expression (Figure 5B, r=-0.33, P=0.020), IL-1β expression (Figure 5D, r=-0.33, P=0.017), and IL-8 expression (Figure 5F, r=-0.28, P=0.048).
GPX7 expression is inversely correlated with TNF-α, IL-1β and IL-8 expression in esophageal tissues. qRT-PCR analysis was performed to determine mRNA expression levels of TNF-α, IL-1β, IL-8, and GPX7 in a panel of human esophageal adenocarcinoma and normal esophageal tissue samples. The relative fold mRNA expression is shown for TNF-α (A), IL-1β (C), and IL-8 (E) in normal and EAC samples. The Spearman rank correlation analysis is shown for each gene (right panels). An inverse correlation between GPX7 and TNF-α (B, r=-0.33, P=.020), IL-1β (D, r=-0.33, P=.017), and IL-8 (F, r=-0.28, P=.048) is shown.
Discussion
In this study, we have shown that both acidic (pH4) and neutral (pH7) bile salts induce significant up-regulation of pro-inflammatory cytokines in esophageal cells through activation of NF-κB pathway. We also show that GPX7 can suppress the up-regulation of these cytokines through inhibiting the bile salts-induced phosphorylation and activation of NF-κB-p65. These findings suggest that bile salts components, and not acid, are the critical factors that mediate the pro-inflammatory signaling in esophageal cells in GERD conditions. For the first time, our data demonstrated that GPX7 can suppress bile salts-induced inflammatory responses and expression of cytokines and chemokines associated with Barrett's carcinogenesis.
Chronic inflammation has long been associated with carcinogenesis through sustaining pro-inflammatory oncogenic signaling, angiogenesis, and immune suppression [9, 10]. This has been further supported by the observed reduced risk of many cancers following the long-term use of non-steroidal anti-inflammatory drugs (NSAIDs) [32]. EAC is developed from Barrett's esophagus resulting from GERD-associated chronic mucosal injury and inflammation in the distal region of the esophagus [2, 3]. Epidemiological studies implied a decrease in the risk of cancer development in patients with BE with the use of aspirin and NSAIDs [33, 34]. We have observed a significant up-regulation of pro-inflammatory cytokines such as TNF-α, IL-1β and IL-8 in human EAC tissue samples. We also found that acidic (pH 4.0) and neutral (pH 7.0) bile salts could induce up-regulation of pro-inflammatory cytokines and chemokines (TNF-α, IL-1β, IL-6, IL-8, CXCL-1, and CXCL-2) in esophageal normal squamous, Barrett's, and adenocarcinoma cells, albeit at lower levels at pH 7.0. These genes are known to promote carcinogenesis in several organs [9, 11, 35]; therefore they may participate in initiating BE or promoting the progression to EAC [2, 36]. These results also suggest that anti-acid therapy such as administering proton pump inhibitors to GERD patients [37-39] alone may not be sufficient to reduce the risk associated with bile salts components and calls for novel treatment that could alter the composition of the refluxate. In fact, in spite of the widespread use of proton pump inhibitors, the incidence of EAC has been steadily rising during the last three decades [40-42]. Recent studies have suggested that hydrophilic urodeoxycholic acid (UDCA) treatment can alter the composition of bile salts in the reflux and block bile-induced NF-κB; therefore UDCA could have a therapeutic window in treatment of BE and/or prevention of Barrett's-associated carcinogenesis [43].
NF-κB is a key regulator of the inflammatory process that has been shown to be activated in EAC [8, 15, 31]. Because the above cytokines and chemokines are known target genes of activated NF-κB, we sought to identify if the bile salts-induced up-regulation of cytokines and chemokines is attributable to the activation of NF-κB. Our data show that both acidic (pH4) and neutral (pH7) bile salts could rapidly induce activation of NF-κB as demonstrated by increased p-p65 (S536) after only 5-10 minutes exposure to the bile salts. Although exposure to acidic bile salts induced higher levels of gene expression of the majority of cytokines and chemokines than neutral bile salts, we did not observe a marked difference in the p-p65 (S536) protein level between the two treatments. These results suggest that, in addition to NF-κB, other molecular signaling such as STAT3 may also be selectively activated by acidic bile salts which could account for the up-regulation of some cytokines and chemokines as compared to neutral bile salts.
Bile salts have been shown to induce intracellular ROS [12, 20], and ROS could induce activation of NF-κB [28, 44]. We investigated if the observed bile salts-induced activation of NF-κB is mediated by ROS. Our data showed that Tiron (a ROS scavenger) alone reduced p-p65 protein level in esophageal cells, indicating that ROS is capable of inducing activation of NF-κB. However, pretreatment of cells with Tiron failed to block the activation of NF-κB by bile salts. These results clearly indicate that ROS plays a minimal role, if any, in the bile salts-induced activation of NF-κB. Of note, in our cell model, we applied short time exposure (5-10 min) of bile salts (100 µM) to the cells to mimic the pathophysiological condition of GERD. However, our results cannot exclude the possibility that ROS plays a larger role in activation of the NF-κB when treating cells with bile salts at higher concentration or longer time. To elucidate the molecular mechanisms by which bile salts activate the NF-κB pathway further investigation is required.
GPX7 is a recent member of the antioxidant enzyme family, glutathione peroxidases. We have recently reported that GPX7 protects esophageal epithelia from acidic bile salts-induced oxidative DNA damage and double strand breaks through modulating intracellular ROS [20]. We also demonstrated that GPX7 has a tumor suppressor function that is silenced through location specific DNA hypermethylation in Barrett's carcinogenesis [23]. In the present study, we show that GPX7 can suppress the bile salts-induced activation of NF-κB and up-regulation of pro-inflammatory, pro-tumor cytokines and chemokines, further supporting a tumor suppressor function of GPX7 in EAC. Interestingly, a recent study has shown GPX3 hypermethylation and underexpression in human colon cancer. This study demonstrated that GPX3-deficient mouse model exhibited increased inflammation and tumor number, suggesting that loss of GPX3 is involved in inflammatory colonic tumorigenesis [45]. Of major importance, the fact that neutral bile salts are still capable of activating NF-κB pathway and up-regulating pro-inflammatory cytokines and chemokines in esophageal epithelium, strongly suggests that strategies focusing on reducing acids only in GERD patients may not be sufficient to prevent the development of BE and/or progression to EAC. This may also explain the sustained increase in the incidence of EAC in the Western population [46, 47]. In this context, novel strategies that can restore and/or induce GPX7 expression need to be developed to abrogate the reflux-induced activation of NF-κB and pro-inflammatory signaling by bile salts.
In summary, our studies suggest that the down-regulation of GPX7 expression in Barrett's tumorigenesis may trigger bile salts-induced activation of pro-inflammatory NF-κB signaling and Barrett's tumorigenesis.
Abbreviations
GPX, Glutathione peroxidase; NF-κB, Nuclear factor-kappa B; EAC, esophageal adenocarcinoma; BE, Barrett's esophagus; GERD, gastro-esophageal reflux disease; qRT-PCR, quantitative real-time PCR (qRT-PCR); ROS, reactive oxygen species; NSAID, non-steroidal anti-inflammatory drug; TNF-α, tumor necrosis factor α; IL-6, interleukin-6; IL-8, interleukin-8; IL-1β, interleukin-1β; CXCL-1, chemokine (C-X-C motif) ligand 1; CXCL-2, chemokine (C-X-C motif) ligand 2; STAT3, signal transducer and activator of transcription 3; MOI, multiplicity of infection.
Acknowledgements
This study was supported by grants from the National Institute of Health; R01CA106176, Vanderbilt SPORE in Gastrointestinal Cancer (P50 CA95103), Vanderbilt Ingram Cancer Center (P30 CA68485) and the Vanderbilt Digestive Disease Research Center (DK058404); and Department of Veterans Affairs. The contents of this work are solely the responsibility of the authors and do not necessarily represent the official views of the National Cancer Institute, Department of Veterans Affairs, or Vanderbilt University.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Parkin DM, Pisani P, Ferlay J. Global cancer statistics. CA Cancer J Clin. 1999;49:33-64
2. Picardo SL, Maher SG, O'Sullivan JN, Reynolds JV. Barrett's to oesophageal cancer sequence: a model of inflammatory-driven upper gastrointestinal cancer. Dig Surg. 2012;29:251-60 doi:10.1159/000341498
3. Shaheen NJ, Richter JE. Barrett's oesophagus. Lancet. 2009;373:850-61 doi:10.1016/S0140-6736(09)60487-6
4. O'Connor JB, Falk GW, Richter JE. The incidence of adenocarcinoma and dysplasia in Barrett's esophagus: report on the Cleveland Clinic Barrett's Esophagus Registry. Am J Gastroenterol. 1999;94:2037-42 doi:10.1111/j.1572-0241.1999.01275.x
5. Spechler SJ, Goyal RK. Barrett's esophagus. N Engl J Med. 1986;315:362-71 doi:10.1056/NEJM198608073150605
6. Gilbert EW, Luna RA, Harrison VL, Hunter JG. Barrett's esophagus: a review of the literature. J Gastrointest Surg. 2011;15:708-18 doi:10.1007/s11605-011-1485-y
7. Zhang HY, Spechler SJ, Souza RF. Esophageal adenocarcinoma arising in Barrett esophagus. Cancer Lett. 2009;275:170-7 doi:10.1016/j.canlet.2008.07.006
8. Abdel-Latif MM, O'Riordan J, Windle HJ, Carton E, Ravi N, Kelleher D. et al. NF-kappaB activation in esophageal adenocarcinoma: relationship to Barrett's metaplasia, survival, and response to neoadjuvant chemoradiotherapy. Ann Surg. 2004;239:491-500
9. Coussens LM, Werb Z. Inflammation and cancer. Nature. 2002;420:860-7 doi:10.1038/nature01322
10. Balkwill F, Mantovani A. Inflammation and cancer: back to Virchow? Lancet. 2001;357:539-45 doi:10.1016/S0140-6736(00)04046-0
11. Karin M. Nuclear factor-kappaB in cancer development and progression. Nature. 2006;441:431-6 doi:10.1038/nature04870
12. Katsha A, Soutto M, Sehdev V, Peng D, Washington MK, Piazuelo MB. et al. Aurora Kinase A Promotes Inflammation and Tumorigenesis in Mice and Human Gastric Neoplasia. Gastroenterology. 2013 doi:10.1053/j.gastro.2013.08.050
13. Soutto M, Belkhiri A, Piazuelo MB, Schneider BG, Peng D, Jiang A. et al. Loss of TFF1 is associated with activation of NF-kappaB-mediated inflammation and gastric neoplasia in mice and humans. J Clin Invest. 2011;121:1753-67 doi:10.1172/JCI43922
14. Karin M. NF-kappaB and cancer: mechanisms and targets. Mol Carcinog. 2006;45:355-61 doi:10.1002/mc.20217
15. Barnes PJ, Karin M. Nuclear factor-kappaB: a pivotal transcription factor in chronic inflammatory diseases. N Engl J Med. 1997;336:1066-71 doi:10.1056/NEJM199704103361506
16. Huo X, Juergens S, Zhang X, Rezaei D, Yu C, Strauch ED. et al. Deoxycholic acid causes DNA damage while inducing apoptotic resistance through NF-kappaB activation in benign Barrett's epithelial cells. Am J Physiol Gastrointest Liver Physiol. 2011;301:G278-86 doi:10.1152/ajpgi.00092.2011
17. Debruyne PR, Witek M, Gong L, Birbe R, Chervoneva I, Jin T. et al. Bile acids induce ectopic expression of intestinal guanylyl cyclase C Through nuclear factor-kappaB and Cdx2 in human esophageal cells. Gastroenterology. 2006;130:1191-206 doi:10.1053/j.gastro.2005.12.032
18. Naugler WE, Karin M. NF-kappaB and cancer-identifying targets and mechanisms. Curr Opin Genet Dev. 2008;18:19-26 doi:10.1016/j.gde.2008.01.020
19. Brigelius-Flohe R, Maiorino M. Glutathione peroxidases. Biochim Biophys Acta. 2013;1830:3289-303 doi:10.1016/j.bbagen.2012.11.020
20. Peng D, Belkhiri A, Hu T, Chaturvedi R, Asim M, Wilson KT. et al. Glutathione peroxidase 7 protects against oxidative DNA damage in oesophageal cells. Gut. 2012;61:1250-60 doi:10.1136/gutjnl-2011-301078
21. Utomo A, Jiang X, Furuta S, Yun J, Levin DS, Wang YC. et al. Identification of a novel putative non-selenocysteine containing phospholipid hydroperoxide glutathione peroxidase (NPGPx) essential for alleviating oxidative stress generated from polyunsaturated fatty acids in breast cancer cells. J Biol Chem. 2004;279:43522-9 doi:10.1074/jbc.M407141200
22. Wei PC, Hsieh YH, Su MI, Jiang X, Hsu PH, Lo WT. et al. Loss of the oxidative stress sensor NPGPx compromises GRP78 chaperone activity and induces systemic disease. Mol Cell. 2012;48:747-59 doi:10.1016/j.molcel.2012.10.007
23. Peng D, Hu T, Soutto M, Belkhiri A, Zaika A, El-Rifai W. Glutathione peroxidase 7 has potential tumour suppressor functions that are silenced by location-specific methylation in oesophageal adenocarcinoma. Gut. 2013 doi:10.1136/gutjnl-2013-304612
24. Peng DF, Razvi M, Chen H, Washington K, Roessner A, Schneider-Stock R. et al. DNA hypermethylation regulates the expression of members of the Mu-class glutathione S-transferases and glutathione peroxidases in Barrett's adenocarcinoma. Gut. 2009;58:5-15 doi:10.1136/gut.2007.146290
25. Kauer WK, Peters JH, DeMeester TR, Feussner H, Ireland AP, Stein HJ. et al. Composition and concentration of bile acid reflux into the esophagus of patients with gastroesophageal reflux disease. Surgery. 1997;122:874-81
26. Siewert JR, Feith M, Stein HJ. Biologic and clinical variations of adenocarcinoma at the esophago-gastric junction: relevance of a topographic-anatomic subclassification. J Surg Oncol. 2005;90:139-46
27. El-Rifai W, Moskaluk CA, Abdrabbo MK, Harper J, Yoshida C, Riggins GJ. et al. Gastric cancers overexpress S100A calcium-binding proteins. Cancer Res. 2002;62:6823-6
28. Pyo CW, Yang YL, Yoo NK, Choi SY. Reactive oxygen species activate HIV long terminal repeat via post-translational control of NF-kappaB. Biochemical and biophysical research communications. 2008;376:180-5 doi:10.1016/j.bbrc.2008.08.114
29. Han YH, Park WH. Tiron, a ROS scavenger, protects human lung cancer Calu-6 cells against antimycin A-induced cell death. Oncol Rep. 2009;21:253-61
30. Okamoto T, Sanda T, Asamitsu K. NF-kappa B signaling and carcinogenesis. Curr Pharm Des. 2007;13:447-62
31. Tak PP, Firestein GS. NF-kappaB: a key role in inflammatory diseases. J Clin Invest. 2001;107:7-11 doi:10.1172/JCI11830
32. Baron JA, Sandler RS. Nonsteroidal anti-inflammatory drugs and cancer prevention. Annu Rev Med. 2000;51:511-23 doi:10.1146/annurev.med.51.1.511
33. Wang F, Lv ZS, Fu YK. Nonsteroidal anti-inflammatory drugs and esophageal inflammation - Barrett's esophagus - adenocarcinoma sequence: a meta-analysis. Dis Esophagus. 2010; doi:10.1111/j.1442-2050. 2010 01153.x
34. Corley DA, Kerlikowske K, Verma R, Buffler P. Protective association of aspirin/NSAIDs and esophageal cancer: a systematic review and meta-analysis. Gastroenterology. 2003;124:47-56 doi:10.1053/gast.2003.50008
35. Aivaliotis IL, Pateras IS, Papaioannou M, Glytsou C, Kontzoglou K, Johnson EO. et al. How do cytokines trigger genomic instability? Journal of biomedicine & biotechnology. 2012;2012:536761. doi:10.1155/2012/536761
36. Poehlmann A, Kuester D, Malfertheiner P, Guenther T, Roessner A. Inflammation and Barrett's carcinogenesis. Pathol Res Pract. 2012;208:269-80 doi:10.1016/j.prp.2012.03.007
37. Kastelein F, Spaander MC, Steyerberg EW, Biermann K, Valkhoff VE, Kuipers EJ. et al. Proton pump inhibitors reduce the risk of neoplastic progression in patients with Barrett's esophagus. Clin Gastroenterol Hepatol. 2013;11:382-8 doi:10.1016/j.cgh.2012.11.014
38. Lada MJ, Nieman DR, Han M, Timratana P, Alsalahi O, Peyre CG. et al. Gastroesophageal reflux disease, proton-pump inhibitor use and Barrett's esophagus in esophageal adenocarcinoma: Trends revisited. Surgery. 2013;154:856-66 doi:10.1016/j.surg.2013.07.020
39. Nguyen DM, Richardson P, El-Serag HB. Medications (NSAIDs, statins, proton pump inhibitors) and the risk of esophageal adenocarcinoma in patients with Barrett's esophagus. Gastroenterology. 2010;138:2260-6 doi:10.1053/j.gastro.2010.02.045
40. Goldman A, Condon A, Adler E, Minnella M, Bernstein C, Bernstein H. et al. Protective effects of glycoursodeoxycholic acid in Barrett's esophagus cells. Diseases of the esophagus: official journal of the International Society for Diseases of the Esophagus / ISDE. 2010;23:83-93 doi:10.1111/j.1442-2050.2009.00993.x
41. Bozikas A, Marsman WA, Rosmolen WD, van Baal JW, Kulik W, ten Kate FJ. et al. The effect of oral administration of ursodeoxycholic acid and high-dose proton pump inhibitors on the histology of Barrett's esophagus. Diseases of the esophagus: official journal of the International Society for Diseases of the Esophagus / ISDE. 2008;21:346-54 doi:10.1111/j.1442-2050.2007.00782.x
42. Jurgens S, Meyer F, Spechler SJ, Souza R. The role of bile acids in the neoplastic progression of Barrett's esophagus - a short representative overview. Z Gastroenterol. 2012;50:1028-34 doi:10.1055/s-0032-1312922
43. Cronin J, Williams L, McAdam E, Eltahir Z, Griffiths P, Baxter J. et al. The role of secondary bile acids in neoplastic development in the oesophagus. Biochemical Society transactions. 2010;38:337-42 doi:10.1042/BST0380337
44. Zhang R, Yin X, Shi H, Wu J, Shakya P, Liu D. et al. Adiponectin Modulates DCA-Induced Inflammation via the ROS/NF-Kappa B Signaling Pathway in Esophageal Adenocarcinoma Cells. Dig Dis Sci. 2013 doi:10.1007/s10620-013-2877-5
45. Barrett CW, Ning W, Chen X, Smith JJ, Washington MK, Hill KE. et al. Tumor suppressor function of the plasma glutathione peroxidase gpx3 in colitis-associated carcinoma. Cancer Res. 2013;73:1245-55 doi:10.1158/0008-5472.CAN-12-3150
46. Devesa SS, Blot WJ, Fraumeni JF Jr. Changing patterns in the incidence of esophageal and gastric carcinoma in the United States. Cancer. 1998;83:2049-53
47. Forman D. Re: The role of overdiagnosis and reclassification in the marked increase of esophageal adenocarcinoma incidence. J Natl Cancer Inst. 2005;97:1013-4
Author contact
![]() Corresponding author: Wael El-Rifai, M.D., Ph.D., Vanderbilt University Medical Center, 760 Preston Research Bldg., 2220 Pierce Avenue, Nashville, TN 37232, E-mail: wael.el-rifaiedu, Phone: 615-322 7934, Fax: 615-322-7852.
Corresponding author: Wael El-Rifai, M.D., Ph.D., Vanderbilt University Medical Center, 760 Preston Research Bldg., 2220 Pierce Avenue, Nashville, TN 37232, E-mail: wael.el-rifaiedu, Phone: 615-322 7934, Fax: 615-322-7852.