Impact Factor ISSN: 1837-9664

 Global reach, higher impact
Global reach, higher impactJ Cancer 2014; 5(7):598-608. doi:10.7150/jca.8052 This issue Cite
Research Paper
Newly Identified Cancer-Associated Role of Human Neuronal Growth Regulator 1 (NEGR1)
Hyejin Kim1, Ji-Sook Hwang1, Bogman Lee2, Jinpyo Hong3, Soojin Lee1 ![]()
1. Department of Microbiology and Molecular Biology, Chungnam National University, Daejeon, Republic of Korea,
2. LG Life Sciences, Ltd., R&D Park, Moonji-dong, Daejeon, Republic of Korea,
3. Department of oral physiology, School of Dentistry, Seoul National University, Seoul, Republic of Korea.
Received 2013-11-5; Accepted 2014-5-16; Published 2014-7-21
Abstract
Neuronal growth regulator 1 (NEGR1) has become a great interest based on the recent findings that its genetic alteration is implicated in human obesity and human dyslexia. By analyzing the gene expression profiles of tumor biopsies and normal tissues, we identified NEGR1 as a commonly down-regulated gene in many types of human cancer tissues. NEGR1 contains a C-terminal GPI anchor attachment site and is primarily localized to cell membrane rafts, especially in cell-to-cell contacting areas. The oncogenic phenotype was clearly attenuated when NEGR1 was overexpressed in the human ovarian cancer cell line SKOV-3. Furthermore, cell aggregation and neurite outgrowth was greatly increased by NEGR1 overexpression. On the contrary, cell migration and invasion was increased in NEGR1-depleted cells, suggesting that NEGR1 may contribute to tumor suppression. Taken together, we suggest that NEGR1 is a raft-associated extracellular protein that may participate in cell recognition and interaction, which is important in growth control and malignant transformation.
Keywords: neuronal growth regulator 1, cancer, lipid raft, cell adhesion, cell-cell interaction.
Introduction
The dynamic nature of the cell membrane plays a crucial roles in cancer development, by participating in cell survival, cell death, endocytosis, signaling, cell division, and cell separation during cancer metastasis [1]. Lipid rafts are sterol- and sphingolipid-enriched membrane subdomains and are known to participate in several important cellular processes such as cell adhesion, cell migration, signaling, protein sorting, and endocytosis [2]. Glycosylphosphatidylinositol (GPI)-anchored proteins are extracellularly tethered to the membrane using a specialized lipid moiety termed as GPI-anchor, which is attached to the C-terminus of the target protein. These proteins are highly enriched in lipid raft domains, and are involved in a number of important cellular activities including signaling processes and cell adhesion [3].
Kilon (kindred of IgLON) is a GPI-anchored protein that was originally identified as a new raft component in rat brain [4]. This protein is homologous to a subset of the immunoglobulin (Ig) superfamily, IgLON, which contains three C2-type Ig-like domains. The IgLON family comprises cell adhesion molecules, including limbic system-associated membrane protein (LAMP), opioid-binding cell adhesion molecule (OBCAM) [5]. The IgLON family is highly expressed in brain tissues, and plays major roles in cell-to-cell recognition and neurite outgrowth via homophilic and heterophilic interactions [6].
Human NEGR1 (neuronal growth regulator 1) is a possible human kilon homologue, exhibiting significant homology with rat kilon. Although recent genome-wide analysis revealed that NEGR1 is associated human body weight regulation [7] and human dyslexia [8], however, no biochemical and functional studies have been reported on human NEGR1 thus far. In this study, in addition to its biochemical characterization, we report that NEGR1 is a commonly downregulated gene in various human cancers and its overexpression substantially reduces the tumorigenicity of SKOV-3 cells.
Materials and Methods
Expression analysis, real-time PCR and northern analysis
The expression values of tumor biopsies and normal tissues were obtained by analyzing GeneExpress Oncology Datasuite™ from Gene Logic (Gaithersburg, USA) obtained written consent from a patient or a from a person legally authorized on behalf of a patient according to ethics standards set by an independent review board. The expression profiles of normal and cancer tissue sets from breast, colon, kidney, liver, lung, esophagus, ovary, pancreas, prostate, rectum, and stomach, were analyzed. Differential expression of genes was determined by statistical methods as previously described [9]. Genes for which the p-value was < 0.05 were included in the analysis.
Quantitative RT-PCR (Q-RT-PCR) was carried out using Taqman™ RT-PCR system (Applied Biosystems, Carlsbad, USA) with paired RNA samples of normal and cancer tissues purchased from Ambion (Austin, USA). The relative expression values were calculated in comparison with C-terminal binding protein 1, which was selected from the database as one of the least regulated genes in cancers [9]. The primers used were: NEGR1 forward: 5'- GGCCCTCGGACAGAGTGAAATCA -3', NEGR1 reverse: 5'-GTACAGTCAGGACTGATAAAATGGG -3'. For Northern blotting analysis, an IMAGE cDNA clone (IMAGE No. 2371021, Invitrogen, Carlsbad, USA) containing the selected NEGR1 specific Affymetrix fragment was purchased and used as the template for the production of Northern blotting probes. After 32P-labeled probes was synthesized using the Prime-It II kit (Stratagene, La Jolla, USA), Northern blotting was conducted using a human multiple tissue blot (BD bioscience, Franklin Lakes, USA) according to the manufacturer's recommendations.
5′-RACE, cloning, and antibody production
To obtain ORF of human NEGR1, we performed 5'-rapid amplification of the cDNA ends (RACE) by using an oligo, 5′-ATGTTAGAGTAACAGCATTTTCC-3′, a Marathon cDNA amplification kit and brain cDNAs (Clontech, Mountain View, USA). An amplicon containing the NEGR1 ORF was identified at around 2.7 kb and used for further analysis after cloning. To generate a membrane-targeted enhanced green fluorescent protein (EGFP)-tagged NEGR1 construct, we followed a stepwise cloning strategy. First, the predicted signal sequence region (amino acids 1-39) was inserted into the N-terminus of EGFP by using a NheI site in the pEGFP-C1 vector (Clontech). Next, the rest of the NEGR1gene was fused to the C-terminus of EGFP gene by using the SalI and BamHI sites. We also generated the FLAG-tagged NEGR1 by locating the FLAG epitope sequence right before the GPI-anchoring site expression vector by using the same two-step cloning strategy with the pcDNA3-3FLAG vector [10]. Three Ig-like domain regions (amino acids 1-314) were subcloned onto the N-terminus of FLAG sequence. Next, the rest part of the NEGR1 gene was inserted into the C-terminus of the FLAG epitope. To remove GPI anchoring site, N-terminal region (amino acids 1-314) of NEGR1 was subcloned into the pcDNA3-3FLAG vector using EcoRI and XbaI. The NEGR1 point mutant (G324L/S235P) was generated using TOPchangeTM mutagenesis kit (Enzynomics, Korea). To produce an anti-NEGR1 antibody, the NEGR1 gene (amino acids 34-323) was subcloned into the pET-28a (Novagen, Madison, USA). The 6His-fused recombinant NEGR1 protein was expressed in the Escherichia coli Rosseta™ strain (Novagen) and purified using the Ni-NTA Superflow™ resin (Qiagen, Valencia, USA). Two rats were immunized by Cosmogenetech (Seoul, Korea).
Cell culture, transfection, siRNAs, and immunofluorescence microscopy
293T, SKOV-3, U178, and SHSY-5Y cells were grown in DMEM medium, while Chinese hamster ovary (CHO) cells and CRT-MG human astrocytoma cells were maintained in Ham's F-12 medium at standard culture condition. Transient transfection was carried out using either EffecteneTM (Qiagen) or a polyethylenimine reagent (Sigma-Aldrich, St. Louis, USA). For siRNA treatment, NEGR1 siRNA (CUCGCAUGAUAUUCAGGU) was synthesized from Bioneer (Korea) and transfected to cells using LipofectaminTM (Invitrogen) at a concentration of 100 nM. After immunostaining was performed as previously described [11], the immunostained cells were imaged with an Olympus IX70 fluorescence microscope or an LSM5-Pascal (Zeiss, Germany) confocal system.
In vitro tumorigenicity test
After SKOV-3 cells were transfected with the pcDNA3-NEGR1-FLAG or vector control, stable clones were selected in the presence of G418. Among the isolated stable cells, a high-expressing (HE) and a low-expressing (LE) clones were selected and used for further tumor assays. For the proliferation test, cells (~ 5 × 104) were seeded into 12-well plates and the cell numbers were counted each day by using a hemocytometer in triplicate. A colony-forming assay was performed after seeding cells (~ 1 × 104) in 60-mm dishes. In 14 days, cells were stained with 0.5% crystal violet and the colonies larger than 1 mm in diameter were counted. In soft agar assays, cells (~ 1 × 104) were suspended in a media containing 0.3% Noble agar (Difco, Detroit, USA) and loaded onto the 0.5% base agar in the 6-well plates. After 3 weeks of incubation, colonies greater than 0.5 mm in diameter were counted. For the wound healing assay, cells were cultured until confluent. Then, a wound was created by using a plastic pipette tip. The wounded area was examined under a bright field (at 100× magnification) at 0, 6, and 24 h after the wound scratch. A Matrigel invasion assay was performed as previously described [9]. Briefly, a Boyden chamber was equipped with a polycarbonate filter coated with an even layer of Matrigel. After the lower compartment was filled with SKOV-3 growth medium, a cell suspension (~ 3 × 105 cells/ml in serum-free media) was added to the upper wells. After a 17-h incubation, the cells migrating through the membrane were counted. For the organotypic invasion assay, the rat hippocampal slice were prepared and maintained according to the previously described methods [12]. One microliter of Dil-stained cells (~5,000 cells) were placed on the prepared rat brain slices. After 6 days, the movement of the cells on the slices was detected with an inverted confocal laser scanning microscope (Zeiss LSM5, Carl Zeiss). ImageJ software (NIH) was used to calculate the invasion area of DiI-stained cells. Invasion area (%) = (area of DiI-stained cells at 120 h/area of DiI-stained cells at 1 h) x 100.
Aggregation assay, cross-linking, and production of Fc-NEGR1
For the hanging drop experiments, SKOV-3-NEGR1 stable cells (1.5 × 104) in 30 µl of growth medium were placed as hanging drops from the lid of a 24-well culture dish and allowed to aggregate for 16 h. After pipetting several times, cells were observed under a microscope and a picture was taken for an individual field. Aggregates were measured and counted. For cross-linking, a water-soluble cross linker, BS3 (Pierce, Rockford, USA), was added at a 2 mM concentration onto a monolayer of CHO-NEGR1-FLAG stable cells at room temperature. Cells were lysed and immunoprecipitated with an anti-FLAG antibody, and the cross-linked products were visualized by western blotting. To perform a binding assay with the soluble NEGR1-Fc chimeric protein, the Ig-like domains of NEGR1 (amino acids 1-314) was amplified and fused to a human Fc segment (hFc). Then, 293T cells were transfected with the NEGR1-hFc construct, and the medium was collected after 2 days. NEGR1-hFc chimeric proteins were purified using protein A sepharose (GE Healthcare, UK). The purified NEGR1-hFc (5 mg/ml) or hFc protein were added to cells and incubated for 1 h at 4°C. After 3 washes, cells were fixed and the hFc proteins were visualized with an Alexta595-conjugated anti-human Fc antibody (1:500) (Molecular Probes, Carlsbad, USA)
Lipid raft fractionation, PNGase F digestion, and neurite outgrowth
Lipid raft fractionation was conducted following the previous method [13] using OptiPrep (Sigma) after 293T cells were transfected with the EGFP-NEGR1. After lysates were adjusted to 40% OptiPrep, the samples was loaded into a centrifuge tube, which was serially overlaid with 2.5 ml of 28% OptiPrep in 1 × PBS and 0.6 ml of 1 × PBS. After centrifuging for 3 h at 36,400 rpm in a Beckman SW 55 Ti rotor, 13-300 μl fractions were collected from the top. A horseradish peroxidase-conjugated cholera toxin B subunit (Sigma) was used to detect Ganglioside GM1, a raft marker. The crude membrane was obtained according to a previously described method [14]. For enzyme digestion, samples were incubated with 500 U of PNGase F or EndoH (New England Biolabs, Ipswich, USA) at 37°C for 1 h. A neurite outgrowth assay was performed using undifferentiated SHSY-5Y human neuroblastoma cells. At 24 h after seeding of 5 × 104 cells into 6-well cell culture plate, 100 ng/ml of either NEGR1-hFc or hFc were added to the culture media and incubated for five days. Cells were also treated with retinoic acid (10 µM) as a positive control. Microtubule-associated protein 2 (MAP2), a marker of neuronal differentiation, was also detected using anti-MAP2 antibody (Sigma-Aldrich).
Results
Human NEGR1 is a downregulated cancer gene
To discover the differentially regulated genes in cancer, we analyzed the mRNA expression profiles of tumor cells by using a commercially available oncology database, which contains the expression profiles of about 300 tumor or normal samples from various human tissues. An Affymetrix fragment from an expressed sequence tag (EST) was identified as a commonly downregulated gene (with p-values < 0.01) in many tissues, especially in ovary (3.0-fold), lymph node (2.6-fold), and colon (2.2-fold) tissues (Fig. 1A). Quantitative RT-PCR was performed to confirm the expression profiles of this gene by using commercial normal/tumor paired RNA samples. Significant down-regulation was observed in the ovary, stomach, and colon tissues, supporting the previous findings from expression analyses (Fig. 1B).
Identification of cancer-downregulated gene, NEGR1 (A) mRNA expression profile in various normal (open bar) and tumor (closed bar) tissues from the Oncology DatasuiteTM of Gene Logic Inc. (B) Quantitative real-time PCR analysis with a tumor/normal paired RNA sample using Taqman™ RT-PCR system (Applied Biosystems). The error bar represents ± S.D. values between three separate reactions from the same RNA. *p <0.05;***p <0.001. (C) Northern blot analysis of the human NEGR1gene using a human multiple tissue blot. Northern blotting was conducted using a human multiple tissue blot (BD bioscience) with NEGR1 specific probes synthesized using Prime-It II kit (Stratagene). (D) Structure of human NEGR1. Three loops represent the Ig-like domains and the predicted N-glycosylation sites are indicated by the small open circles.
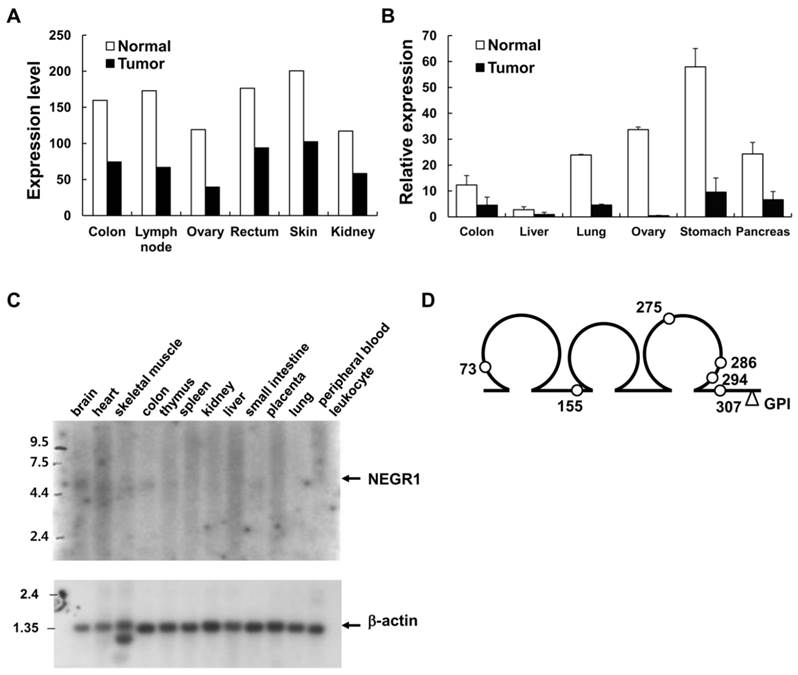
Northern analysis was carried out to identify the transcript size of the target gene by using a human multiple tissue blot (BD Biosciences). An IMAGE clone (IMAGE No. 2371021) containing the selected Affymetrix fragment was purchased and used for the production of northern blotting probes. Although an intense β-actin mRNA signal was clearly observed under the same experimental conditions (Fig. 1C), the transcript of the target gene was vaguely detected in the brain and colon tissues, indicating that the gene is expressed at low levels with a size of approximately 5 kb (Fig. 1C). To obtain the protein-coding region of this selected gene, we performed 5′-RACE by using human brain cDNA library. From the PCR-amplified product of the 2.7kb RACE product, we identified a putative ORF that encodes a 354-amino acid protein showing as much as 94% homology with rat kilon. Consecutive protein domain analysis revealed that this protein comprises three consecutive N-glycosylated C2-type Ig-like domains with an N-terminal signal sequence (Fig. 1D). A putative GPI-attachment site was also identified at Gly324 located close to the C-terminal end. By the time we obtained this gene, it was categorized as a Unigene, and was later named “NEGR1” in the National Center for Biotechnology Information (NCBI) website.
Human NEGR1 is enriched in membrane rafts at cell junctions
NEGR1 showed strong homology with rat kilon, which was identified as a GPI-anchored lipid raft protein. To identify if human NEGR1 is also a component of membrane rafts, we conducted two-step cloning to generate an EGFP-NEGR1 construct, in which EGFP is inserted in-frame between the putative signal peptide and the rest of the protein. Then, membrane raft fractionation was carried out using CHO cells transiently transfected with EGFR-NEGR1. The fractionation results revealed that human NEGR1 is exclusively localized in the floating lipid raft fraction (Fig. 2A). A separately performed dot blot clearly verified the raft fractionation by detecting the lipid raft marker, GM1 ganglioside.
Subcellular localization of human NEGR1. (A) Lipid raft fractionation. After transient transfection of EGFP-NEGR1 in CHO cells, lipid raft fractions are isolated by centrifugation for 3 h at 36,400 rpm using Optiprep (Sigma). Thirteen fractions were collected from the top and the fraction 1 represents the top. GM1 raft marker was also visualized by dot blotting. (B) N-linked glycosylation analysis. The membrane and cytosolic protein fraction of SKOV-3-NEGR1 stable cells were digested with PNGase F (500 U) for 1 h. The total cell lysate (30 μg) was incubated with or without Endo H (500 U) prior to Western blotting. (C) Localization of NEGR1 in CRT-MG stable cells. The NEGR1 protein was captured by confocal microscopy after immunostaining CRT-MG cells stably expressing NEGR1-FLAG. The lower panel illustrates the line intensity profile of the upper image using Zen imaging software (Carl Zeiss) in the direction indicated by the red arrow. (D) Subcellular localization of NEGR1 in various cells. SKOV-3-NEGR1-FLAG stable cells were immunostained using anti-FLAG antibody with or without 0.1% saponin. Endogenous NEGR1 protein was also visualized in U178 and CRT-MG neuroblastoma cells using an anti-NEGR1 antibody. (E) Raft association of NEGR1 GPI anchor mutant proteins. After NEGR1 deletion mutant (ΔGPI, amino acids 1 - 314) or site-specific mutant (G324L/S325P) at ω and ω -1 site were generated, the sub-membrane localization of each protein was examined by lipid raft fractionation method using Optiprep. (F) Membrane targeting of GPI-site mutants. NEGR1 mutant proteins were expressed and their extracellular display was visualized by immunostaining with anti-FLAG antibody under cell-permeable or -impermeable condition.
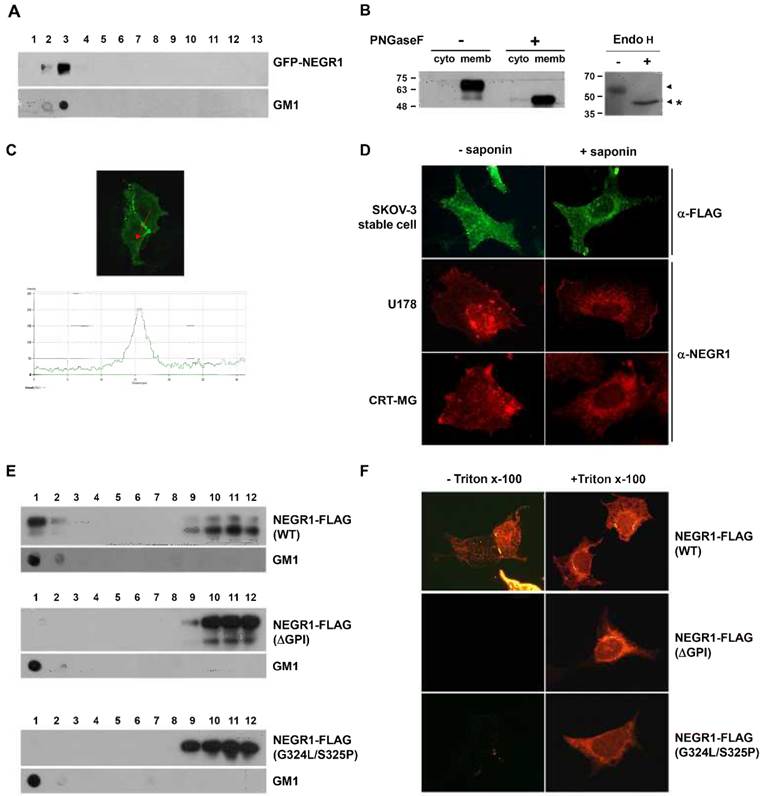
We also tested the presence of an N-glycosyl moiety on NEGR1 using NEGR1-FLAG-expressing SKOV-3 ovarian cancer cells. We first fractionated the membrane compartment from SKOV-3-NEGR1 stable cells, and incubated samples with peptide-N-glycosidase (PNGase) F. Although two differently migrating bands of NEGR1-FLAG were normally detected in Western blotting, the high-molecular weight NEGR-FLAG protein was clearly processed to a lower molecular weight unmodified form by PNGase F digestion (Fig. 2B). When cell lysates were incubated with Endo H, a lower molecular weight form was also identified (Fig. 2B). These data may demonstrate that NEGR1 is N-glycosylated in human cells.
Next, we examined the distribution of NEGR1 by immunostaining without cell permeabilization using CRT-MG astrocytoma cells stably expressing FLAG-tagged NEGR1 protein. Human NEGR1 was visualized in the cell membrane, and interestingly, the strongest signal for NEGR1 was obtained in cell-to-cell contacting area. NEGR1 is likely enriched in the lateral cell membrane around in the cell junction area, which is well demonstrated by the scanning confocal image in Fig. 2C. To demonstrate the membrane localization more clearly, we compared images after staining cells with or without the cell permeabilizing detergent, saponin. When SKOV-3-NEGR1 stable cells were stained using an anti-FLAG antibody without saponin, the protein was observed in the cell membrane, being enriched in the outermost border of the cells (1st row of Fig. 2D, left). However, when saponin was added, as numerous speckles dispersed from the nucleus (1st row of Fig. 2D, right), showing possible localization in the ER or endosomal vesicles. Finally, we examined the distribution of endogenous NEGR1 protein in human U178 glioma cells and CRT-MG cells using anti-NEGR1 antiserum obtained from rats. Endogenous NEGR1 was observed in two astrocytoma cell lines and was fundamentally the same as the NEGR1 in SKOV-3 stable cells (2nd and 3rd row of Fig. 2D). Collectively, these results indicated that human NEGR1is primarily localized in a membrane raft, especially enriched in the lateral cell border or cell junction between juxtaposed cells.
To find out if GPI anchor is important for the proper localization of NEGR1, we generated several GPI anchor mutants. Since Gly324 was predicted as a putative GPI-attachment site (ω site), we first constructed a tailless deletion mutant (ΔGPI) which contains amino acids 1-314. Given that GPI anchor is added to the ω residue lying a particular consensus sequence of ω, ω+1, ω+2 [15], We also replaced the consensus sequences of GPI-anchoring region (ω and ω+1 site) with the least favorable amino acids (G324L/S325P). Lipid raft fractionation results showed that the raft association of both GPI site mutant proteins were completely abrogated (Fig. 2E). Moreover, the extracellular targeting of these mutant proteins was also clearly impaired (Fig. 2F), indicating that the GPI attachment is prerequisite factor for the proper membrane targeting and raft association of NEGR1 protein.
NEGR1 overexpression suppresses proliferation and anchorage-independent growth of SKOV-3 cells
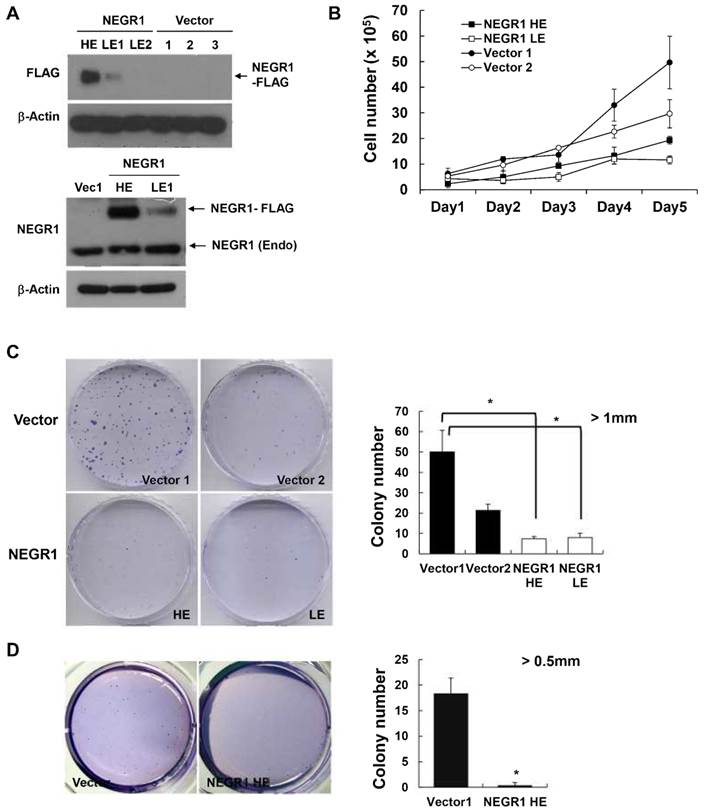
In order to investigate if human NEGR1 functions in tumor biogenesis, we chose NEGR1 stable SKOV-3 cells, the ovarian adenocarcinoma cell line, based on the previous findings that NEGR1 expression is the most dysregulated in ovarian cancer. We examined the expression level of selected stable clones using Western blotting with anti-FLAG antibody (upper panel of Fig. 3A) or anti-NEGR1 antibody (lower panel of Fig. 3A), and we chose a high expressor (HE) and a low expressor (LE1), along with two of the vector-transfected clones, for further cancer assays. We first examined the proliferation rate of these stable cells by counting cells every day. The NEGR1-expressing cells showed a decreased growth compared to vector-transfected controls (Fig. 3B). A colony forming assay also revealed that NEGR1-expressing cells proliferate slower than controls, suggesting that NEGR1 expression suppresses cell growth (Fig. 3C). Cell growth in soft agar medium is one of the hallmarks of cancer cells which are capable of anchorage-independent growth. We monitored the growth properties of NEGR1 stable cells (HE) in soft agar medium and found that the anchorage-independent growth of SKOV-3 cells was significantly suppressed by NEGR1 overexpression (Fig. 3D), suggesting that NEGR1 attenuates the tumorigenic property of cancer cells.
Proliferation and anchorage-independence of NEGR-overexpressing SKOV-3 cells. (A) Isolation of SKOV-3-NEGR1-FLAG stable cells. After SKOV-3-NEGR1 stable clones were generated, high expressing (HE) and low expressing (LE) clones were isolated. Western blotting was performed using either an anti-FLAG (upper panel) or anti-NEGR1 antibody (lower panel). (B) Cell proliferation assay. After seeding 5 × 104 cells into 12-well plates, the cells were counted each day using a hemocytometer. The mean values of triplicates were plotted and the error bars represent standard deviation (SD) between three samples. (C) Colony forming assay. After seeding cells (~ 1 × 104) in 60-mm dishes, cells were stained with crystal violet 14 days after seeding. Colonies greater than 1mm in diameter were counted. Data are presented as the mean ± SD (*p <0.05). (D) Colony formation in soft agar. About 1 × 104 cells in 0.3% agar were plated onto 0.5% base agar solidified in 6-well plates. After three weeks, colonies larger than 0.5 mm in diameter were counted in triplicate. The error bar represents ± SD values between samples. *p <0.05.
The role of NEGR1 in tumor migration and invasion
Given that tumor malignancy is closely related to the ability of cancer cells to migrate and invade, we first tested if NEGR1 affects cell migration by performing wound healing assay. A scratch wound was generated by scraping the cell mono-layer and the healing rate of the injured region was monitored by microscopy. NEGR1-overexpressing cells showed an ~ 35% slower migration rate than control at 24h after the scratch wound was made (Fig. 4A), suggesting that NEGR1 expression contributes to inhibiting cell migration. We also examined the cell invasiveness of SKOV-3-NEGR1 cells by using a Matrigel-coated filter in a Boyden chamber. We placed cells in the upper chamber with serum-free media and allowed them to invade the Matrigel layer in the lower wells filled with serum. We could observe that NEGR1 expression in SKOV-3 cells greatly reduced the cell invasiveness according to the Matrigel invasion assay (Fig. 4B). The penetration rate of NEGR1-overexpressing cells significantly decreased to only ~ 20% of the vector-transfected controls. Next, we examined cancer phenotype of NEGR1-suppressed SKOV-3 cells following treatment with NEGR1-specific siRNA. Treatment of siRNA reduced mRNA level of NEGR1 by ~50% compared to control siRNA (data not shown). The results of wound healing assay and Matrigel invasion assay indicated that a reduction of intracellular NEGR1 protein promoted both cell migration (Fig. 4C) and invasion (Fig. 4D), which suggests that NEGR1 could inhibit the cell migration and invasion.
Both cell migration and invasion are influenced by NEGR1 expression. (A) Wound healing assay with NEGR1-overexpressed SKOV-3 cells. After wounds were made by scratching the confluent cell surface, the wounded area was examined at 0, 6, and 24 h. The error bar represents ± SD values between samples at different time points. *p <0.05. (B) Matrigel cell invasion assay with NEGR1-overexpressing cells. Fifty microliters of cell suspension (~ 3 × 105 cells/ml in DMEM) was placed in the upper section of a Boyden chamber equipped with a Matrigel-coated filter. After 17 h, the filter was stained and the migrating cells were counted under microscope with10 x magnification. Data are presented as the mean ± SD. (**p <0.005) (C) Cell migration assay in NEGR1-knockdown cells. SKOV-3 cells were transfected with either control siRNA or NEGR1-specific siRNA (100 nM) for 48h, and cell migration activity was examined after scratch wounding. The data represent the mean ± SD. **p <0.005. (D) Cell invasion activity of NEGR1-suppressed cells. After siRNA treatment, cells were placed on the Boyden chamber and invasiveness of the cell through a Matrigel-coated filter was monitored. The data represent the mean ± SD. **p <0.005. (E) Organotypic invasion assay. One microliter of Dil-stained cells (~5,000 cells) was overlaid on the rat brain slices. Cell migration was detected after 6 days incubation using Zeiss LSM5 confocal scanning microscope. Data are represented as the mean ± SEM. *p <0.05.
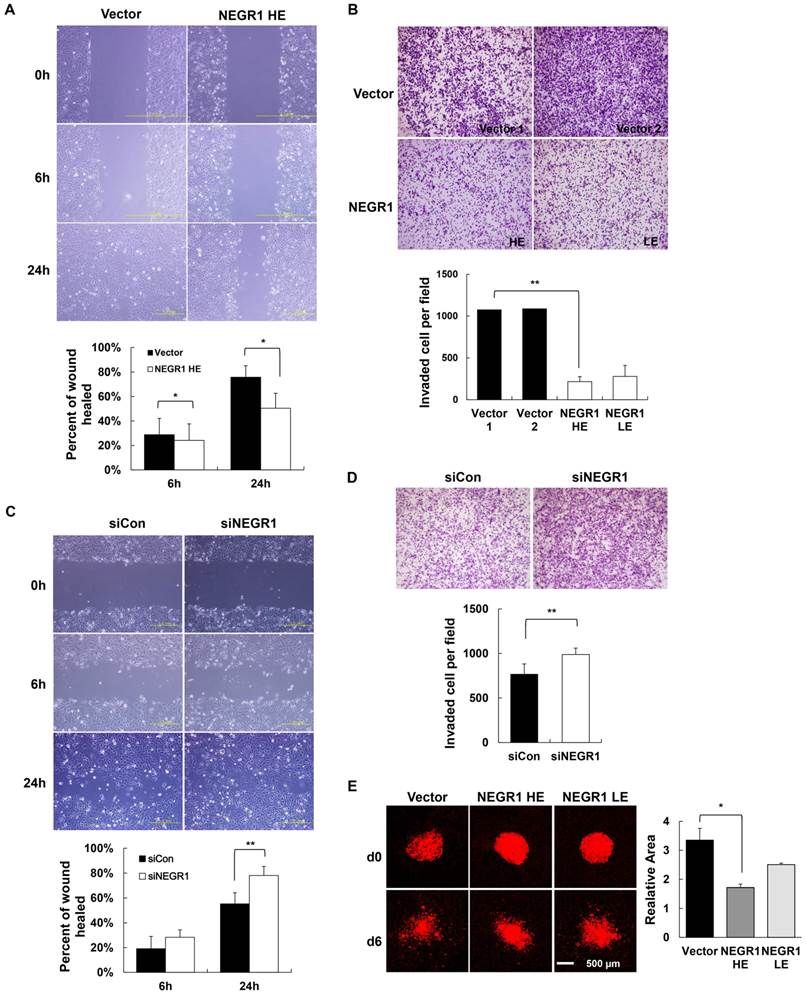
Finally, to test the anti-migrating activity of NEGR1 using an ex vivo culture system, we performed the organotypic invasion assay [16]. The hippocampal slices were prepared from early postnatal rat brain and NEGR1 stable cells were gently overlaid onto the slices. After incubation for 6 days, cell migration was examined under fluorescence microscope. The dispersed area of the NEGR1-expressing cells (HE) were calculated as ~51% of control cells on brain slice, indicating that NEGR1 expression suppress the cell invasiveness on the brain slice (Fig. 4E). Taken together, these results demonstrated that NEGR1 overexpression may suppress cell migration and invasiveness of SKOV-3 cancer cells.
NEGR1 may function in cell-cell adhesion
Given that NEGR1 showed high homology with the IgLON family, which is known to function in neural cell communication, we tested if NEGR1 might participate in cell-cell interactions. For this experiment, we generated a soluble NEGR1-hFc fusion protein and examined if NEGR1 mediated hemophilic binding using a purified NEGR1-hFc protein. After we placed CHO-NEGR1 stable cells in culture plates, the soluble NEGR1-hFc fusion protein or hFc control protein was added to the culture media. After thorough washing, immunostaining with an anti-hFc antibody revealed that NEGR1-hFc fusion proteins bound more tightly to the NEGR1-expressing cells (Fig. 5A), suggesting homophilic binding between NEGR1proteins. To confirm the homophilic interaction of NEGR1 proteins, we performed a cross-linking experiment using a bis(sulfosuccinimidyl) suberate, BS3, a water-soluble, non-permeable cross-linker. After addition of BS3 on SKOV-3-NEGR1 stable cells, we harvested the cells and carried out immunoprecipitation. Western blotting analysis showed that high molecular weight bands, possibly corresponding to the dimer and tetramer of NEGR1, were increased by BS3 treatment over time (Fig. 5B), indicating that NEGR1 may interact homophilically outside of the cell membrane.
Role of NEGR1 in cell-to-cell adhesion. (A) NEGR1 may mediate homophilic binding. Soluble NEGR1-hFc or hFc protein (5 mg/ml) was incubated with CHO-NEGR1 stable cells for 1 h, and bound hFc proteins were detected with an anti-human Fc antibody. (B) BS3 cross-linking assay. After SKOV-3-NEGR1 stable cells were treated with BS3, cells were harvested and immunoprecipitation was performed with an anti-FLAG antibody. (C) Hanging-drop aggregation assay. Cells were loaded as hanging drops from the lid of a 24-well culture dish and allowed to aggregate for 16 h. Aggregated cells were measured by analyzing the pictures. The data represent the mean ± SD. **p <0.005; ***p <0.001. (D) Aggregation assay with GPI anchor mutants. Cell aggregation was monitored using cells expressing NEGR1 mutants defective in the GPI-anchoring after cells were loaded in the lid of the culture dish. The data as shown as mean ± SD. (***p <0.001) (E) Soluble NEGR1 proteins promote neurite outgrowth. After undifferentiated SHSY-5Y cells were incubated with purified NEGR1-hFc protein or hFc control (100 ng/ml) for 5 days, morphological changes were monitored under a microscope. Retinoic acid (10 μM) was used as positive control for the neurite outgrowth inducer. Neuronal differentiaion marker proteins, MAP-2 were also examined to support cell differentiation.
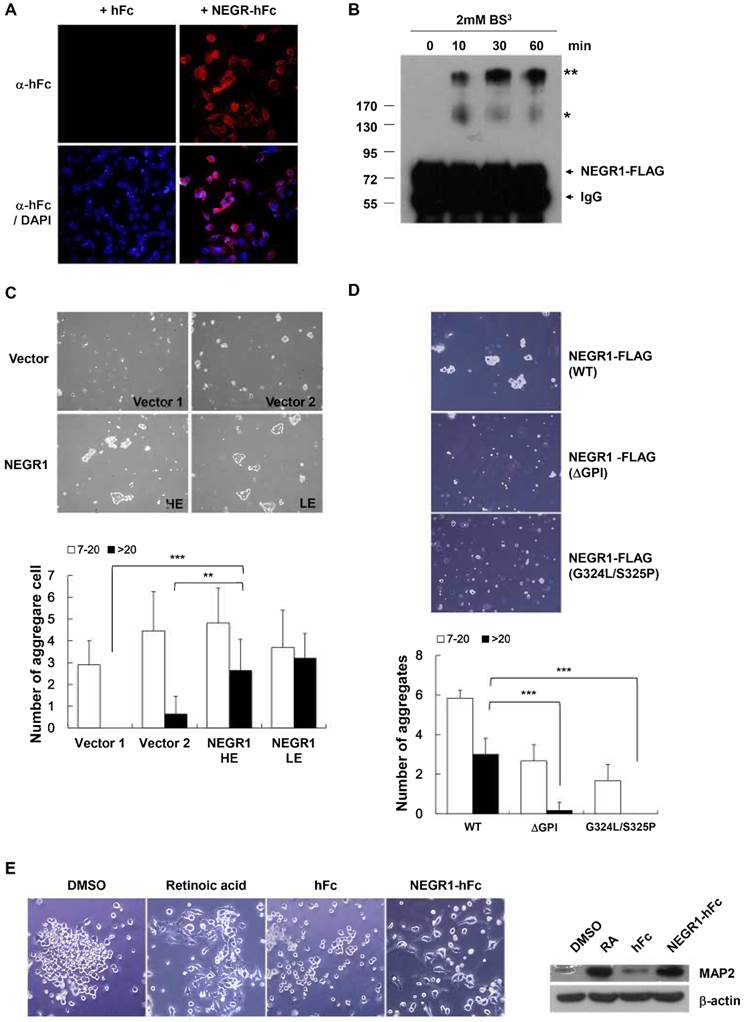
Considering that numerous cell adhesion molecules play key roles in cell-cell contact, aggregation, and are regulated in human cancer, we wanted to determine if NEGR1 expression affects cell-cell adhesion. We monitored the cell aggregation states in the hanging drops from a culture plate lid. The hanging drop aggregation assay showed that NEGR1-overexpressing cells were more likely to congregate, especially forming clumps containing more than 20 cells (Fig. 5C), suggesting the NEGR1 may promote cell-to-cell attachment and aggregation. To further investigate this observation, we tested non-GPI anchor mutant proteins which showed defective targeting to cell membrane. Cell aggregation activity was highly reduced when cells expressed non-GPI anchored mutant proteins (Fig.5D), supporting that NEGR1 truly functions in cell-to-cell attachment, and the GPI anchor is crucial for NEGR1-mediated cell adhesion.
Next, we examined whether NEGR1 regulates neuronal cell growth as its name implies. Undifferentiated SHSY-5Y human neuroblastoma cells were prepared and incubated for 5 days with purified NEGR1-hFc protein, along with the control the hFc protein. Since retinoic acid is well known to be a potent inducer of neurite outgrowth, we treated retinoic acid as a positive control. As shown in Fig. 5E, neurite outgrowth of SHSY-5Y cells was substantially stimulated by the NEGR1 protein. In addition to the morphological change, the increased protein level of the neuronal differentiation marker, microtubule-associated protein-2 (MAP-2) also indicated that NEGR1 is capable of promoting neurite growth. Collectively, these results indicated that the raft-associated membrane protein, NEGR1 may function in cell-to-cell recognition and cell adhesion via homophilic or possibly heterophilic protein interactions.
Discussion
Cell adhesion molecules (CAM) participate to a great extent in a variety of cell functions including signal transduction, cell growth, differentiation, morphogenesis, motility, and inflammation [17]. Since alterations in the adhesion properties of tumor cells are crucial in tumor invasion and metastasis, diverse CAMs play key roles in the development of cancer by regulating intercellular and cell-matrix interactions during cancer progression [17]. Cell adhesion molecules from the Ig supergene family (Ig-CAM) are expressed in various cell types and play roles in a broad spectrum of cellular activities, including brain development, and epithelial morphogenesis by promoting cell-cell interactions through hemophilic or heterophilic protein interactions [18]. Changes in Ig-CAM expression also have been observed in a variety of human malignancies. Neuronal CAM (NCAM), which plays an important function in neurite outgrowth and long-term potentiation, is markedly downregulated in colon carcinomas, pancreatic cancer, and astrocytomas, while it is up-regulated in neuroblastomas [18]. Various Ig-CAM proteins have a mixed dysregulation depending on the cell tissue type or the tumor developmental stage [18], which might imply that these proteins are involved in multiple steps of tumorigenesis during malignant cell development.
In an attempt to discover differentially regulated tumor-associated genes, we identified NEGR1 as a commonly downregulated gene in various tumor biopsies. Although NEGR1 is identified as a downregulated gene in cancer, it seems to be expressed at low level even in normal tissues, based on the normalized expression values of microarray data and northern results (Fig. 1A & C). Classically, the loss of cell to matrix adhesion is an essential prerequisite factor for active tumor cell migration, and invasion during metastasis. When NEGR1 is over-expressed, the SKOV-3 cells become less proliferative, and NEGR1-expressing cells showed clear suppression of invasiveness in SKOV-3 tumor cells in the Matrigel (Fig. 4A and B). On the contrary, the tumorigenic activity was increased in NEGR1-knockdown cells (Fig. 4C and D). These results may suggest that NEGR1 functions as a modulator of cell-matrix interaction.
Although the name strongly indicates that NEGR1 may function in neural cell growth, no study has been published to support this function. We have now shown that human NEGR1 functions as a neuronal growth regulator, by observing that this protein substantially promotes neurite outgrowth in SH-SY5Y cells. The recent interest in NEGR1's role in human obesity and dyslexia is also based on the assumption that NEGR1 functions in the development of the central nervous system [7]. These results, along with the findings that NEGR1 promotes cell-cell aggregation, suggest that NEGR1 may participate in the cell-to-cell communication as well as cell-to-matrix interaction during formation of neural circuit.
Taken together, we now report that NEGR1 is commonly downregulated in many human cancers and suggest that NEGR1 may functions in the regulation of cell adhesion that is required for the normal cell growth and cell-to-cell communication.
Acknowledgements
This research was supported by Basic Science Research Program through NRF by the Ministry of Education, Science and Technology, Korea (2013R1A1A3A04005274).
Abbreviations
NEGR1: neuronal growth regulator 1; kilon: kindred of IgLON; GPI: glycosylphosphatidylinositol; ORF: open reading frame; EST: expressed sequence tag; Ig: immunoglobulin; BS3: bis(sulfosuccininmidyl) suberate; PNGase F: peptide-N-glycosidase F; Endo H: Endo-ß-N-acetylglucosaminidase H.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Patra SK. Dissecting lipid raft facilitated cell signaling pathways in cancer. Biochim Biophys Acta. 2008;1785:182-206
2. Viola A, Gupta N. Tether and trap: regulation of membrane-raft dynamics by actin-binding proteins. Nat Rev Immunol. 2007;7:889-896
3. Sharom FJ, Lehto MT. Glycosylphosphatidylinositol-anchored proteins: structure, function, and cleavage by phosphatidylinositol-specific phospholipase C. Biochem Cell Biol. 2002;80:535-549
4. Funatsu N, Miyata S, Kumanogoh H. et al. Characterization of a novel rat brain glycosylphosphatidylinositol-anchored protein (Kilon), a member of the IgLON cell adhesion molecule family. J Biol Chem. 1999;274:8224-8230
5. Hashimoto T, Maekawa S, Miyata S. IgLON cell adhesion molecules regulate synaptogenesis in hippocampal neurons. Cell Biochem Funct. 2009;27:496-498
6. Reed J, McNamee C, Rackstraw S. et al. Diglons are heterodimeric proteins composed of IgLON subunits, and Diglon-CO inhibits neurite outgrowth from cerebellar granule cells. J Cell Sci. 2004;117:3961-3973
7. Willer CJ, Speliotes EK, Loos RJ. et al. Six new loci associated with body mass index highlight a neuronal influence on body weight regulation. Nat Genet. 2009;41:25-34
8. Veerappa AM, Saldanha M, Padakannaya P, Ramachandra NB. Family-based genome-wide copy number scan identifies five new genes of dyslexia involved in dendritic spinal plasticity. J Hum Genet. 2013;58:539-547
9. Jung J, Kim JM, Park B. et al. Newly identified tumor-associated role of human Sharpin. Mol Cell Biochem. 2010;340:161-167
10. Lee S, Gang J, Jeon SB. et al. Molecular cloning and functional analysis of a novel oncogene, cancer-upregulated gene 2 (CUG2). Biochem Biophys Res Commun. 2007;360:633-639
11. Chun Y, Park B, Koh W. et al. New centromeric component CENP-W is an RNA-associated nuclear matrix protein that interacts with nucleophosmin/B23 protein. J Biol Chem. 2011;286:42758-42769
12. De Simoni A, Yu LM. Preparation of organotypic hippocampal slice cultures: interface method. Nat Protoc. 2006;1:1439-1445
13. Macdonald JL, Pike LJ. A simplified method for the preparation of detergent-free lipid rafts. J Lipid Res. 2005;46:1061-1067
14. Schimmel SD, Kent C, Bischoff R, Vagelos PR. Plasma membranes from cultured muscle cells: isolation procedure and separation of putative plasma-membrane marker enzymes. Proc Natl Acad Sci U S A. 1973;70:3195-3199
15. White IJ, Souabni A, Hooper NM. Comparison of the glycosyl-phosphatidylinositol cleavage/attachment site between mammalian cells and parasitic protozoa. J Cell Sci. 2000;113( Pt 4):721-727
16. Eyupoglu IY, Hahnen E, Buslei R. et al. Suberoylanilide hydroxamic acid (SAHA) has potent anti-glioma properties in vitro, ex vivo and in vivo. J Neurochem. 2005;93:992-999
17. Okegawa T, Pong RC, Li Y, Hsieh JT. The role of cell adhesion molecule in cancer progression and its application in cancer therapy. Acta Biochim Pol. 2004;51:445-457
18. Cavallaro U, Christofori G. Cell adhesion and signalling by cadherins and Ig-CAMs in cancer. Nat Rev Cancer. 2004;4:118-132
Authors Biography
Dr. Soojin Lee is currently an associate professor in the Department of Microbiology and Molecular Biology at Chungnam National University in South Korea. With her colleagues, she initiated this study by using the GeneExpress Oncology Datasuite™ from Gene Logic (Gaithersburg, MD, USA) when she worked in the LG Biomedical Institute (San Diego, CA, USA). Later on, she developed a functional study for the previously unknown cancer-related genes. So far, her efforts have uncovered many unidentified cancer-associated genes leading to several publications (Jung et al. Mol Cell Biochem, 2010, 340:161-7; Chun et al., J Biol Chem, 2011, 286(49):42758-69; Chun et al., J Biol Chem. 2013, 288(38):27208-19).
Author contact
![]() Corresponding author: Soojin Lee, Department of Microbiology and Molecular Biology, Chungnam National University, Daejeon, 305-764, Korea. Phone: 82-42-821-6414; Fax: 82-42-822-7367; E-mail: leesoojinac.kr.
Corresponding author: Soojin Lee, Department of Microbiology and Molecular Biology, Chungnam National University, Daejeon, 305-764, Korea. Phone: 82-42-821-6414; Fax: 82-42-822-7367; E-mail: leesoojinac.kr.