Impact Factor ISSN: 1837-9664

 Global reach, higher impact
Global reach, higher impactJ Cancer 2014; 5(8):670-678. doi:10.7150/jca.9688 This issue Cite
Research Paper
Preferential Effect of Akt2-Dependent Signaling on the Cellular Viability of Ovarian Cancer Cells in Response to EGF
Dineo Khabele1, Syeda M Kabir2, Yuanlin Dong2, Eunsook Lee3, Valerie Montgomery Rice4, Deok-Soo Son2 ![]()
1. Department of Obstetrics and Gynecology, Vanderbilt University Medical Center, Nashville, TN, USA;
2. Department of Biochemistry and Cancer Biology, Meharry Medical College, Nashville, TN, USA;
3. Department of Physiology, Meharry Medical College, Nashville, TN, USA;
4. Department of Medicine, Morehouse College of Medicine, Atlanta, GA, USA.
Received 2014-5-19; Accepted 2014-8-13; Published 2014-9-5
Abstract
Objective: Overexpression of the epidermal growth factor receptor (EGFR) is associated with the malignant phenotype in many cancers including ovarian cancer, which leads to increased cell proliferation and survival. In spite of emerging EGFR inhibitors as a potentially useful agent, they are largely ineffective in patients with advanced or recurrent ovarian cancers. Since Akt as a key downstream factor of EGFR is highly activated in some high grade serous ovarian tumors, the augmented Akt activation may attribute to irregular EGFR-mediated signaling observed in ovarian cancer. Here we investigated the differential effect of Akt on the EGF-induced cell viability in a panel of ovarian cancer cell lines.
Methods: Cellular viability assay and western blot analysis were used to measure cell viability and expression levels of proteins, respectively. Knockdown of Akt was achieved with siRNA and stable transfection of expression vectors was performed.
Results: Cellular viability increased in OVCAR-3 ovarian cancer cells exposed to EGF, but little to no difference was observed in the 5 other ovarian cancer cells including SKOV-3 cells despite of the expression of EGFR. In OVCAR-3 cells, EGF activated Erk and Akt, but an Erk inhibitor had no impact on cellular viability. On the other hand, the EGFR and PI3K inhibitors decreased EGF-induced cellular viability, indicating the involvement of Akt signaling. Although EGF activated Erk in SKOV-3 cells, the Akt activation was very weak as compared to OVCAR-3 cells. Furthermore, we observed a different expression of Akt isoforms: Akt1 was constitutively expressed in all tested ovarian cancer cells, while Akt3 was little expressed. Interestingly, Akt2 was highly expressed in OVCAR-3 cells. Knockdown of Akt2 blocked EGF-induced OVCAR-3 cell viability whereas knockdown for Akt1 and Erk1/2 had no significant effect. Stable transfection of Akt2 into SKOV-3 cells phosphorylated more Akt and enhanced cell viability in response to EGF.
Conclusions: Akt2-dependent signaling appears to play an important role in EGFR-mediated cellular viability in ovarian cancer and targeting specific Akt isoform may provide a potential therapeutic approach for EGFR-expressing ovarian cancers.
Keywords: EGFR, Akt, Erk, cell viability, ovarian cancer.
Introduction
Ovarian cancer is the commonest cause of gynecological cancer-associated death and is an insidious disease because it typically is asymptomatic until tumors have spread far beyond the ovaries [1-3]. Furthermore, the 5-year survival rate of ovarian cancer has changed little over several decades despite intensive surgical treatment and advances in the use of novel therapeutic agents [2]. Genetic alterations including overexpression of epidermal growth factor receptor (EGFR) are frequently observed in high-grade malignant ovarian cancer [4, 5]. The ErbB/EGFR family of receptors consists of four structurally-related type 1 transmembrane tyrosine kinase receptors as follows: EGFR (ErbB1, HER1), ErbB2 (HER2, neu), ErbB3 (HER3) and ErbB4 (HER4) [6, 7]. Activation of ErbB/EGFR tyrosine kinase receptors recruits multiple signaling proteins, such as phospholipase Cγ, intracellular kinase Src, phosphoinositide 3-kinase (PI3K), serine-threonine specific protein kinase B (PKB/Akt) and mitogen-activated protein kinase (MAPK) [6]. These signal transduction pathways play important roles in cell proliferation, survival, adhesion, motility, invasion and angiogenesis [6, 7]. Because of the multiple signaling processes originating from EGFR activation, EGFR overexpression has been correlated with a poor prognosis and a decreased therapeutic responsiveness in patients with ovarian cancer [6]. Thus EGFR inhibitors may be potentially useful as a therapy in select patients with advanced or recurrent ovarian cancer [8-10]. However, clinical studies have revealed that EGFR inhibitors or antibodies to EGRF are largely ineffective against ovarian cancer [11, 12]. This limits the potential of EGFR inhibitors as therapeutic agents. Thus clarifying the response to EGF in ovarian cancer cells would provide a better understanding of an EGFR-targeted antitumor therapeutic. Since Akt is a key downstream factor of EGFR frequently observed in high-grade malignant ovarian cancers [4], we investigated the involvement of Akt signaling on the differential response to EGF in a panel of ovarian cancer cell lines.
In this study, we found that EGF differentially contributed to cell viability in EGFR-expressing ovarian cancer cell lines. The differential effect of EGF indicates the functional role of specific components of the downstream signaling pathways elicited by EGFR activation. Here we demonstrated the impact of Akt signaling, and particularly the contribution of the Akt2 isoform on EGF-induced cell viability in ovarian cancer.
Materials and Methods
Reagents
Recombinant human EGF was obtained from R&D Systems (Minneapolis, MN). PD98059 [extracellular signal-regulated kinases (Erk) inhibitor] was purchased from EMD Chemicals (Billerica, MA). SB203580 (p38 MAPK inhibitor), SP600125 [stress activated protein kinase/c-jun terminal kinase (SAPK/JNK) inhibitor], AG-1478 (EGFR inhibitor) and wortmannin (PI3K inhibitor) were purchased from Enzo Life Sciences (Farmingdale, NY). LY294002 (PI3K inhibitor) was purchased from Cayman Chemical (Ann Arbor, MI). Small interfering RNAs (siRNA) for control and transfection reagents were obtained from Dharmacon (Thermo Scientific, Pittsburgh, PA). The siRNAs for Akt1 and Erk1/2 and antibodies for ErbB isoforms and β-actin were obtained from Santa Cruz Biotechnology (Dallas, TX). Antibodies for MAPK/Akt isoforms and human-specific/mouse-specific Akt2 siRNAs were obtained from Cell Signaling Technology (Danvers, MA). Chemiluminescent detection kits came from GE Healthcare (Piscataway, NJ).
Cell culture
The human ovarian cancer cell lines (OVCAR-3, SKOV-3, CaOV-3 and TOV-21G), human colon adenocarcinoma cell line SW480, and human breast cancer cell lines MCF7 and MB231 were purchased from the American Type Culture Collection (Manassas, VA) and the A2780 human ovarian cancer cell line was provided by Dr. Andrew Godwin (University of Kansas, Kansas City, KS). The mouse ovarian surface epithelial cancer cell line (ID8) was provided by Drs. Katherine Roby and Paul Terranova (University of Kansas, Kansas City, KS). Cells (approximately 5 X 104 cells/ml) were cultured at 37°C in a water-saturated atmosphere of 95% air and 5% CO2 in 24- or 6-well plates with appropriate media: Dulbecco's Modified Eagles Medium with penicillin/streptomycin and 4% FBS for ID8 cells, and RPMI Medium 1640 with penicillin/streptomycin and 10% FBS for other cells. After an overnight culture to allow cellular attachment to the plates, the medium was removed and fresh medium without FBS was added along with the various agents as described in the Results.
Cellular viability assay
The cellular viability assay was performed by monitoring the cleavage of 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) to a colored product. Briefly, after culture in a 24-well plate, each well was washed twice with phenol red-free media. After removing the media, 250 µL of MTT solution (1 mg/mL of phenol red-free media:PBS = 4:1) was added and the plates were incubated for 3 hours at 37 °C under 5 % CO2 in air with protection from light. The MTT solution in each well was removed and 500 µL of isopropanol were added. The plates were placed on a shaker for 10 minutes at room temperature to thoroughly dissolve the MTT color product. The optical density was measured at 595 nm using a microplate reader (Bio-Rad). Values were normalized to controls.
Western blot
Cell lysates were prepared, fractionated on SDS-polyacrylamide gels and transferred to nitrocellulose membranes according to established procedures. Blocking of nonspecific proteins was performed by incubation of the membranes with 5% nonfat, dry milk in Tris buffered saline Tween-20 (TBST containing 10 mmol/L Tris, 150 mmol/L phosphate buffered saline, 0.05% Tween 20, pH 8.0) for 2 hours at room temperature. Blots were incubated with primary antibodies at 1:1,000 dilution in blocking solution overnight at 4ºC. The membranes were washed three times with TBST for 10 minutes followed by incubation for 1 hour with horseradish peroxidase conjugated secondary antibody (according to the primary antibody used) at a 1:2,500 in 5% milk/TBST. The membranes were then rinsed 3 times with TBST for 10 minutes and the bands visualized by enhanced chemiluminescence. After membrane stripping for 10 minutes with methanol containing 3% H2O2, β-actin was detected in order to serve as an internal loading control for cell lysates.
Gene silencing with small interfering RNA transfection
Expression levels of Akt1, Akt2 and Erk1/2 were selectively knocked down by using predesigned siRNAs. NT1 was used as a non-targeting control siRNA. OVCAR-3 cells were transiently transfected with 10 nmol/L siRNA (final concentration for maximum knock-down effect) using the transfection reagent for 48 hours and then treated with the agents as described under Results. In addition, a western blot was performed to validate the silencing of protein expression.
Akt2 stable cell line
SKOV3 cells were stably transfected with pcDNA3 based Akt2 expression vector which was a kind gift from Dr. Joseph R. Testa (Fox Chase Cancer Center, Philadelphia, PA) followed by G418 (Sigma) selection. Cell transfection was performed following the protocol of Lipofectamine 2000 (Invitrogen). The empty vector transfected cells were used as controls. After 12 days, resistant colonies were selected and confirmed Akt2 expression.
Statistical analysis
Data were analyzed by the paired Student's t-test and one-way analysis of variance (ANOVA) as appropriate. If a statistical significance (p ≤ 0.05) was determined by ANOVA, the data were further analyzed by Tukey's pairwise comparisons to detect specific differences between treatments.
Results
EGF stimulation induces variable effects on cell viability in ovarian cancer cells
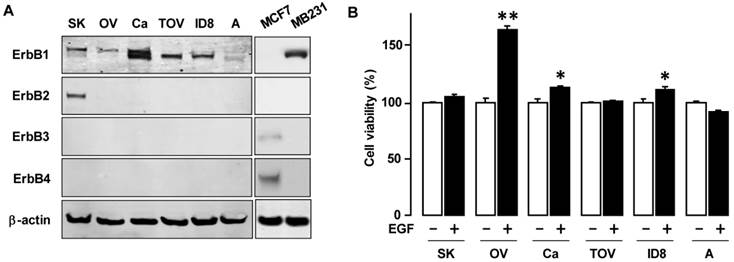
To determine the basal expression of EGFR isoforms, we examined a panel of ovarian cancer cells: one mouse ovarian cancer cell line (ID8) and five human ovarian cancer cell lines (OVCAR-3, SKOV-3, A2780, CaOV-3 and TOV-21G) (figure 1A). ErbB1 was highly expressed in OVCAR-3, SKOV-3, CaOV-3, TOV-21G and ID8 ovarian cancer cells while A2780 was less expressed. ErbB1 were expressed in two bands: probably phosphorylated from for upper bands and non-phosphorylated from for lower bands.ErbB2 was intensively expressed in SKOV-3 cells, while Erb3 and Erb4 showed minimal to no expression in the ovarian cancer cell lines. To evaluate the responsiveness to EGF, we tested the effects of EGF on cell viability. EGF increased cell viability in OVCAR-3 cells (and to a modest degree in CaOV-3 and ID8 cells), but had no significant effect on cell viability in the other cells (figure 1B).
EGF differentially affects cell viability in EGFR-expressing ovarian cancer cells. (A) Expression pattern of EGFR subtypes in a mouse and five human ovarian cancer cell lines. Whole cell lysates were prepared from each cell line and a western blot was carried out using antibodies specific to ErbB1, ErbB2, ErbB3 and ErbB4. Abbreviations used for ovarian cancer cell lines = SK (SKOV-3); OV (OVCAR-3); Ca (CaOV-3); TOV (TOV-21G); ID8 (ID8 mouse cells) and A (A2780). β-actin was used as a loading control. As positive controls for ErbB3 and ErbB4, MCF7 cells were used. MB231 cells are used as a positive control for ErbB1. (B) Differential effect of EGF on cell viability in various ovarian cancer cell lines. Cells were treated for 48 h with EGF (10 ng/mL). The cell viability assay was performed using MTT; values were normalized to untreated controls for each cell line. * indicates significance (p≤0.05) between EGF treated versus untreated cells by the paired Student's t-test. ** indicates significance (p≤0.05) between the cell lines having EGF-induced viability. Experiments were performed in triplicate and all data are shown as mean ± SEM.
The MAPK pathway is not a contributing factor to cell viability in OVCAR-3 and SKOV-3 ovarian cancer cells
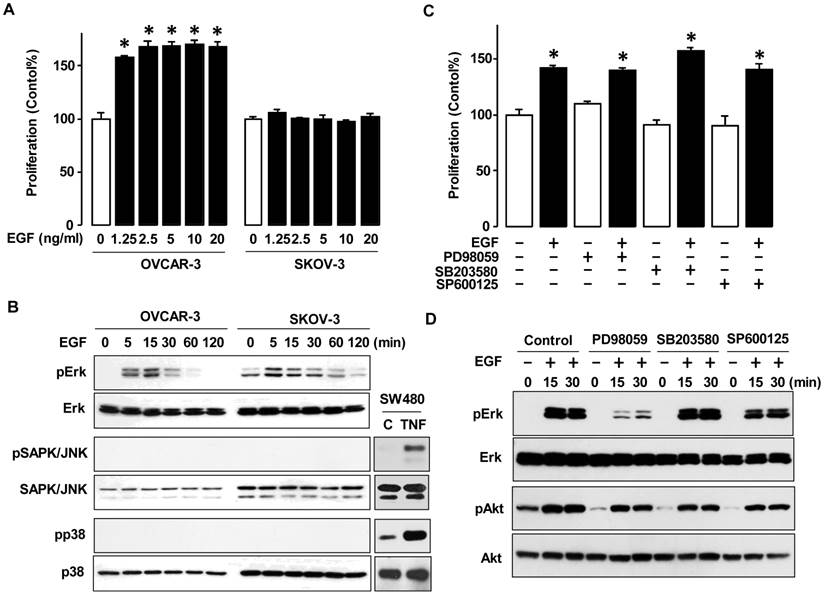
To investigate differences in response to EGF in a dose-dependent manner, we selected OVCAR-3 and SKOV-3 cells as representatives of frequently studied ovarian cancer cell lines. Both cell lines are known to have the PI3K/Akt pathway [11]. EGF increased cell viability in OVCAR-3 cells, but not SKOV-3 cells even at high concentrations (figure 2A). Next, we investigated MAPK activity as a main downstream of EGFR activation (figure 2B). In both OVCAR-3 and SKOV-3 cells, EGF activated Erk within the first 5-15 min followed by a decline in activation. However, EGF did not activate SAPK/JNK or p38 MAPK. We then used specific inhibitors for the MAPK pathway and re-evaluated cell viability. PD98059 (Erk inhibitor), SB203580 (p38 inhibitor) and SP600125 (SAPK/JNK inhibitor) had no significant effect on EGF-induced OVCAR-3 cell viability (figure 2C). We also determined if these MAPK inhibitors blocked the early Erk activation response in OVCAR-3 cells. The Erk inhibitor PD98059 blocked the EGF-activated Erk as anticipated, whereas neither SB203580 nor SP600125 significantly inhibited EGF-activated Erk (when normalized to expression levels of Erk as a loading control) (figure 2D). None of the MAPK inhibitors had a significant effect on EGF-activated Akt (figure 2D).
EGF activates Erk in ovarian cancer cells without affecting cell proliferation. (A) Differential effect of EGF on cell viability in OVCAR-3 versus SKOV-3 ovarian cancer cells. Cells were treated for 48 h with EGF in a dose-dependent manner (0, 1.25, 2.5, 5, 10 and 20 ng/mL). Blackened bars were EGF treated and * indicate a significant increase (p≤0.05) relative to controls by ANOVA and Tukey's pairwise comparisons. (B) Effect of EGF on the MAPK activation in OVCAR-3 versus SKOV-3 ovarian cancer cells. Cells were treated with EGF (10 ng/mL) in a time-dependent manner (0, 5, 15, 30, 60 and 120 min). As positive controls for phosphorylated p38 and SAPK/JNK, SW480 cells were used at 30 minute TNF (10 ng/ml) post-treatment. (C) Effects of MAPK inhibitors on EGF-induced cell viability in OVCAR-3 cells. Cells were treated for 48 h with EGF (10 ng/mL) alone or in combination with PD98059 (Erk inhibitor, 10 µmol/L), SB203580 (p38 inhibitor, 10 µmol/L) or SP600125 (SAPK/JNK inhibitor, 1 µmol/L). The blackened bars were EGF treated and * indicates a significant increase (p≤0.05) relative to paired samples without EGF by ANOVA and Tukey's pairwise comparisons. (D) Effects of MAPK inhibitors on Erk and Akt activation in OVCAR-3 cells. Cells were treated with EGF (10 ng/mL) in a time-dependent manner (0, 15 and 30 min) after a 30 min pretreatment with PD98059 (10 µmol/L), SB203580 (10 µmol/L) or SP600125 (1 µmol/L). For western blots, whole cell lysates were prepared and immunoblots were carried out using antibodies specific to Erk, SAPK/JNK, p38, Akt and their phosphorylated forms (pErk, pSAPK/JNK, pp38 and pAkt). Non-phosphorylated forms were used as loading controls. A cell viability assay was performed using MTT and values were normalized to untreated controls. Experiments were performed in triplicate and data are shown as a mean ± SEM.
EGF-induced cell viability in OVCAR-3 ovarian cancer cells is closely associated with Akt signaling
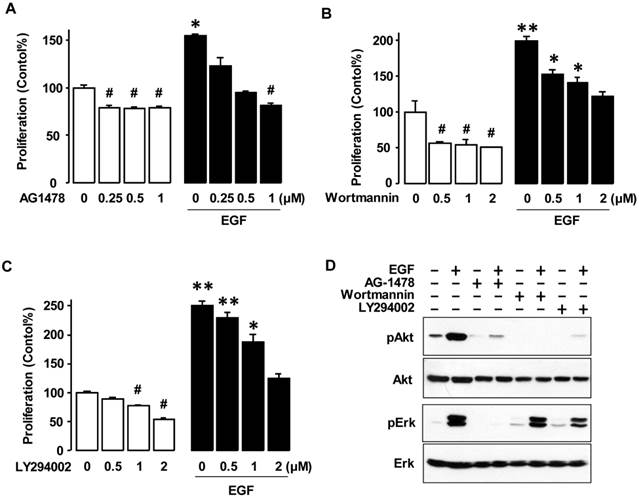
Since EGF-activated Erk had no significant effect on cell viability (Figure 2), we investigated the Akt pathway, which is another key downstream signaling linked to EGFR activation. The tyrosine kinase inhibitor associated with EGFR (AG-1478) significantly decreased both the basal level of cell viability in OVCAR-3 cells and blocked EGF-induced cell viability in a dose-dependent manner as expected (figure 3A). The PI3K inhibitors wortmannin and LY294002 decreased both the basal and EGF-induced cell viability in a dose-dependent manner (figures 3B and 3C). These PI3K inhibitors blocked EGF-activated Akt, but not EGF-activated Erk (figure 3D). AG-1478 also suppressed both EGF-activated Akt and Erk (figure 3D).
EGFR and Akt Inhibitors decrease EGF-induced cell viability by blocking Akt activation. (A) Dose-dependent effects of EGFR inhibitor AG-1478, the Akt inhibitors (B) wortmannin and (C) LY294002 on EGF-induced cell viability in OVCAR-3 cells. Cells were treated for 48 h with EGF (10 ng/mL) alone or in combination of AG-1478 (0, 0.25, 0.5 and 1 µmol/L), wortmannin (0, 0.5, 1 and 2 µmol/L) or LY294002 (0, 0.5, 1 and 2 µmol/L). A cell viability assay was performed using MTT and values were normalized to untreated controls (no EGF, no inhibitors). Blackened bars were EGF treated. *, ** indicate a significant increase (p≤0.05) and # indicates a significant decrease (p≤0.05) between groups by ANOVA and Tukey's pairwise comparisons. Experiments were performed in triplicate and all data are shown as mean ± SEM. (D) Effects of EGFR and Akt inhibitors on Erk and Akt activation in OVCAR-3 cells. Cells were treated for 15 min with EGF (10 ng/mL) after a 30 min pretreatment with AG-1478 (1 µmol/L), wortmannin (2 µmol/L) or LY294002 (2 µmol/L). Whole cell lysates were prepared and a western blot was carried out using antibodies specific to Erk and Akt and their phosphorylated forms (pErk and pAkt). Non-phosphorylated forms were used as loading controls.
The Akt2 isoform is an important contributor to cell proliferation in OVCAR-3 ovarian cancer cells
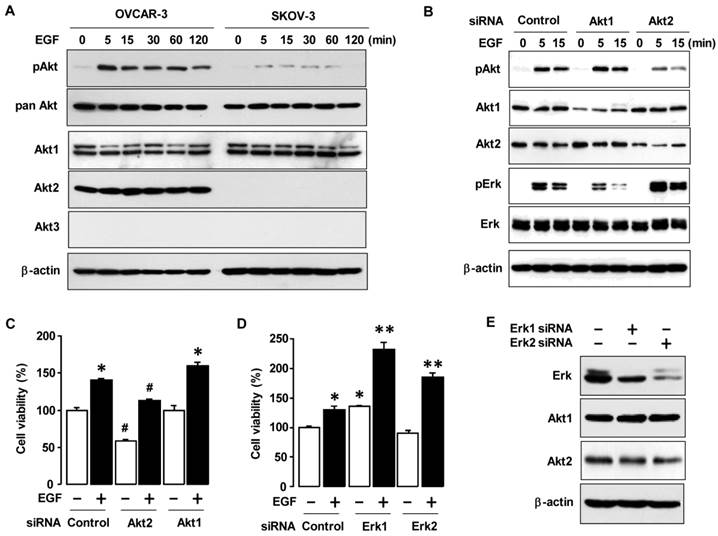
To determine if there is a difference in Akt signaling in OVCAR-3 and SKOV-3 cells, we stimulated the cells with EGF. OVCAR-3 cells responded to EGF with a robust activation of Akt as compared to SKOV-3 cells (figure 4A). Since Akt has three known isoforms (Akt1, Akt2 and Akt3) [13], we then investigated the expression patterns of Akt isoforms in OVCAR-3 versus SKOV-3 cells (figure 4A). The Akt1 isoform was expressed in both OVCAR-3 and SKOV-3 cells. Interestingly, Akt2 was specifically expressed in OVCAR-3 cells but was absent or less in SKOV-3 cells. Compared to Akt1 and Akt2, Akt3 was quite less expressed in either cell line. These findings suggest that the presence of Akt2 in OVCAR-3 cells could contribute to the differential effects of EGF on cell viability in OVCAR-3 versus SKOV-3 cells. Using Akt1 and Akt2 siRNAs to knockdown Akt1 and Akt2, we validated the critical effects of Akt1 and Akt2 silencing on EGF-activated Akt and Erk in OVCAR-3 cells (figure 4B). Akt2 siRNA reduced EGF-activated Akt and potentiated Erk activation. On the other hand, Akt1 siRNA did not alter EGF-activated Akt, but reduced Erk activation (in contrast to Akt2 siRNA). Furthermore, Akt1 siRNA induced Akt2 expression, probably via some compensatory mechanism. Next, we asked if Akt2 knockdown could impair EGF-induced cell viability. Indeed, Akt2 siRNA decreased both basal and EGF-induced cell viability in OVCAR-3 cells whereas Akt1 siRNA had no effect (figure 4C). On the other hand, knockdown of Erk1 or Erk2 with siRNAs did not decrease basal or EGF-induced cell growth (figure 4D). Interestingly, knockdown of Erk1 potentiated basal and EGF-induced cell viability (figure 4D). We validated knockdown of Erk1/2 which had no effect on Akt1 and Akt2 expression (figure 4E).
Akt2 signaling involves in EGF-activated cell viability. (A) Effects of EGF on Akt activation and status of Akt isoforms between OVCAR-3 and SKOV-3 ovarian cancer cells. Cells were treated with EGF (10 ng/mL) in a time-dependent manner (0, 5, 15, 30, 60 and 120 min). (B) Effect of siRNAs for Akt1 and Akt2 on EGF-activated Akt and Erk in OVCAR-3 cells. Cells were transiently transfected with siRNAs for Akt1 and Akt2 (final concentration 10 nmol/L) using the transfection reagent for 48 h followed by treatment with EGF (10 ng/mL) in a time-dependent manner (0, 5 and 15 min). (C) Effects of Akt1/2 siRNAs on EGF-induced cell viability in OVCAR-3 cells. Cells were transiently transfected with Akt1/2 siRNAs (final concentration 10 nmol/L) using the transfection reagent for 48 h followed by treatment for 48 h with EGF (10 ng/mL). (D) Effects of s Erk1/2 iRNAs on EGF-induced cell viability in OVCAR-3 cells. Cells were transiently transfected with Erk1/2 siRNAs (final concentration 10 nmol/L) for 48 h followed by treatment for 48 h with EGF (10 ng/mL). Blackened bars were EGF treated. * and ** indicate a significant increase (p≤0.05) between treatments by ANOVA and Tukey's pairwise comparisons. (E) The validation of Erk1 and Erk2 silencing. For western blot, whole cell lysates were prepared and immunoblots were carried out using antibodies specific for phosphorylated Akt (pAkt) and pErk, pan Akt and the three Akt isoforms (Akt1, Akt2 and Akt3). Pan Akt and β-actin were used as loading controls. Experiments were performed in duplicate and a representative result is shown. The cell viability assay was performed by using MTT, and values were normalized to untreated controls. Blackened bars were EGF treated. * and # indicate a significant increase and decrease (p≤0.05), respectively, by ANOVA and Tukey's pairwise comparisons. Experiments were performed in triplicate and all data are shown as mean ± SEM.
Reconstitution of Akt2 expression increases EGF responsiveness and increases cell proliferation in SKOV-3 cells
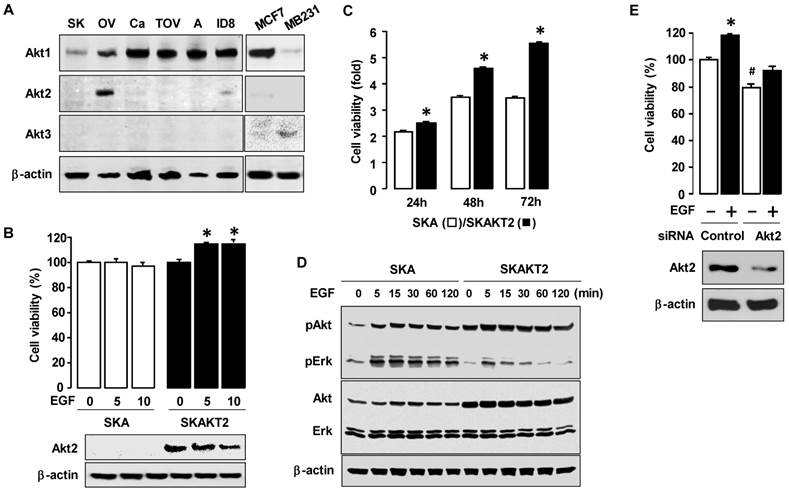
We examined the expression patterns of the Akt isoforms in ovarian cancer cell lines (ID8, OVCAR-1, SKOV-3, A2780, CaOV-3 and TOV-21G cells) (figure 5A). The Akt2 isoform was highly expressed in OVCAR-3 cells and low in ID8 cells, whereas this isoform was not clearly demonstrable in the other cell lines. Akt1 was highly expressed in all ovarian cancer cell lines, whereas Akt3 had a minimal expression as compared to other isoforms. To test the effects of Akt2 expression on cell viability, we generated stably transfected Akt2 SKOV-3 cells (SKAKT2). EGF increased cell proliferation in SKAKT2 cells whereas proliferation in SKA cells with empty vector demonstrated no effect (figure 5B). In addition, SKAKT2 cells grew more quickly than SKA cells even by 24 h (figure 5C). SKAKT2 cells induced more Akt activation basally and when induced by EGF than SKA cells with no AKT2 expression (figure 5D). Interestingly, SKAKT2 cells showed lower Erk activation in response to EGF than SKA cells (figure 5D). In addition, based on the expression of Akt2 in ID8 cells (figure 5A), we confirmed the involvement of Akt2 on EGF-induced cell viability in ID8 cells. Akt2 siRNA decreased both basal and EGF-induced cell viability also in ID8 cells (figure 5E) as demonstrated in OVCAR-3 cells. We also validated the knockdown of Akt2 expression in ID8 cells (figure 5E).
EGF markedly induces cell viability in Akt2-expressing ovarian cancer cells. (A) The expression pattern of Akt isoforms in mouse and five human ovarian cancer cells. The abbreviations of cells used were: ID8 (mouse epithelial ovarian cancer cells); OV (OVCAR-3); SK (SKOV-3); A (A2780); Ca (CaOV-3); and TOV (TOV-21G). MB231 cells are used as a positive control for Akt3. (B) Comparative effect of EGF on cell viability in SKA versus SKAKT2 cells. Cells were treated for 48 h with EGF (0, 5 or 10 ng/mL). Blackened bars were SKAKT2 cells and * indicates a significant increase (p≤0.05) relative to control by ANOVA and Tukey's pairwise comparisons. The validation of Akt2 expression was confirmed by western blot. (C) Comparison of cell viability in SKA versus SKAKT2 cells. Cells were cultured for 24, 48 and 72 h. Blackened bars were SKAKT2 cells and * indicates a significant increase (p≤0.05) relative to SKA cells by Student's t-test. (D) Effects of EGF on the Akt and Erk activation in SKA versus SKAKT2 cells. Cells were treated with EGF (10 ng/mL) in a time-dependent manner (0, 5, 15, 30, 60 and 120 min). For western blot, whole cell lysates were prepared and immunoblot was carried out using antibodies specific to phosphorylated Akt (pAkt) and Erk (pErk), Akt1/2/3 and Erk1/2. β-actin was used as a loading control. Experiments were performed in duplicate and a representative result is shown. (E) Effects of Akt2 siRNA on EGF-induced cell viability in ID8 cells. Cells were transiently transfected with Control or Akt2 siRNAs (final concentration 10 nmol/L) using the transfection reagent for 48 h followed by treatment for 48 h with EGF (10 ng/mL). The validation of Akt2 silencing was confirmed by western blot. The cell viability assay was performed by using MTT, and values were normalized to controls. Experiments were performed in triplicate and all data are shown as mean ± SEM.
Discussion
One of the primary findings in this study is that the existence of Akt2 appears to play an important role in EGF-induced cell viability in EGFR-expressing ovarian cancers.
Different response to EGF on the cell viability between OVCAR-3 and SKOV-3 cells indicates the involvement of specific EGFR downstream signaling links and the different therapeutic outcome to EGFR inhibitors. As example, cetuximab, an EGFR-directed monoclonal antibody, inhibited cell growth in OVCAR-3 cells, whereas SKOV-3 cell growth was not significantly affected [15]. The MAPK signaling pathway including Erk, SAPK/JNK and p38 MAPK has been well characterized and plays an important role in the regulation of proliferation, survival and apoptosis in response to external stimuli in ovarian cancer [16]. In spite of similar Erk activation between OVCAR-3 and SKOV-3 cells in response to EGF, Erk1/2 signaling is unlikely to be involved in EGF-induced cell viability. This fact is based on having no effect of MAPK inhibitors and Erk1/2 siRNA on EGF-induced cell viability. In addition, Erk1/2 silencing further enhanced the EGF-induced cell viability. In contrast, silencing Erk1/2 protein expression led to the suppression of cell proliferation in HeyC2 ovarian epithelial cancer cells and KGN granulosa tumor cells [17]. Because these two cell lines activate Erk1/2 constitutively, different responses in cell viability may exist between constitutive and inducible Erk1/2 activated cells. On the other hand, the ERK1/2 pathway is involved in the antiproliferative effect of type II gonadotropin-releasing hormone in OVCAR-3 cells [18]. Although endocrine-disrupting chemicals activate Erk1/2 and p38 MAPK, they are unlikely to be involved in estrogen-responsive ovarian cancer cell growth [19]. Interestingly, in prostate cancer cells Akt has a pivotal role in EGFR-mediated cell migration by activating epithelial-mesenchymal transition rather than Erk signaling [20]. Further studies are necessary to explore the possible roles of MAPK activation in ovarian cancer.
Akt plays pivotal roles in many responses including growth, survival, migration, invasion and metastasis. EGF strongly activated Akt in OVCAR-3 cells compared to SKOV-3 cells and specific EGFR/PI3K inhibitors decreased EGF-induced cell viability by blocking Akt activation and maintaining Erk1/2 activation. These results indicate the impact of Akt signaling on EGF-induced cell viability as supported in several reports. Ascites is commonly found in advanced ovarian cancer patients. Malignant ascites prevents TRAIL-induced apoptosis by activating Akt without affecting Erk phosphorylation in human ovarian carcinoma cells [21]. Akt genes are frequently observed in high-grade malignant ovarian cancers [4, 22]. Since the Akt pathway is often aberrantly regulated in various cancers, targeting Akt may be a novel, and more effective anticancer therapy [23, 24]. Actually, because Akt activation promotes cisplatin-resistance, inhibition of Akt activity indicates be a useful therapeutic approach for the treatment of cisplatin-resistant ovarian cancer [25, 26]. Three members of the Akt family have been identified in mammals: Akt1, Akt2 and Akt3, which are ubiquitously expressed in all cell types and tissues [13, 27]. In particular, Akt2 was highly expressed in OVCAR-3 cells as previously reported [28]. Although other authors indicated a low expression of Akt2 in SKOV-3 cells, OVCAR-3 cells expressed higher Akt2 as compared to other ovarian cancer cells [29]. These facts indicate that Akt2 may be a significant regulator of EGF-induced cell viability. Akt2 siRNA decreased basal cell growth whereas Akt1 siRNA had no effect as observed by others [30]. Silencing Akt2 blocked EGF-induced Akt phosphorylation and cell viability. On the other hand, overexpression of Akt2 increased the response to EGF and enhanced cell viability. Supportably, other authors indicated that activated Akt2 might increase ovarian cancer cell survival via inhibition of apoptosis [31]. Frequent activation of Akt2 is a common occurrence in human ovarian cancer [32] (particularly in undifferentiated tumors) and is often followed by a poor prognosis [33]. Although silencing Akt1 had no significant effect on cell viability, it is likely to be involved in cell migration and invasion [34]. Akt1, for instance, was specifically involved in the migration of lysophosphatidic acid (LPA)-induced mouse embryo fibroblast cells [35]. In addition, a point mutation (E17K) in the Akt1 gene was described in ovarian cancer cells and was found to induce leukemia in mice [36]. The Akt1 transfected NIH3T3 cells resulted in a malignant phenotype. These data indicate that Akt1 may be a determinant of oncogenesis [37]. Also, specific silencing of Akt3 inhibited proliferation of ovarian cancer OVCA429, DOV13 and ES2 cells which have high Akt3 expressions [28, 38]. On the contrary, in lung cancer, Akt1 ablation delayed initiation of lung tumor growth, Akt2 deficiency dramatically accelerated the growth rate of lung tumors and Akt3 ablation had a small, not statistically significant [39]. But several reports in other cancer types support in part our results. In the androgen-independent prostate cell line PC-3, Akt1 ablation induced loss of cell adhesion whereas Akt2 (but not Akt1) ablation promoted cell cycle arrest at G0/G1 [40]. Suppression of Akt2 expression in highly metastatic colorectal cancer cells inhibited metastasis and even overexpression of Akt1 did not restore the metastatic potential [41]. Also Akt2 depleted MDA-MB-231 breast cancer cells showed a marked reduction in metastasis to mouse lungs [42]. As described in other cancer types, a subset of ovarian cancers are likely to differentially regulate Akt1 [37], Akt2 [32] and Akt3 [28, 38] in Akt isoform specific manner. Although potential mechanisms for how Akt2 reduces the phosphorylated Erk1/2 are still unknown, Akt2-dependent signaling may encroach on Erk activation in constant response to EGF.
Regulation of each Akt isoform in ovarian cancer is widely unknown. Therefore, it is important that the specific roles of each Akt isoform be further clarified in ovarian tumors. We have shown that Akt2 dependent signaling plays an important role in the cellular viability in some EGFR-expressing ovarian cancer cells like OVCAR-3, which may provide a potential therapeutic approach for a sub-set of ovarian cancers.
Acknowledgements
The authors thank Dr. Diana Marver (Meharry Medical College, Nashville, TN) for her editorial suggestions. The authors are grateful to Dr. Andrew Godwin (University of Kansas, Kansas City, KS) for the A2780 human ovarian cancer cell line, Drs. Katherine Roby and Paul Terranova (University of Kansas, Kansas City, KS) for the mouse ovarian surface epithelial cancer cell line (ID8) and Dr. Joseph R. Testa (Fox Chase Cancer Center, Philadelphia, PA) for the Akt2 expression vector. Grant support: This research was supported by NIH grants U54CA091408 (DK & DS), G12RR003032 (DK & DS), K08CA148887 (DK), SC1 089630 (EL) and SC1AI089073 (DS).
Conflicts of Interest
No potential conflicts of interest were disclosed.
References
1. Jayson GC, Kohn EC, Kitchener HC. et al. Ovarian cancer. Lancet. 2014 [Epub ahead of print]
2. Rooth C. Ovarian cancer: risk factors, treatment and management. Br J Nurs. 2013;22:S23-30
3. Sudo T. Molecular-targeted therapies for ovarian cancer: prospects for the future. Int J Clin Oncol. 2012;17:424-9
4. Landen CN Jr, Birrer MJ, Sood AK. Early events in the pathogenesis of epithelial ovarian cancer. J Clin Oncol. 2008;26:995-1005
5. Siwak DR, Carey M, Hennessy BT. et al. Targeting the epidermal growth factor receptor in epithelial ovarian cancer: current knowledge and future challenges. J Oncol. 2010;2010:568938
6. Normanno N, De Luca A, Bianco C. et al. Epidermal growth factor receptor (EGFR) signaling in cancer. Gene. 2006;366:2-16
7. Lafky JM, Wilken JA, Baron AT. et al. Clinical implications of the ErbB/epidermal growth factor (EGF) receptor family and its ligands in ovarian cancer. Biochim Biophys Acta. 2008;1785:232-65
8. Palayekar MJ, Herzog TJ. The emerging role of epidermal growth factor receptor inhibitors in ovarian cancer. Int J Gynecol Cancer. 2008;18:879-90
9. Gui T, Shen K. The epidermal growth factor receptor as a therapeutic target in epithelial ovarian cancer. Cancer Epidemiol. 2012;36:490-6
10. Wilken JA, Badri T, Cross S. et al. EGFR/HER-targeted therapeutics in ovarian cancer. Future Med Chem. 2012;4:447-69
11. Moscova M, Marsh DJ, Baxter RC. Protein chip discovery of secreted proteins regulated by the phosphatidylinositol 3-kinase pathway in ovarian cancer cell lines. Cancer Res. 2006;66:1376-83
12. Sheng Q, Liu J. The therapeutic potential of targeting the EGFR family in epithelial ovarian cancer. Br J Cancer. 2011;104:1241-5
13. Franke TF. PI3K/Akt: getting it right matters. Oncogene. 2008;27:6473-88
14. Bull Phelps SL, Schorge JO, Peyton MJ. et al. Implications of EGFR inhibition in ovarian cancer cell proliferation. Gynecol Oncol. 2008;109:411-7
15. Bijman MN, van Berkel MP, Kok M. et al. Inhibition of functional HER family members increases the sensitivity to docetaxel in human ovarian cancer cell lines. Anticancer Drugs. 2009;20:450-60
16. Choi KC, Auersperg N, Leung PC. Mitogen-activated protein kinases in normal and (pre)neoplastic ovarian surface epithelium. Reprod Biol Endocrinol. 2003;1:71
17. Steinmetz R, Wagoner HA, Zeng P. et al. Mechanisms regulating the constitutive activation of the extracellular signal-regulated kinase (ERK) signaling pathway in ovarian cancer and the effect of ribonucleic acid interference for ERK1/2 on cancer cell proliferation. Mol Endocrinol. 2004;18:2570-82
18. Kim KY, Choi KC, Park SH. et al. Extracellular signal-regulated protein kinase, but not c-Jun N-terminal kinase, is activated by type II gonadotropin-releasing hormone involved in the inhibition of ovarian cancer cell proliferation. J Clin Endocrinol Metab. 2005;90:1670-7
19. Park SH, Kim KY, An BS. et al. Cell growth of ovarian cancer cells is stimulated by xenoestrogens through an estrogen-dependent pathway, but their stimulation of cell growth appears not to be involved in the activation of the mitogen-activated protein kinases ERK-1 and p38. J Reprod Dev. 2009;55:23-9
20. Gan Y, Shi C, Inge L. et al. Differential roles of ERK and Akt pathways in regulation of EGFR-mediated signaling and motility in prostate cancer cells. Oncogene. 2010;29:4947-58
21. Lane D, Robert V, Grondin R. et al. Malignant ascites protect against TRAIL-induced apoptosis by activating the PI3K/Akt pathway in human ovarian carcinoma cells. Int J Cancer. 2007;121:1227-37
22. de Graeff P, Crijns AP, Ten Hoor KA. et al. The ErbB signalling pathway: protein expression and prognostic value in epithelial ovarian cancer. Br J Cancer. 2008;99:341-9
23. Steelman LS, Stadelman KM, Chappell WH. et al. Akt as a therapeutic target in cancer. Expert Opin Ther Targets. 2008;12:1139-65
24. Chen Y, Wang BC, Xiao Y. PI3K: a potential therapeutic target for cancer. J Cell Physiol. 2012;227:2818-21
25. Yang X, Fraser M, Abedini MR. et al. Regulation of apoptosis-inducing factor-mediated, cisplatin-induced apoptosis by Akt. Br J Cancer. 2008;98:803-8
26. Peng DJ, Wang J, Zhou JY. et al. Role of the Akt/mTOR survival pathway in cisplatin resistance in ovarian cancer cells. Biochem Biophys Res Commun. 2010;394:600-5
27. Hers I, Vincent EE, Tavaré JM. Akt signalling in health and disease. Cell Signal. 2011;23:1515-27
28. Cristiano BE, Chan JC, Hannan KM. et al. A specific role for AKT3 in the genesis of ovarian cancer through modulation of G(2)-M phase transition. Cancer Res. 2006;66:11718-25
29. Hanrahan AJ, Schultz N, Westfal ML. et al. Genomic complexity and AKT dependence in serous ovarian cancer. Cancer Discov. 2012;2:56-67
30. Noske A, Kaszubiak A, Weichert W. et al. Specific inhibition of AKT2 by RNA interference results in reduction of ovarian cancer cell proliferation: increased expression of AKT in advanced ovarian cancer. Cancer Lett. 2007;246:190-200
31. Huang Q, Lan F, Zheng Z. et al. Akt2 kinase suppresses glyceraldehyde-3-phosphate dehydrogenase (GAPDH)-mediated apoptosis in ovarian cancer cells via phosphorylating GAPDH at threonine 237 and decreasing its nuclear translocation. J Biol Chem. 2011;286:42211-20
32. Yuan ZQ, Sun M, Feldman RI. et al. Frequent activation of AKT2 and induction of apoptosis by inhibition of phosphoinositide-3-OH kinase/Akt pathway in human ovarian cancer. Oncogene. 2000;19:2324-30
33. Bellacosa A, de Feo D, Godwin AK. et al. Molecular alterations of the AKT2 oncogene in ovarian and breast carcinomas. Int J Cancer. 1995;64:280-5
34. Meng Q, Xia C, Fang J. et al. Role of PI3K and AKT specific isoforms in ovarian cancer cell migration, invasion and proliferation through the p70S6K1 pathway. Cell Signal. 2006;18:2262-71
35. Kim EK, Yun SJ, Do KH. et al. Lysophosphatidic acid induces cell migration through the selective activation of Akt1. Exp Mol Med. 2008;40:445-52
36. Carpten JD, Faber AL, Horn C. et al. A transforming mutation in the pleckstrin homology domain of AKT1 in cancer. Nature. 2007;448:439-44
37. Sun M, Wang G, Paciga JE. et al. AKT1/PKBalpha kinase is frequently elevated in human cancers and its constitutive activation is required for oncogenic transformation in NIH3T3 cells. Am J Pathol. 2001;159:431-7
38. Liby TA, Spyropoulos P, Buff Lindner H. et al. Akt3 controls vascular endothelial growth factor secretion and angiogenesis in ovarian cancer cells. Int J Cancer. 2012;130:532-43
39. Linnerth-Petrik NM, Santry LA, Petrik JJ, Wootton SK. Opposing functions of akt isoforms in lung tumor initiation and progression. PLoS One. 2014;9:e94595
40. Cariaga-Martinez AE, López-Ruiz P, Nombela-Blanco MP. et al. Distinct and specific roles of AKT1 and AKT2 in androgen-sensitive and androgen-independent prostate cancer cells. Cell Signal. 2013;25:1586-97
41. Rychahou PG, Kang J, Gulhati P. et al. Akt2 overexpression plays a critical role in the establishment of colorectal cancer metastasis. Proc Natl Acad Sci USA. 2008;105:20315-20
42. Wang J, Wan W, Sun R. et al. Reduction of Akt2 expression inhibits chemotaxis signal transduction in human breast cancer cells. Cell Signal. 2008;20:1025-34
Author contact
![]() Corresponding author: DS Son, Department of Biochemistry and Cancer Biology, Meharry Medical College, 1005 Dr. D.B. Todd Jr. Blvd, Nashville, TN 37208, USA, Tel:+1-615-327-6501; Fax: +1-615-327-6042, Email: dsonedu.
Corresponding author: DS Son, Department of Biochemistry and Cancer Biology, Meharry Medical College, 1005 Dr. D.B. Todd Jr. Blvd, Nashville, TN 37208, USA, Tel:+1-615-327-6501; Fax: +1-615-327-6042, Email: dsonedu.