Impact Factor ISSN: 1837-9664

 Global reach, higher impact
Global reach, higher impactJ Cancer 2014; 5(9):745-753. doi:10.7150/jca.9696 This issue Cite
Research Paper
cMET Activation and EGFR-Directed Therapy Resistance in Triple-Negative Breast Cancer
Joohyuk Sohn1,2, Shuying Liu1, Napa Parinyanitikul1, Jangsoon Lee1, Gabriel N. Hortobagyi1, Gordon B. Mills3, Naoto T. Ueno1, Ana M. Gonzalez-Angulo1,3 ![]()
1. Department of Breast Medical Oncology, The University of Texas MD Anderson Cancer Center, Houston, TX, USA
2. Division of Medical Oncology, Department of Internal Medicine, Breast Cancer Clinic, Severance Hospital, Yonsei University College of Medicine, Seoul, Korea
3. Department of Systems Biology, The University of Texas MD Anderson Cancer Center, Houston, TX, USA
Received 2014-5-20; Accepted 2014-6-15; Published 2014-10-15
Abstract
Background: EGFR expression and pathway activation are common in triple-negative breast cancer (TNBC). However, anti-EGFR therapies have not been effective in these patients. We aimed to study the efficacy of targeting MET in overcoming resistance to EGFR therapy in TNBC cell lines.
Methods: TNBC lines (MDA-MB-468, HCC-1395, and MDA-MB-231), and a hormone receptor-positive breast cancer line (T47D) were stimulated with epidermal growth factor (EGF) and hepatocyte growth factor (HGF). Lines were then treated with different concentrations of EGFR inhibitors (gefitinib or cetuximab), with or without a MET tyrosine kinase inhibitor (EMD 1214063). Proliferation was measured by MTS assay, in soft agar and with a matrigel assay. Synergy was measured with Calcusyn. Protein expression and signaling were examined with immunoblotting.
Results: There was activation of ligand-receptor-downstream signaling pathways in MDA-MB-468 and HCC-1395 upon stimulation with EGF and HGF. In these cell lines, we observed synergism when combining EGFR and MET inhibitors. These results were observed across assays. In western blotting, combination therapy resulted in abrogation of pAKT and pMAPK while monotherapy did not.
Conclusion: Our data demonstrate that dual EGFR/MET inhibition is synergistic in TNBC. Targeting both EGFR and MET receptors may provide an effective therapeutic strategy in TNBC.
Keywords: cMET, EGFR, Therapy resistance, Triple-negative breast cancer
Introduction
Triple-negative breast cancer (TNBC) comprises 15% of breast cancers, and today has the poorest survival outcome of all breast cancer subtypes. Due to its heterogeneity, TNBC lacks validated therapeutic targets compared with other breast cancer subtypes [1]. Several molecular targets including the epidermal growth factor receptor (EGFR), the poly ADP ribose polymerase (PARP), and angiogenesis ligands and receptors, are currently under clinical investigation for the treatment of this disease [2].
In breast cancer, EGFR has been reported to be overexpressed in approximately in 50% of TNBC and observed to be an independent predictor of poor prognosis [3,4]. Genomic amplification of EGFR is also reported in 6% of breast cancers and correlates with increased protein expression [5]. However, the use of anti-EGFR therapies, either with EGFR catalytic domain inhibitors or monoclonal antibodies has not proven to be effective in this disease. [6,7].
The receptor tyrosine kinase MET acts as an oncogene in many human cancers [8]. It is overexpressed in 20-30% of breast cancer and seems to be associated with a worse prognosis [8-11]. MET amplification and overexpression leads to resistance to anti-EGFR therapies in non-small cell lung cancer [12] and contributes to inefficiency of gefitinib in brain tumors through receptor co-activation with EGFR [13]. We hypothesized that MET activation contributes to the intrinsic resistance to anti-EGFR therapies in TNBC.
Materials and Methods
Cell lines
TNBC cell lines (MDA-MB-468, HCC-1395, and MDA-MB-231) and the hormone receptor positive cell line (T47D) were used. Cell lines were obtained from MD Anderson Cancer Center (MDACC)'s cell line core. Cells were grown in Roswell Park Memorial Institute (RPMI)-1640 medium supplemented with 5% fetal bovine serum (FBS) and penicillin/streptavidin, and maintained at 37°C in a humidified atmosphere at 5% CO2.
Chemicals
Gefitinib was.purchased from LC laboratories (Woburn, MA). EMD 1214063 (MSC2156119J) was provided by EMD Serono (Rockland, MA). Both drugs were dissolved in DMSO. Cetuximab purchased from ImClone Systems, Inc. (Bridgewater, NJ) was provided by the clinical pharmacy at MDACC. Compounds were stored as 10mM aliquots at -80°C. EGF and HGF were purchased from R&D system (Minneapolis, MN).
Cell viability assays
Cells were seeded in 96-well microplates in medium supplemented with 5% FBS and penicillin/streptavidin. The optimal cell number for each cell line was determined to ensure that each was in log growth phase at the end of the assay (~70% confluency). Cells were allowed to attach for 24 hours. The media was changed to low FBS (2%) and drugs with different combinations were added (cetuximab 200ug/mL, gefitinib 0.25-8 umol/L and EMD 121463 2-10 umol/L). In terms of determination of drug concentration for combination assays, twofold serial dilution was conducted for gefitinib and dose was increased by 2 umol/L in EMD 121463 with the upper limit of 10 umol/L because of poor solubility. Cells were incubated at 37°C for 72 hours. Growth was determined with Cell Titer Blue (Promega, Madison, WI) at a 72-hour time point according to the manufacturer's instructions. Quantification of fluorescent signal intensity was performed using a fluorescent plate reader at excitation and emission wavelengths of 530/604.
Soft agar assay
Cells were suspended in complete RPMI medium containing 0.3% soft agar and seeded in triplicate on 35-mm dishes precoated with 0.6% agar in complete growth medium mixed with the corresponding drug and incubated at 37°C, 5% CO2. Drug concentrations were decided from the results of cell viability assay (cetuximab 200ug/mL, gefitinib 0.25-8 umol/L and EMD 121463 2-10 umol/L). The same drug concentration was used both as a single agent and in combinations. Medium containing different drugs or combinations were added to each well every 3-4 days. After 10 days, colonies were photographed and counted in 10 randomly chosen fields and expressed as means of triplicates. For staining of the colonies, 0.005% crystal violet in 20% methanol solution was added to the dishes for 30 minutes and washed until the background became clear.
Matrigel assay
3D matrices were reconstituted from Matrigel (BD Biosciences, San Jose, CA). Thawed 45ul Matrigel on ice were placed in each well of a pre-cooled 8-well glass chamber slide (BD Falcon, San Jose, CA). After 30 minutes of solidification, TNBC cell lines and T47D mixed in ice-cold medium with 2% Matrigel and drugs were loaded in each well of the 8-chamber slide and incubated at 37°C. Media with 2% Matrigel and drugs were changed every three days. Drug concentrations were decided from the results of cell viability assay (cetuximab 200ug/mL, gefitinib 0.25-8 umol/L and EMD 121463 2-10 umol/L).
Western blot
Cells were starved with RPMI 1640 medium overnight and next morning, they were stimulated with EGF 20ng/mL and/or recombinant human HGF 75ng/mL for 10 minutes before cell lysis. Cells were lysed in ice-cold X-100 lysis buffer (1% Triton X-100, 50mM Hepes, pH 7.4, 150mM NaCl, 1.5mM MgCl2, 1mM EGTA, 10% glycerol, 100mM NaF, 10mM sodium pyrophosphate, and 1mM Na3VO4). Before use, protease and phosphatase inhibitors (Roche Applied Science, Indianapolis, IN) were added. Cell lysates were resolved by SDS/PAGE, transferred to Immobilon (polyvinylidene difluoride), and immunoblotted with antibodies following the protocols provided by the manufacturers. Immunocomplexes were visualized with an enhanced chemiluminescence detection kit (Amersham Bioscience, Pittsburgh, PA) using horseradish peroxidase-conjugated secondary antibodies (Bio-Rad, Hercules, CA). The following antibodies were used for immunoblotting: Anti-phospho-Akt (T308, S474), anti-total Akt, anti-EGFR, anti-pEGFR (Y992, Y1068), anti-MET, anti-pMET (Y1235), anti-pMAPK (T202/Y204), anti-pS6 (S235/236), anti-S6, anti-pSTAT3 (Y1289) were from Cell Signaling Technology (Beverly, MA). Anti-total Erk was purchased from Santa Cruz Biotechnology (Santa Cruz, CA) and anti-β actin from Sigma (St. Louis, MO).
Immunocomplexes were visualized with an enhanced chemiluminescence detection kit (Amersham Biosciences) using horseradish peroxidase-conjugated secondary antibodies (Bio-Rad, Herculus, CA).
Antibody arrays
MDA-MB-468 in 100mm dish was harvested as described in western blotting and Phospho-Receptor Tyrosine Kinase (RTK) antibody arrays (R&D System, #ARY-001, Minneapolis, MN) were performed as recommended but with 450ug protein lysate per array.
DNA transfection
AKT and MEK inserted in pCMV5-expression vector with a selection marker of ampicilline (100ug/ml) were kindly provided by Dr. Ueno at MDACC. DNA was gently diluted (0.01ug/ul) with serum free media. Diluted DNA (100ul) was mixed with X-tremeGENE DNA transfection reagent (3ul) (Roche Applied Science) and incubated for 15 minutes at room temperature. These mixtures were added into the 60mm dish culturing MDA-MB-468 on day 1 and incubated overnight and drugs including gefitinib (1uM) and EMD 121463 (5uM) alone or in combination were treated with serum-free media on day 2. Cells were harvested for western blotting after treatment of EGF 20ng/mL and/or recombinant human HGF 75ng/mL for 10 minutes before cell lysis.
Statistical Methods
Student's t test was used to compare unpaired variables and P<0.05 (2-sided) was considered statistically significant. Synergism for combination therapies was defined using Calcusyn software (Biosoft, Ferguson, MO) and applied to MTS assays. The fraction of cells affected (Fa) was determined and combination indices (CI) were produced by the software that utilizes the methodology applied by Chou and Talalay [14].
Results
Combination of EGFR and MET inhibition is synergistic in triple-negative breast cancer cell lines
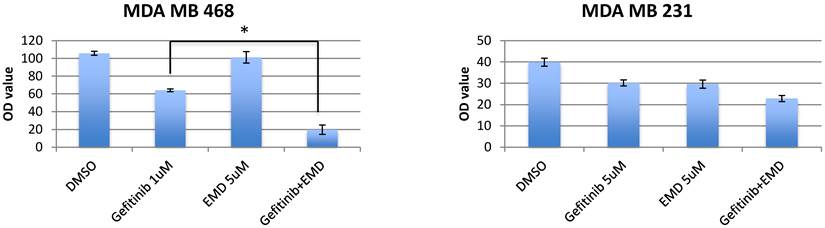
To determine the sensitivity of TNBC cell lines and T47D to EGFR inhibition, MET inhibition, and combined therapy inhibition, breast cancer cell lines were treated with gefitinib (0.25-8 umol/L), EMD 121463 (2-10 umol/L), or both agents given at the same concentration in combination for 72 hours. GI 50 of these cell lines were 1-5 uM to gefitinib and 4uM- ≥10uM to EMD 121463. These cell lines were essentially resistant to both gefitinib and EMD 121463 as single agents. The inhibitory effect of combined treatment with gefitinib and EMD 121463 was significantly enhanced compared with single agent therapy in MDA-MB-468 cells (P=0.002) but not in the other cell lines (MDA-MB-231, HCC1395, and T47D) (Fig.1). However, when data were analyzed in Calcusyn software, synergism was also documented at different concentration combinations in the other cell lines (Table 1). These experiments indicate a dose dependent synergistic interaction between gefitinib and EMD 121463 in suppressing growth of triple-negative breast cancer cells.
Combination index (CI)
| Cell line | Gefitinib (uM) | EMD 121463 (uM) | CI |
|---|---|---|---|
| MDA-MB-468 | 1 | 6 | 0.87 |
| HCC 1395 | 0.5 | 2 | 0.39 |
| HCC 1395 | 1 | 4 | 0.15 |
| HCC 1395 | 2 | 6 | 0.29 |
| MDA-MB-231 | 0.5 | 2 | 0.59 |
| T47D | 0.5 | 2 | 0.55 |
| T47D | 1 | 4 | 0.73 |
Effects of combination of EGFR inhibitors with cMET inhibitor, EMD 1214063 in MTS assay. Cells were seeded in 96-well microplates in medium supplemented with 5% FBS and penicillin/streptavidin. The optimal cell number for each cell line was determined to ensure that each was in growth phase at the end of the assay (~70% confluency). Cells were allowed to attach for 24 hours. The media was changed to low FBS (2%) and drugs with different combinations were added (cetuximab 200ug/mL, gefitinib 0.25-8 umol/L and EMD 121463 2-10 umol/L). In terms of determination of drug concentration, twofold serial dilution was conducted for gefitinib and dose was increased by 2 umol/L in EMD 121463 with the upper limit of 10 umol/L because of poor solubility. Cells were incubated at 37°C for 72 hours. Growth inhibition was determined with Cell Titer Blue (Promega, Madison, WI) at a 72-hour time point according to the manufacturer's instructions. Quantification of fluorescent signal intensity was performed using a fluorescent plate reader at excitation and emission wavelengths of 530/604. Drug concentrations were as follows; Gefitinib 1uM and EMD 121463 5uM in MDA-MB-468; Gefitinib 5uM and EMD 121463 5uM in MDA-MB-231. There was no statistical difference in T47D and HCC1395 (data not shown). The data are mean ± standard deviations of triplicates (*, P<0.001).
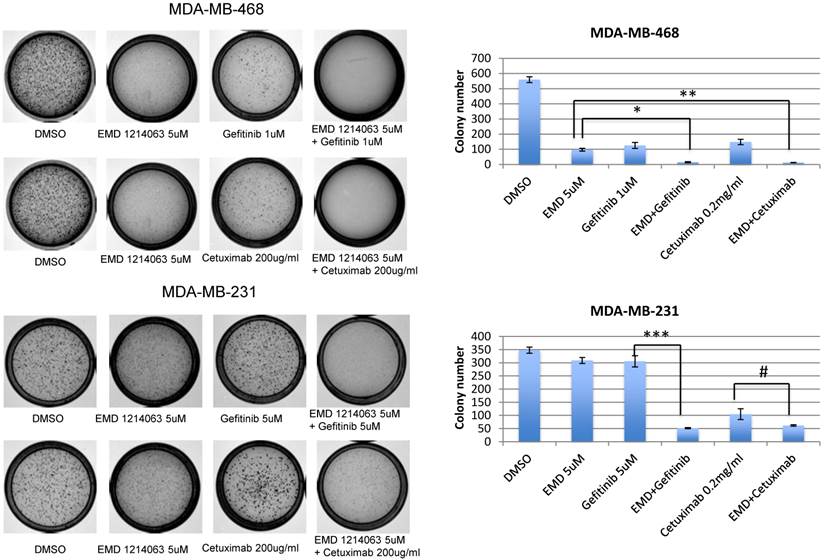
Effects of EGFR and MET inhibition in soft agar and 3D culture
In order to evaluate effects of drug combinations on anchorage independent growth, cells were seeded in soft agar and EGFR inhibitors (gefitinib or cetuximab), EMD 121463, or combinations were added. After 10-14 days, the colony size and number were significantly reduced in the MDA-MB-468 cell line when gefitinib or cetuximab was combined with EMD 121463 compared to single agents. In MDA-MB-231 cell line, there was a significant reduction of colony number when EMD 121463 was combined with gefitinib but not with cetuximab compared to single agents (Fig. 2).
Effects of combination of EGFR inhibitors with cMET inhibitor EMD 1214063 on soft agar assay. Cells were suspended in complete RPMI medium containing 0.3% soft agar and seeded in triplicate on 35-mm dishes precoated with 0.6% agar in complete growth medium mixed with corresponding drug and incubated at 37°C, 5% CO2. After 10 days, colonies were photographed and counted in 10 randomly chosen fields and expressed as means of triplicates. For staining of the colonies, 0.005% crystal violet in 20% methanol solution were added to the dishes for 30 minutes and washed until the background becomes clear. Drug concentrations were as follows; Gefitinib 1uM, EMD 121463 5uM, and Cetuximab 200 ug/ml, in MDA-MB-468; Gefitinib 5uM, EMD 121463 5uM, and Cetuximab 200 ug/ml in MDA-MB-231. Colonies were not formed in HCC 1395 and there were no difference between single agent and combination in T47D (data not shown). The data are mean ± standard deviations of triplicates (*, P<0.001 vs EMD 121463; **, P<0.05 vs EMD 121463; ***, P<0.001 vs gefitinib; #, P>0.05 vs cetuximab).
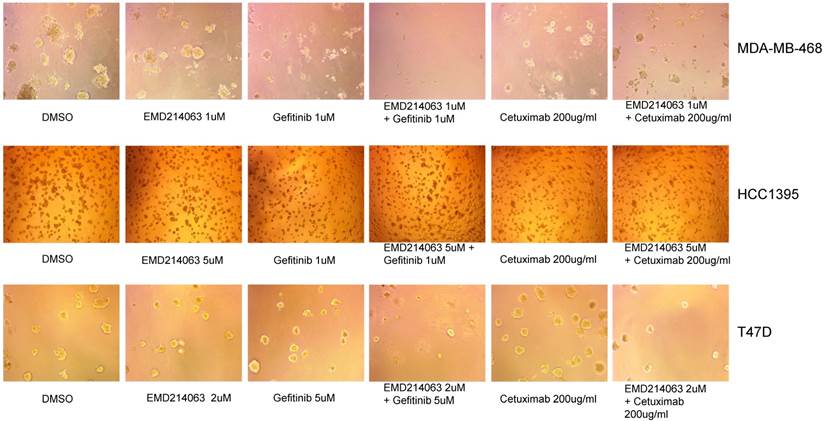
We also observed three-dimensional morphology of the cell lines in Matrigel with the drugs as described above. Again, MDA-MB-468 showed remarkable regression when the drug combination was applied but not with the other cell lines assessed (Fig. 3).
Matrigel assay in TNBC cell lines, and T47D. Thawed 45ul Matrigel on ice were placed in each well of a pre-cooled 8-well glass chamber slide. After 30 minutes of solidification, TNBC cell lines and T47D mixed in ice-cold medium with 2% Matrigel and drugs were loaded in each well of the 8-chamber slide and incubated at 37°C. Media with 2% Matrigel and various concentrations of drugs were changed every three days. Drug concentrations were as follows; Gefitinib 1um, EMD 121463 1uM and Cetuximab 200ug/ml in MDA-MB-468, Gefitinib 1um, EMD 121463 5uM and Cetuximab 200ug/ml in HCC 1395, Gefitinib 5um, EMD 121463 2uM and Cetuximab 200ug/ml in T47D.
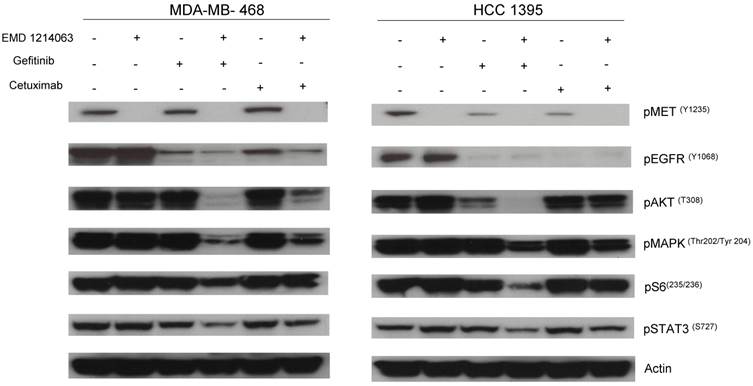
Effects of EGFR and MET inhibition on downstream signaling
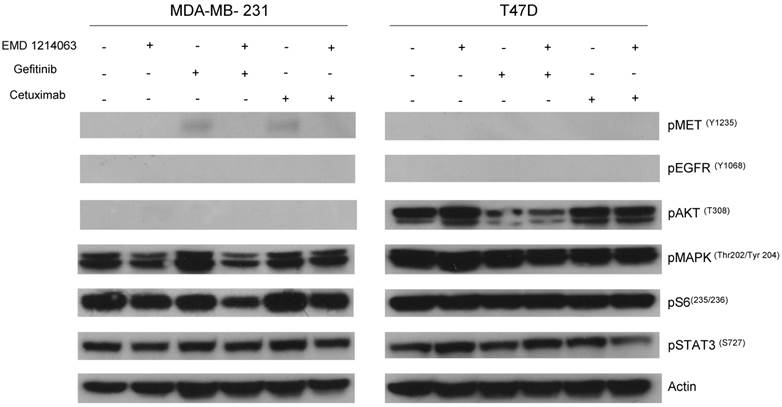
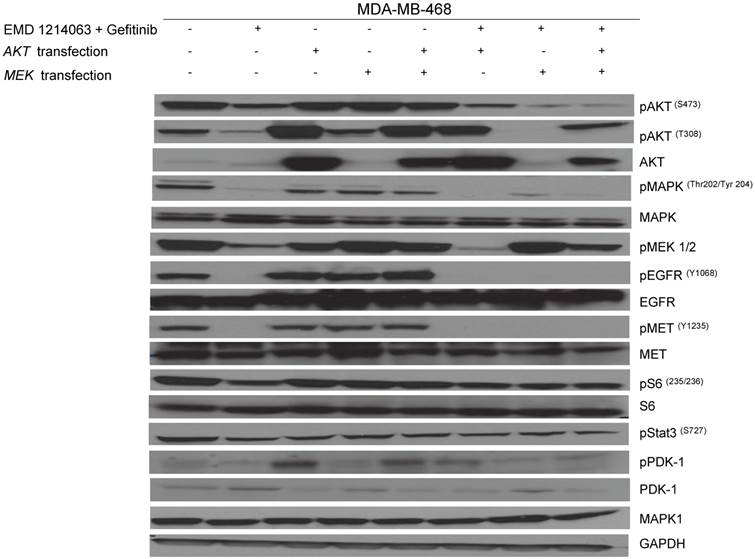
Then, we investigated the changes in downstream signaling for each cell line before and after treatment of drug alone and in combination. In western blotting, both gefitinib and cetuximab reduced pEGFR and EMD1214063 abrogated pMET, respectively in MDA- MB-468 as well as in HCC-1395. Either drug alone was insufficient to alter pAKT, pMAPK and pS6 levels. Strikingly, however the combination of these two drugs significantly reduced or abrogated the expression of downstream signaling molecules. These results suggest that each inhibitor has effects on their respective direct targets but that these effects are not translated into blocking of survival and proliferative pathways. However, the combination of EGFR and MET inhibition could abrogate the activation of essential molecules required for cell growth control. Interestingly, in MDA-MB-231 and T47D, there were no effects on PI3K or MAPK signaling not only after the single agent therapy but also even after combination treatment (Fig. 4). Next, we checked if we could reverse the inhibition of these drugs on downstream molecules when we transfect either AKT or MEK into the cells. As shown in Figure 5, transfection of wild type AKT and/or wild type MEK into MDA-MB-468 cells increased pAKT and pMEK respectively (Fig 5). Surprisingly, in transfected cells, the combination of EGFR or MET inhibition failed to decrease pAKT in AKT transfected cells and pMEK in MEK transfected cells. This effect was also seen when AKT and MEK were cotransfected (Fig. 5).
Evaluation of receptors and downstream pathways after different treatments in TNBC cell lines and T47D. Treated cells were starved with RPMI 1640 medium overnight and next morning, they were stimulated with EGF 20ng/mL and/or recombinant human HGF 75ng/mL for 10 minutes before cell lysis. Thereafter, cell lysates were collected and loaded for Western blot with antibodies as indicated. Actin was used as a loading control. D; DMSO, G; Gefitinib, E; EMD 121463, C; Cetuximab. EGF 20ng/ml, HGF 75ng/ml were treated 10 min before cell lysis.
Rescue experiment evaluating AKT and MEK transfection after therapy inhibition. AKT and MEK inserted in pCMV5-expression vector with a selection marker of ampicillin (100ug/ml) was gently diluted (0.01ug/ul) with serum-free media. Diluted DNA (100ul) was mixed with X-tremeGENE DNA transfection reagent (3ul) and incubated for 15 minutes at room temperature. These mixtures were added into the 60mm dish culturing MDA-MB-468 on day 1 and incubated overnight with drugs including gefitinib (1uM) and EMD 121463 (5uM) alone or in combination; they were treated with serum-free media on day 2. Cells were harvested for western blotting after treatment of EGF 20ng/mL and/or recombinant human HGF 75ng/mL for 10 minutes before cell lysis.
Determination of active EGFR/MET pathways in cell lines
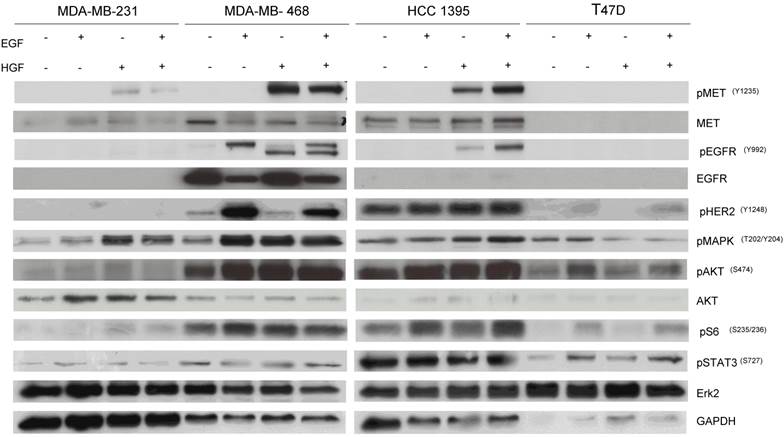
To determine the potential mechanisms involved with the pathway inhibition, cells were treated with EGF and/or HGF after overnight starvation with RPMI-1640 without FBS and lysed to perform western blotting. MDA-MB-468 and HCC-1395 were responsive to both EGF and HGF stimulation consistent with the presence of functional cell surface receptors able to link to downstream signals including pMAPK and pAKT. However, MDA-MB-231 and T47D showed faint or negligible expression of pEGFR and pMET even after stimulation with growth factors and there were little changes in levels of downstream targets including pMAPK and pAKT (Fig. 6). Accordingly, we designated MDA-MB-468 and HCC-1395 as cell lines that have active EGFR and MET pathways and MDA-MB-231 and T47D as cell lines with inactive EGFR and MET pathways. These results are consistent with MDA-MB-468 and HCC-1395 responding to gefitinib and EMD 121463 and with MDA-MB-231 and T47D not responding or poorly responding.
Western blot results after stimulation with EGF, HGF, and their combination in TNBC cell lines, and T47D. Cells were starved with RPMI 1640 medium overnight and next morning, they were stimulated with EGF 20ng/mL and/or recombinant human HGF 75ng/mL for 10 minutes before cell lysis. Thereafter, cell lysates were collected and loaded for Western blot with antibodies as indicated. Actin was used as a loading control.
Phospho-Receptor Tyrosine Kinase (Phospho-RTK) arrays
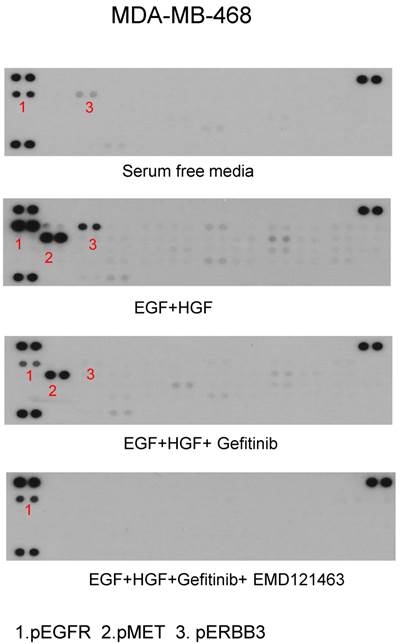
Using phospho-receptor tyrosine kinase array, we investigated whether alternative growth factor receptors responded to EGF or HGF and whether gefitinib or the combination of gefitinib and EMD 121463 would alter the activity. In MDA MB-468 cells, EGF increased levels of pEGFR and pErbB3 whereas EGF and HGF together increased phosphorylated MET, EGFR and ErbB3. ErbB3 plays a critical role in escaping from EGFR TKI inhibition via Akt negative feedback [15]. Geifitinb and EMD 121463 abrogated pMET as well as pErbb3 (Fig. 7). The effects of the combination of gefitinib and EMD 121463 do not seem to be compensated by activation of other RTKs in MDA MB-468 cells (Fig.7). Similar findings were obtained with HCC-1395 (data not shown).
Phospho-receptor tyrosine kinase array results in MDA-MB-468. After stimulation of EGF and HGF and after treatment of gefitinib and gefitinib plus EMD 121463: MDA-MB-468 in 100mm dish was harvested as described in western blotting and Phospho-Receptor Tyrosine Kinase (RTK) antibody arrays (R&D System, #ARY-001, Minneapolis, MN) were performed as recommended but with 450ug protein lysate per array.
Discussion
In this study, we demonstrate that MET and EGFR inhibition are synergistic in TNBC cell lines especially when cell lines have active ligand-receptor-downstream signaling loops. This combination could be helpful to overcome intrinsic EGFR resistance in TNBC.
Overexpression of EGFR is one of the most common aberrations in breast cancer [3,4,15,16]. Constitutively activating mutations like EGFR in non-small cell lung cancer or V600K BRAF mutations in melanoma are rarely found in breast cancer [5,16,17] . In terms of MET, ligand-independent MET activation, by mutation or overexpression, has been detected in a minority of cancers. More commonly solid tumors are ligand-responsive and require either autocrine or paracrine HGF for malignant transformation [18-20]. Therefore, it was assumed that auto/paracrine loop with the activating ligands such as EGF and HGF would play a greater role in TNBC in which both EGFR and MET amplifications occur [21] and where elevated tyrosine phosphorylation of both receptors was found in reverse phase protein array [22]. However, the data demonstrate that EGF and HGF activated downstream signaling in MDA-MB-468 and HCC 1395 but not the other breast cancer cell lines assessed. Whether this represents the underlying genetic aberrations in the lines with for example MD MB 231 cells have both RAS and RAF mutations remains to be ascertained.
With this perspective, we report that the combination of EGFR and MET inhibitors is effective in cell lines that have functionally active ligand-receptor-downstream signaling pathways in TNBC. Failure to induced inhibition of Akt has been reported as a major cause of resistance to EGFR inhibitors [6,23]. Especially, PTEN mutation in MDA-MB-468 is like to contribute to the inability of inhibition of EGFR to down-regulate downstream Akt activity [20,24,25]. In TNBC MDA-MB-468 cell lines that respond to EGF and HGF, the combination of gefitinib and EMD 1214063 shows not only enhanced cytotoxicity but also reduced or abrogated downstream activation of signaling molecules such as Akt, MAPK, S6 and STAT3.
Previous studies of the interactions between EGFR and MET inhibitors suggested that cSRC and MET cooperate to cope with the effects of EGFR inhibitors in breast cancers [26]. Synergistic effects between EGFR and MET inhibitors were also seen in HGF transgenic severe combined immunodeficient mice [27]. In this study, we focused on determining the effectiveness of combined treatment in triple negative breast cell lines to investigate the potential translational relevance of EGFR-MET combination therapy in TNBC.
Combined therapy with EGFR and MET inhibitors are being studied in other disease including colorectal cancer, head and neck cancer and lung cancer [12,20,28]. Triple negative breast cancer is now considered to represent a heterogenous group of diseases based on gene expression profiles of TNBC cell lines [29]. MDA-MB-468 is categorized as Basal-Like subtype and MDA-MB-231 is grouped as Mesenchymal-Like subtype while HCC 1395 is regarded as Unclassified. Therefore, in order to develop biomarkers of the treatment of EGFR and MET inhibition, TNBC should not be studied as a whole entity, rather it should be studied based on biomarkers such as a presence of active EGFR and MET pathways. In contrast to oncogenic addictions like activating mutation or gene rearrangement followed by constitutive signaling, receptor overexpression can contribute to activation of additional receptors or so called receptor co-activation [13,30]. In this case, it would be desirable to block both pathways after identification of the active receptor co-activation processes in future clinical trials.
Acknowledgements
This work was supported in part by National Cancer Institute 1K23CA121994 (AMG) ASCO Career Development Award (AMG), Komen for the Cure Catalystic Award KG090341 (AMG), Komen for the Cure SAC 1000004, American Cancer Society Research Scholar Grant 121329-RSG-11-187-01-TBG (AMG), The Commonwealth Foundation for Cancer research (AMG), and EMD Serono (Billerica, MA).
Competing Interests
AMG has received compound donation from EMD Serono and NTU has research funding from EMD Serono. EMD Serono has reviewed the publication. The views and opinions described in the publication do not necessarily reflect those of EMD Serono.
References
1. Foulkes WD, Smith IE, Reis JS. Triple-Negative Breast Cancer. N Engl J Med. 2010;363:1938-48
2. Cleator S, Heller W, Coombes RC. Triple-negative breast cancer: therapeutic options. Lancet Oncol. 2007;8:235-44
3. Nielsen TO, Hsu FD, Jensen K. et al. Immunohistochemical and clinical characterization of the basal-like subtype of invasive breast carcinoma. Clinical Cancer Research. 2004;10:5367-74
4. Burness ML, Grushko TA, Olopade OI. Epidermal growth factor receptor in triple-negative and basal-like breast cancer: promising clinical target or only a marker? Cancer J. 2010;16:23-32
5. Bhargava R, Gerald WL, Li AR. et al. EGFR gene amplification in breast cancer: correlation with epidermal growth factor receptor mRNA and protein expression and HER-2 status and absence of EGFR-activating mutations. Modern Pathol. 2005;18:1027-33
6. Baselga J, Albanell J, Ruiz A. et al. Phase II and tumor pharmacodynamic study of gefitinib in patients with advanced breast cancer. J Clin Oncol. 2005;23:5323-33
7. Carey LA, Rugo HS, Marcom PK. et al. TBCRC 001: Randomized Phase II Study of Cetuximab in Combination With Carboplatin in Stage IV Triple-Negative Breast Cancer. J Clin Oncol. 2012;30:2615-23
8. Gherardi E, Birchmeier W, Birchmeier C. et al. Targeting MET in cancer: rationale and progress. Nature Rev Cancer. 2012;12:89-103
9. Gastaldi S, Comoglio PM, Trusolino L. The Met oncogene and basal-like breast cancer: another culprit to watch out for? Breast Cancer Res. 2010;12:208
10. Blumenschein GR, Mills GB, Gonzalez-Angulo AM. Targeting the Hepatocyte Growth Factor-cMET Axis in Cancer Therapy. J Clin Oncol. 2012;30:3287-96
11. Raghav KP, Wang WT, Liu SY. et al. cMET and Phospho-cMET Protein Levels in Breast Cancers and Survival Outcomes. Clin Cancer Res. 2012;18:2269-77
12. Engelman JA, Zejnullahu K, Mitsudomi T. et al. MET amplification leads to gefitinib resistance in lung cancer by activating ERBB3 signaling. Science. 2007;316:1039-43
13. Stommel JM, Kimmelman AC, Ying HQ. et al. Coactivation of receptor tyrosine kinases affects the response of tumor cells to targeted therapies. Science. 2007;318:287-290
14. Chou TC, Talalay P. The mechanism of S-adenosyl-L-methionine synthesis by purified preparations of bakers' yeast. Biochemistry. 1972;11:1065-73
15. Sergina NV, Rausch M, Wang DH. et al. Escape from HER-family tyrosine kinase inhibitor therapy by the kinase-inactive HER3. Nature. 2007;445:437-41
16. Corso S, Migliore C, Ghiso E. et al. Silencing the MET oncogene leads to regression of experimental tumors and metastases. Oncogene. 2008;27:684-93
17. Reis JS, Milanezi F, Carvalho S. et al. Metaplastic breast carcinomas exhibit EGFR, but not HER2, gene amplification and overexpression: immunohistochemical and chromogenic in situ hybridization analysis. Breast Cancer Res. 2005;7:R1028-35
18. Trusolino L, Comoglio PM. Scatter-factor and semaphorin receptors: Cell signalling for invasive growth. Nature Rev Cancer. 2002;2:289-300
19. Birchmeier C, Birchmeier W, Gherardi E. et al. Met, metastasis, motility and more. Nature Reviews Mol Cell Biol. 2003;4:915-25
20. Liska D, Chen CT, Bachleitner-Hofmann T. et al. HGF Rescues Colorectal Cancer Cells from EGFR Inhibition via MET Activation. Clin Cancer Res. 2011;17:472-82
21. Koboldt DC, Fulton RS, McLellan MD. et al. Comprehensive molecular portraits of human breast tumours. Nature. 2012;490:61-70
22. Hochgrafe F, Zhang LX, O'Toole SA. et al. Tyrosine Phosphorylation Profiling Reveals the Signaling Network Characteristics of Basal Breast Cancer Cells. Cancer Res. 2010;70:9391-401
23. Spector NL, Xia WL, Burris H. et al. Study of the biologic effects of lapatinib, a reversible inhibitor of ErbB1 and ErbB2 tyrosine kinases, on tumor growth and survival pathways in patients with advanced malignancies. J Clin Oncol. 2005;23:2502-12
24. Li J, Yen C, Liaw D. et al. PTEN, a putative protein tyrosine phosphatase gene mutated in human brain, breast, and prostate cancer. Science. 1997;275:1943-47
25. Moasser MM, Basso A, Averbuch SD. et al. The tyrosine kinase inhibitor ZD1839 ("Iressa") inhibits HER2-driven signaling and suppresses the growth of HER2-overexpressing tumor cells. Cancer Res. 2001;61:7184-8
26. Mueller KL, Hunter LA, Ethier SP. et al. Met and c-Src cooperate to compensate for loss of epidermal growth factor receptor kinase activity in breast cancer cells. Cancer Res. 2008;68:3314-22
27. Zhang YW, Staal B, Essenburg C. et al. MET Kinase Inhibitor SGX523 Synergizes with Epidermal Growth Factor Receptor Inhibitor Erlotinib in a Hepatocyte Growth Factor-Dependent Fashion to Suppress Carcinoma Growth. Cancer Res. 2010;70:6880-90
28. Xu H, Stabile LP, Gubish CT. et al. Dual Blockade of EGFR and c-Met Abrogates Redundant Signaling and Proliferation in Head and Neck Carcinoma Cells. Clin Cancer Res. 2011;17:4425-38
29. Lehmann BD, Bauer JA, Chen X. et al. Identification of human triple-negative breast cancer subtypes and preclinical models for selection of targeted therapies. J Clin Invest. 2011;121:2750-67
30. Xu AM, Huang PH. Receptor Tyrosine Kinase Coactivation Networks in Cancer. Cancer Res. 2010;70:3857-60
Author contact
![]() Corresponding author: Ana M. Gonzalez-Angulo M.D., Department of Breast Medical Oncology, The University of Texas MD Anderson Cancer Center, Unit 1354, 1515 Holcombe Boulevard, Houston, TX 77030-4009; Tel: (713) 792-2817; Fax: (713) 794-4385; E-mail: agonzalezorg
Corresponding author: Ana M. Gonzalez-Angulo M.D., Department of Breast Medical Oncology, The University of Texas MD Anderson Cancer Center, Unit 1354, 1515 Holcombe Boulevard, Houston, TX 77030-4009; Tel: (713) 792-2817; Fax: (713) 794-4385; E-mail: agonzalezorg