Impact Factor ISSN: 1837-9664

 Global reach, higher impact
Global reach, higher impactJ Cancer 2014; 5(9):790-796. doi:10.7150/jca.9794 This issue Cite
Research Paper
Amplification of Chromosome 1q Genes Encoding the Phosphoinositide Signalling Enzymes PI4KB, AKT3, PIP5K1A and PI3KC2B in Breast Cancer
Mark G. Waugh ![]()
Lipid and Membrane Biology Group, Institute for Liver and Digestive Health, UCL, Royal Free Campus, Rowland Hill Street, London, NW3 2PF United Kingdom.
Received 2014-6-2; Accepted 2014-9-11; Published 2014-10-28
Abstract
Little is known about the possible oncogenic roles of genes encoding for the phosphatidylinositol 4-kinases, a family of enzymes that regulate an early step in phosphoinositide signalling. To address this issue, the mutational status of all four human phosphatidylinositol 4-kinases genes was analyzed across 852 breast cancer samples using the COSMIC data resource. Point mutations in the phosphatidylinositol 4-kinase genes were uncommon and appeared in less than 1% of the patient samples however, 62% of the tumours had increases in gene copy number for PI4KB which encodes the phosphatidylinositol 4-kinase IIIbeta isozyme. Extending this analysis to subsequent enzymes in the phosphoinositide signalling cascades revealed that the only PIP5K1A, PI3KC2B and AKT3 genes exhibited similar patterns of gene copy number variation. By comparison, gene copy number increases for established oncogenes such as EGFR and HER2/Neu were only evident in 20% of the samples. The PI4KB, PIP5K1A, PI3KC2B and AKT3 genes are related in that they all localize to chromosome 1q which is often structurally and numerically abnormal in breast cancer. These results demonstrate that a gene quartet encoding a potential phosphoinositide signalling pathway is amplified in a subset of breast cancers.
Keywords: Breast cancer, phosphatidylinositol, 4-kinase, gene copy number.
Introduction
The starting point for this study was the increasing number of reports describing possible roles for the PtdIns (PtdIns) 4-kinase enzymes in cancer [1-11]. PtdIns 4-kinases catalyze the phosphorylation of PtdIns on the D4 position to produce PtdIns4P. This lipid product can be subsequently utilized by downstream phosphoinositide kinases and phosphatases to generate lipids such as PtdIns(4,5)P2 and PtdIns(3,4,5)P3 which have important signalling and trafficking functions. Therefore the synthesis of PtdIns4P is necessary for almost all of the cellular functions ascribed to the phosphoinositide lipids including the regulation of cell proliferation, survival and motility. Moreover, phosphoinositide-dependent deregulation of these functions is well documented in cancer; and this has mainly been attributed to activating point mutations in genes for particular PtdIns 3-kinase isoforms such as PIK3CA, or the reduced expression of phosphoinositide phosphatases such as PTEN. These genetic mutations result in elevated concentrations of particular phosphoinositide species such as PtdIns(3,4,5)P3 and trigger the activation of downstream effecter proteins such as PDK and Akt [12, 13]. In addition to activating mutations and gene deletions more recent work has indicated that increased levels of individual phosphoinositide metabolizing enzymes such as PIK3CA may be sufficient to drive pro-oncogenic signalling [14, 15].
While there is some evidence that PtdIns 4-kinase expression is important in breast cancer [1, 3, 4, 8, 16] the genetic basis for this remains unexplored. To address this knowledge gap the mutational status of the genes encoding for each PtdIns 4-kinase isozyme was analyzed across 852 breast cancer genomes collated in the catalogue of somatic mutations in cancer (COSMIC) database. This strategy was then extended to downstream components of the phosphoinositide signalling cascades in order to establish if any particular enzymatic pathway was upregulated at the genomic level. This approach culminated in the identification of four genes localized to chromosome 1q which exhibited similar copy number increases in over 60% of the tumour samples analyzed and which could potentially constitute a novel pro-oncogenic signalling cascade in breast cancer.
Methods
Genomic analysis. The catalogue of somatic mutations in cancer (COSMIC) bioinformatics resource (http://www.sanger.ac.uk/cosmic) [17, 18] established and maintained by Wellcome Trust Sanger Institute was used in order to identify changes in a panel of genes encoding for enzymes along the phosphoinositide signalling pathway. The initial genomic breast cancer data was derived from 852 individual tumour samples collated from two independent studies - the breast cancer (UK) project and breast invasive carcinoma data from the Cancer Genome Atlas (USA). For these samples, in the COSMIC V69 database, gene copy number analysis was via ASCAT (Allele-Specific Copy number Analysis of Tumors) algorithm software [19] available at http://heim.ifi.uio.no/bioinf/Projects/ASCAT. For this analysis the average copy number for the genome is 1.90 and a reduction in total copy number to a value of 1.30 was taken to demarcate a loss. Copy number gain was specified when average ploidy ≥ 1.8 and gene copy number ≥ 3. For a more stringent assessment of copy number amplification taking into account changes arising from changes to ploidy, the ASCAT criteria used in COSMIC V70 were employed. In this analysis copy number gain is defined as either average genome ploidy <= 2.7 and total gene copy number >= 5 or in the case of genome duplication average genome ploidy > 2.7 and gene copy number >= 9. Note that the copy number data calculated from COSMIC V70 was from an increased cohort of 939 patient samples.
Protein expression analysis using a web-based database. The Human Protein Atlas [20, 21] (www.proteinatlas.org) on-line resource was employed to investigate the expression of candidate proteins identified from the genomic analyses. These immunohistochemical data were obtained from duplicate, paraffin-embedded 1 mm cores from patient surgical samples arranged on tissue microarrays. A full description of the procedures and antibodies used in these studies is available on the Human protein Atlas web-site.
Results & Discussion
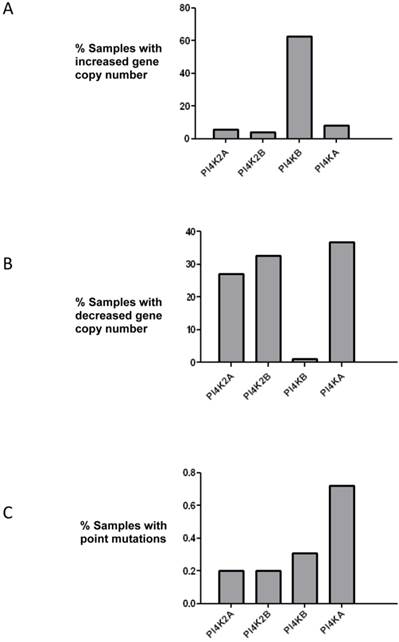
PtdIns 4-kinases catalyze the phosphorylation of PtdIns to generate PtdIns4P and this is the first committed energy consuming step in the pathway that produces the signalling molecules PtdIns(4,5)P2 and PtdIns(3,4,5)P3. The initial aim of this study was to ascertain if any PtdIns 4-kinase gene was mutated in breast cancer. The COSMIC database which collates genomic sequencing results from different studies was searched, and of the four mammalian PtdIns 4-kinases only the PI4KB gene which encodes PtdIns 4-kinase IIIβ was found to be commonly mutated (Figure 1). In over 60% of the samples surveyed there was an increase in PI4KB gene copy number. Moreover, there were concomitant decreases in gene copy number for the other PtdIns 4-kinase genes indicating that in breast cancer PI4KB is the dominant PtdIns 4-kinase. Across all four human PtdIns 4-kinase genes less than 1% exhibited point mutations indicating that this type of genetic alteration was unlikely to significantly contribute to breast cancer tumourigenesis.
Analysis of mutations in the genes encoding for the four human PtdIns 4-kinases in the 852 breast cancer samples collated in the COSMIC database. (a) Over 60% of breast cancer samples have increased copy numbers of the PI4KB gene. (b) Analysis of PtdIns 4-kinase gene copy number loss and (c) frequency of point mutations in the same breast cancer samples.
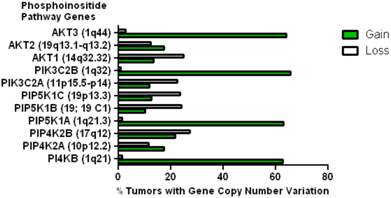
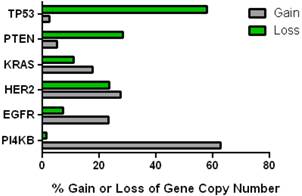
This data mining strategy was extended to encompass copy number variation for genes encoding proteins involved in subsequent steps in phosphoinositide signalling (Figure 2). Investigating the copy number status of genes for the PtdIns4P 5-kinases which produce PtdIns(4,5)P2 by D5 phosphorylation of PtdIns4P and the PtdIns5P 4-kinases which produce the same lipid product via phosphorylation of PtdIns5P on the D4 position, revealed that of the five candidate genes examined, only PI4P5K1A demonstrated increased copy number in breast cancer. A similar approach focused on the phosphoinositide 3-kinase gene family, identified a substantial gene copy number increase for PIK3C2B which encodes a PtdIns4P 3-kinase. To complete this analysis the status of all three human AKT genes was assessed and copy number gains were most noteworthy for the gene encoding the Akt3 isoform and again this was in over 60% of the patient samples. Hence in the majority of the breast cancer cases represented on the COSMIC database there was an elevation in gene copy number for PI4KB, PIP5K1A, PIK3C2B and AKT3. Interestingly, this phosphoinositide gene quartet maps to chromosome 1q - a chromosome which is numerically abnormal in up to 50-60% of breast cancers [22-25]. Comparisons with established oncogenes (Figure 3) indicated that the copy number increase for PI4KB was 2-3 fold higher than for tumour promoter genes such as EGFR and HER2/NEU. Furthermore the magnitude of PI4KB copy number amplification mirrored the scale of copy number loss for tumour suppressor genes such as TP53 and PTEN.
Copy number increases in genes encoding for enzymes downstream of the PtdIns 4-kinases in the COSMIC breast cancer database. The chromosomal localization of each gene is given in brackets.
Comparison of the changes in gene copy number variation in PI4KB with more established oncogenes and tumour suppressor genes in breast cancer.
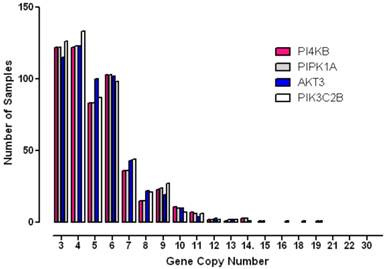
To further analyse the degree to which these genes were amplified in each breast cancer case, the copy number status of each gene was investigated in individual patient samples (Figure 4). These results showed that gene copy number amplification for this pathway ranged from 3 -16 additional copies but that for most tumours the range was 3 - 6 extra copies. Since gene copy number increases can arise from either an increase in ploidy - a common event associated with multinucleation in many cancers, or from numerical increases in specific chromosomes, a more stringent algorithm was employed in order to minimize the contribution of copy number variation caused solely by changes to complete genome duplication. Application of these more rigorous thresholds revealed that all four genes of interest were amplified in approximately 10% of breast cancer samples (10% for PI4KB, 11.4% for AKT3, 11.3% for PIK3C2B and 10.2% for PIP5K1A). These values are consistent with copy number co-amplification arising from numerical increases to chromosome 1q and not solely by mitotic defects leading to genome duplication.
Histogram demonstrating the distribution of copy number increases in individual breast tumours for each of the amplified phosphoinositide pathway genes identified.
To further probe the specificity of this finding for the quartet of phosphoinositide metabolizing genes, the copy number status of functionally unconnected and randomly chosen genes mapping elsewhere to chromosome 1 were assessed. The genes used in this test were CRP banding at 1q23.2 encoding C-reactive protein, LMNA at 1q22 which encodes nuclear laminar protein lamin A/C and PSEN2 at 1q42.13 which encodes presenilin 2, a protein which is sometimes mutated in inherited forms of Alzheimer's disease. The level of high copy number amplification for all three of these genes was similar to that observed for the quartet of phosphoinositide signalling enzymes at 8.5% for CRP, 7.88% for LMNA and 10.33% for PSEN2. These results indicate that the genes in question are amplified due to their co-localization to chromosome 1q and furthermore that this effect is not specific for phosphoinositide pathway genes.
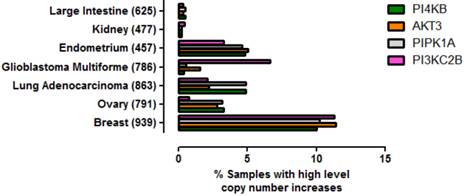
To ascertain whether amplification of this gene quartet occurs in cancers affecting tissues other than breast, an assessment of copy number variation across multiple tissues was performed (Figure 5). From this analysis it was clear that co-amplification of PI4KB/PIPK1A/AKT3/PIK3C2B was most pronounced for breast carcinoma but also occurred in about 5% of endometrial cancers, lung adenocarcinomas and ovarian cancer. Mutations in these four genes were also found more rarely in less than 1% of cancers affecting the large intestine and kidney. These results demonstrate that amplification of this gene set is most commonly detected in invasive breast carcinoma but can also be found in other malignancies but at much lower frequencies.
Prevalence of high level gene copy number increases for PI4KB, AKT3, PIP5K1A and PIK3C2B in a range of cancers affecting different tissues. The data were derived from the COSMIC V70. The number of patient samples used for each analysis is given in brackets after the tissue name. Breast refers to invasive breast carcinoma, ovary to ovarian serous cyst adenocarcinoma, kidney to clear cell carcinoma, endometrium to uterine corpus endometrial cancer, large intestine to colon adenocarcinoma and lung to lung adenocarcinoma.
One possible selective pressure to drive amplification of PtdIns4P and PtdIns(4,5)P2 generation could be the co-presence of constitutively active oncogenic PIK3CA requiring an increased input of phosphoinositide substrate to produce PtdIns(3,4,5)P3. Moreover, activating point mutations of PIK3CA have previously been reported in breast cancer and this therefore prompted an assessment of the frequency and types of PIK3CA point mutations in each of the patient samples with PI4KB amplification. This analysis revealed that activating PIK3CA amino acid point mutations were present in 22/93 (23.7%) of the high copy number PI4KB samples which was similar to 26.44% frequency for point mutations in this gene for all samples in the database. The vast majority of PIK3CA point mutations in high PI4KB samples were H1074R substitutions (13/24), followed by E545K (6/24), G118D (1/24) and C240R (1/24) activating mutations. Inactivating PTEN mutations were rarer and found in just 3 of the high PI4KB tumours, and in only one instance was this co-incident with a PIK3CA activating point mutation. Hence it is possible that about a quarter of the samples in question had a potential selective pressure for increased PtdIns 4-kinase and PtdIns4P 5-kinase activity to supply phosphoinositide substrate to a constitutively active PIK3CA however, this was not the case for the majority of the tumours with amplified PI4KB.
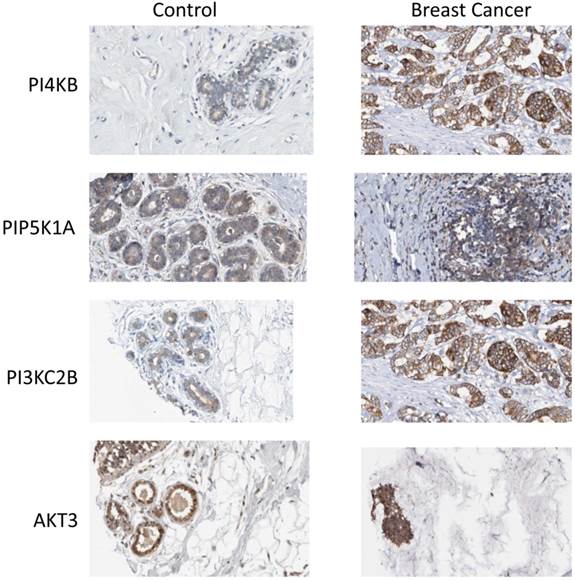
Finally, the Human Protein Atlas [20, 21] (www.proteinatlas.org) was surveyed to search if there was any evidence for upregulated expression of the enzyme products of the four identified genes in breast cancer tissues. In normal breast tissue (Figure 6), the strongest intensity of immunohistochemical staining for PtdIns 4-kinase IIIβ, PtdIns4P 5-Kinase 1α, Akt3 and phosphoinositide 3-kinase C2β was in the ductal tissue. Increased expression of all four proteins was observed in ductal carcinomas and this was most pronounced for the PtdIns 4-kinase IIIβ and phosphoinositide 3-kinase C2β proteins. For the eleven antibody-stained breast cancer tumour samples in the human protein atlas, the intensity of anti-PtdIns 4-kinase IIIβ staining was classified as moderate or strong in 8/11 cases. Similarly for the PIK3C2B gene product, 8/11 patient samples exhibited strong expression. Immunohistochemical staining for PIP5K1A was less strong with only low level immunoreactivity detected in 7/11 tumours. This mirrored the immunohistochemical data for Akt3 where 8/12 samples were determined to have low expression of this protein and only one sample exhibited moderate Akt3 overexpression. However, while these immunohistochemical results partly support the genomic data they need to be treated with caution as they derive from a much lower number of patient samples and from tumours where the chromosome 1q status was not known.
Expression of phosphoinositide signalling proteins in normal breast tissue and in ductal carcinoma. Images were obtained by immunohistochemical staining of paraffin-embedded tissue samples using isoform-specific antibodies directed against the protein products of PI4KB, PIP5K1A, PIK3C2B and AKT3. Antibody binding appears as brown/black staining on a background of blue hematoxylin counterstain. All images are from the Human Protein Atlas.
In conclusion, the genomic analyses presented here reveal that a significant proportion of breast cancer tumours possess gene copy number increases for a quartet of enzymes along a D4 phosphoinositide signalling cascade. The exact functional relationship between gene copy number variation and disease is still being established [26-28] but increased gene dosage and downstream protein expression are very likely to define molecular gene expression signatures in several cancer subtypes [28]. Since the four identified phosphoinositide pathway genes are all located on chromosome 1q it is likely that their increased copy number variation reflects the frequent chromosome 1 abnormalities that have been established for several years in breast cancer [22, 24, 29, 30]. Furthermore, amplification of this specific gene set has the potential to lead to enhanced phosphoinositide synthesis potentially giving rise to augmented cell proliferation and motility [31-33] and hence a more aggressive tumour [1, 34-44]. These insights may have implications for the use of small molecule inhibitors of phosphoinositide signalling that are currently in development since with the possible exception of Akt3 [41], none of these four enzymes has previously been considered as a high profile chemotherapeutic target [12, 45, 46]. Moreover, the high degree of functional redundancy for enzymes involved in phosphoinositide synthesis [47, 48] combined with the possible amplification of the particular phosphoinositide signalling pathway identified here, has the potential to give rise to enhanced chemoresistance and survival adaptation. Noteworthy in this context is the recent finding that overexpression of PI4P 5-kinase 1α in prostate cancer cells leads to augmented Akt activation, enhanced invasiveness and a poor prognosis for this disease [49]. Therefore, structural alterations and duplications of chromosome 1q which are common in breast cancer, may lead to aberrant phosphoinositide supply to key oncogenic driver pathways.
Abbreviations
CNV: copy number variation; PtdIns: phosphatidylinositol.
Acknowledgements
This work was supported by a grant from the Royal Free Charity. I thank David Brown and Shane Minogue for many helpful discussions.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Pinke DE, Lee JM. The lipid kinase PI4KIIIbeta and the eEF1A2 oncogene co-operate to disrupt three-dimensional in vitro acinar morphogenesis. Exp Cell Res. 2011;317:2503-11
2. Chu K, Minogue S, Hsuan J, Waugh M. Differential effects of the phosphatidylinositol 4-kinases, PI4KIIalpha and PI4KIIIbeta, on Akt activation and apoptosis. Cell Death Dis. 2010;1:e106
3. Li J, Lu Y, Zhang J, Kang H, Qin Z, Chen C. PI4KIIalpha is a novel regulator of tumor growth by its action on angiogenesis and HIF-1alpha regulation. Oncogene. 2010;29:2550-9
4. Li J, Zhang L, Gao Z, Kang H, Rong G, Zhang X. et al. Dual inhibition of EGFR at protein and activity level via combinatorial blocking of PI4KIIalpha as anti-tumor strategy. Protein Cell. 2014;5:457-68
5. Ilboudo A, Nault JC, Dubois-Pot-Schneider H, Corlu A, Zucman-Rossi J, Samson M. et al. Overexpression of phosphatidylinositol 4-kinase type IIIalpha is associated with undifferentiated status and poor prognosis of human hepatocellular carcinoma. BMC Cancer. 2014;14:7
6. Waugh MG. Phosphatidylinositol 4-kinases, phosphatidylinositol 4-phosphate and cancer. Cancer Lett. 2012;325:125-31
7. Clayton EL, Minogue S, Waugh MG. Mammalian phosphatidylinositol 4-kinases as modulators of membrane trafficking and lipid signaling networks. Prog Lipid Res. 2013;52:294-304
8. Waring MJ, Andrews DM, Faulder PF, Flemington V, McKelvie JC, Maman S. et al. Potent, selective small molecule inhibitors of type III phosphatidylinositol-4-kinase alpha- but not beta-inhibit the phosphatidylinositol signaling cascade and cancer cell proliferation. Chem Commun (Camb). 2014;50:5388-90
9. Tokuda E, Itoh T, Hasegawa J, Ijuin T, Takeuchi Y, Irino Y. et al. Phosphatidylinositol 4-Phosphate in the Golgi Apparatus Regulates Cell-Cell Adhesion and Invasive Cell Migration in Human Breast Cancer. Cancer Res. 2014
10. Carloni V, Mazzocca A, Ravichandran KS. Tetraspanin CD81 is linked to ERK/MAPKinase signaling by Shc in liver tumor cells. Oncogene. 2004;23:1566-74
11. Mazzocca A, Liotta F, Carloni V. Tetraspanin CD81-regulated cell motility plays a critical role in intrahepatic metastasis of hepatocellular carcinoma. Gastroenterology. 2008;135:244-56 e1
12. Klempner SJ, Myers AP, Cantley LC. What a tangled web we weave: emerging resistance mechanisms to inhibition of the phosphoinositide 3-kinase pathway. Cancer Discov. 2013;3:1345-54
13. Wong KK, Engelman JA, Cantley LC. Targeting the PI3K signaling pathway in cancer. Curr Opin Genet Dev. 2010;20:87-90
14. Yuan TL, Wulf G, Burga L, Cantley LC. Cell-to-cell variability in PI3K protein level regulates PI3K-AKT pathway activity in cell populations. Curr Biol. 2011;21:173-83
15. Fidler MJ, Morrison LE, Basu S, Buckingham L, Walters K, Batus M. et al. PTEN and PIK3CA gene copy numbers and poor outcomes in non-small cell lung cancer patients with gefitinib therapy. Br J Cancer. 2011;105:1920-6
16. Chu KM, Minogue S, Hsuan JJ, Waugh MG. Differential effects of the phosphatidylinositol 4-kinases, PI4KIIalpha and PI4KIIIbeta, on Akt activation and apoptosis. Cell Death Dis. 2010;1:e106
17. Forbes SA, Bhamra G, Bamford S, Dawson E, Kok C, Clements J. et al. The Catalogue of Somatic Mutations in Cancer (COSMIC). Curr Protoc Hum Genet. 2008 Chapter 10: Unit 10 1
18. Forbes SA, Bindal N, Bamford S, Cole C, Kok CY, Beare D. et al. COSMIC: mining complete cancer genomes in the Catalogue of Somatic Mutations in Cancer. Nucleic Acids Res. 2011;39:D945-50
19. Van Loo P, Nordgard SH, Lingjaerde OC, Russnes HG, Rye IH, Sun W. et al. Allele-specific copy number analysis of tumors. Proc Natl Acad Sci U S A. 2010;107:16910-5
20. Ponten F, Schwenk JM, Asplund A, Edqvist PH. The Human Protein Atlas as a proteomic resource for biomarker discovery. J Intern Med. 2011;270:428-46
21. Uhlen M, Oksvold P, Fagerberg L, Lundberg E, Jonasson K, Forsberg M. et al. Towards a knowledge-based Human Protein Atlas. Nat Biotechnol. 2010;28:1248-50
22. Chen LC, Dollbaum C, Smith HS. Loss of heterozygosity on chromosome 1q in human breast cancer. Proc Natl Acad Sci U S A. 1989;86:7204-7
23. Kovacs G. Abnormalities of chromosome No. 1 in human solid malignant tumours. Int J Cancer. 1978;21:688-94
24. Cruciger QV, Pathak S, Cailleau R. Human breast carcinomas: marker chromosomes involving 1q in seven cases. Cytogenet Cell Genet. 1976;17:231-5
25. Orsetti B, Nugoli M, Cervera N, Lasorsa L, Chuchana P, Rouge C. et al. Genetic profiling of chromosome 1 in breast cancer: mapping of regions of gains and losses and identification of candidate genes on 1q. Br J Cancer. 2006;95:1439-47
26. Valsesia A, Mace A, Jacquemont S, Beckmann JS, Kutalik Z. The Growing Importance of CNVs: New Insights for Detection and Clinical Interpretation. Front Genet. 2013;4:92
27. Kuiper RP, Ligtenberg MJ, Hoogerbrugge N, Geurts van Kessel A. Germline copy number variation and cancer risk. Curr Opin Genet Dev. 2010;20:282-9
28. Ciriello G, Miller ML, Aksoy BA, Senbabaoglu Y, Schultz N, Sander C. Emerging landscape of oncogenic signatures across human cancers. Nat Genet. 2013;45:1127-33
29. Ciruela A, Hinchliffe KA, Divecha N, Irvine RF. Nuclear targeting of the beta isoform of type II phosphatidylinositol phosphate kinase (phosphatidylinositol 5-phosphate 4-kinase) by its alpha-helix 7. Biochem J. 2000;346(Pt 3):587-91
30. Chin K, DeVries S, Fridlyand J, Spellman PT, Roydasgupta R, Kuo WL. et al. Genomic and transcriptional aberrations linked to breast cancer pathophysiologies. Cancer Cell. 2006;10:529-41
31. Maffucci T, Cooke FT, Foster FM, Traer CJ, Fry MJ, Falasca M. Class II phosphoinositide 3-kinase defines a novel signaling pathway in cell migration. J Cell Biol. 2005;169:789-99
32. Domin J, Harper L, Aubyn D, Wheeler M, Florey O, Haskard D. et al. The class II phosphoinositide 3-kinase PI3K-C2beta regulates cell migration by a PtdIns3P dependent mechanism. J Cell Physiol. 2005;205:452-62
33. Katso RM, Pardo OE, Palamidessi A, Franz CM, Marinov M, De Laurentiis A. et al. Phosphoinositide 3-Kinase C2beta regulates cytoskeletal organization and cell migration via Rac-dependent mechanisms. Mol Biol Cell. 2006;17:3729-44
34. Zinda MJ, Johnson MA, Paul JD, Horn C, Konicek BW, Lu ZH. et al. AKT-1, -2, and -3 are expressed in both normal and tumor tissues of the lung, breast, prostate, and colon. Clin Cancer Res. 2001;7:2475-9
35. Mende I, Malstrom S, Tsichlis PN, Vogt PK, Aoki M. Oncogenic transformation induced by membrane-targeted Akt2 and Akt3. Oncogene. 2001;20:4419-23
36. Stahl JM, Sharma A, Cheung M, Zimmerman M, Cheng JQ, Bosenberg MW. et al. Deregulated Akt3 activity promotes development of malignant melanoma. Cancer Res. 2004;64:7002-10
37. Kirkegaard T, Witton CJ, Edwards J, Nielsen KV, Jensen LB, Campbell FM. et al. Molecular alterations in AKT1, AKT2 and AKT3 detected in breast and prostatic cancer by FISH. Histopathology. 2010;56:203-11
38. Grabinski N, Bartkowiak K, Grupp K, Brandt B, Pantel K, Jucker M. Distinct functional roles of Akt isoforms for proliferation, survival, migration and EGF-mediated signalling in lung cancer derived disseminated tumor cells. Cell Signal. 2011;23:1952-60
39. Banfic H, Visnjic D, Mise N, Balakrishnan S, Deplano S, Korchev YE. et al. Epidermal growth factor stimulates translocation of the class II phosphoinositide 3-kinase PI3K-C2beta to the nucleus. Biochem J. 2009;422:53-60
40. Banerji S, Cibulskis K, Rangel-Escareno C, Brown KK, Carter SL, Frederick AM. et al. Sequence analysis of mutations and translocations across breast cancer subtypes. Nature. 2012;486:405-9
41. Chin YR, Yoshida T, Marusyk A, Beck AH, Polyak K, Toker A. Targeting Akt3 signaling in triple-negative breast cancer. Cancer Res. 2014;74:964-73
42. Yamaguchi H, Yoshida S, Muroi E, Kawamura M, Kouchi Z, Nakamura Y. et al. Phosphatidylinositol 4,5-bisphosphate and PIP5-kinase Ialpha are required for invadopodia formation in human breast cancer cells. Cancer Sci. 2010;101:1632-8
43. Halstead JR, Roefs M, Ellson CD, D'Andrea S, Chen C, D'Santos CS. et al. A novel pathway of cellular phosphatidylinositol(3,4,5)-trisphosphate synthesis is regulated by oxidative stress. Curr Biol. 2001;11:386-95
44. Halstead JR, Savaskan NE, van den Bout I, Van Horck F, Hajdo-Milasinovic A, Snell M. et al. Rac controls PIP5K localisation and PtdIns(4,5)P(2) synthesis, which modulates vinculin localisation and neurite dynamics. J Cell Sci. 2010;123:3535-46
45. Emerling BM, Hurov JB, Poulogiannis G, Tsukazawa KS, Choo-Wing R, Wulf GM. et al. Depletion of a putatively druggable class of phosphatidylinositol kinases inhibits growth of p53-null tumors. Cell. 2013;155:844-57
46. Holand K, Boller D, Hagel C, Dolski S, Treszl A, Pardo OE. et al. Targeting Class IA PI3K Isoforms Selectively Impairs Cell Growth, Survival, and Migration in Glioblastoma. PLoS One. 2014;9:e94132
47. Juss JK, Hayhoe RP, Owen CE, Bruce I, Walmsley SR, Cowburn AS. et al. Functional redundancy of class I phosphoinositide 3-kinase (PI3K) isoforms in signaling growth factor-mediated human neutrophil survival. PLoS One. 2012;7:e45933
48. Foukas LC, Berenjeno IM, Gray A, Khwaja A, Vanhaesebroeck B. Activity of any class IA PI3K isoform can sustain cell proliferation and survival. Proc Natl Acad Sci U S A. 2010;107:11381-6
49. Semenas J, Hedblom A, Miftakhova RR, Sarwar M, Larsson R, Shcherbina L. et al. The role of PI3K/AKT-related PIP5K1alpha and the discovery of its selective inhibitor for treatment of advanced prostate cancer. Proc Natl Acad Sci U S A. 2014;111:E3689-98
Author contact
![]() Corresponding author: E-Mail: m.waughac.uk.
Corresponding author: E-Mail: m.waughac.uk.