Impact Factor ISSN: 1837-9664

 Global reach, higher impact
Global reach, higher impactJ Cancer 2015; 6(1):70-81. doi:10.7150/jca.10478 This issue Cite
Research Paper
ERCC1 Induction after Oxaliplatin Exposure May Depend on KRAS Mutational Status in Colorectal Cancer Cell Line: In Vitro Veritas
A. Orlandi, M. Di Salvatore, C. Bagalà, M. Basso, A. Strippoli, F. Plastino, M.A. Calegari, A. Cassano, A. Astone, C. Barone ![]()
Medical Oncology, Università Cattolica del Sacro Cuore, Rome, Italy.
Received 2014-9-3; Accepted 2014-11-16; Published 2015-1-1
Abstract
Introduction: Oxaliplatin (Oxa) is widely used in metastatic colorectal cancer (mCRC), but currently there are not valid predictors of response to this drug. In the control arms both of OPUS and PRIME studies Oxa seems more active in patients with mCRC with mutated (mt) KRAS than in those with wild type (wt) KRAS. Recently we have retrospectively confirmed this suggestion, therefore we have hypothesized that the mutational status of KRAS could influence the expression of ERCC1, one of the main mechanisms of Oxa resistance.
Material and Methods: We used four cell lines of colorectal cancer: two KRAS wild type (wt) (HCT-8 and HT-29) and two KRAS mt (SW620 and SW480). We evaluated the sensitivity of these cell lines to Oxa by MTT-test as well the ERCC1 levels before and after 24 h exposure to Oxa by Real-Time PCR. We silenced KRAS in a KRAS mt cell line (SW620LV) to evaluate the impact on Oxa sensitivity and ERCC1 levels. Lastly, ERCC1 was also silenced in order to confirm the importance of this protein as an Oxa resistance factor.
Results: The KRAS mt cell lines resulted more sensitive to Oxa (OR 2.68; IC 95% 1.511-4.757 p<0.001). The basal levels of ERCC1 did not show significant differences between KRAS mt and wt cell lines, however, after 24 h exposure to Oxa, only the wt KRAS lines showed the ability to induce ERCC1, with a statistically significant difference (OR 42.9 IC 95% 17.260-106.972 p<0.0005). By silencing KRAS, sensitivity to Oxa was reduced in mt KRAS cell lines and this effect was associated with the acquisition of ability to induce ERCC1. Silencing of ERCC1, in turn, enhanced the sensitivity to Oxa in wt KRAS cell lines and restored sensitivity to Oxa in SW620LV cell line.
Conclusion: KRAS mutated cell lines were more sensitive to Oxa. This feature seems secondary to the inability of these cells to induce ERCC1 after exposure to Oxa. Thus, KRAS mutational status might be a predictor of response to Oxa in CRC surrogating the cell ability to induce ERCC1.
Keywords: Oxaliplatin, KRAS, ERCC1, colorectal cancer cells.
Introduction
Colorectal cancer (CRC) is the third leading cause of cancer-related deaths in the western world (1). Drugs for management of CRC include both cytotoxic and targeted agents; among these Oxa is one of the most frequently used. Oxa is a platinum analogue that differs from Cisplatin for the presence of a diaminocyclohexane ligand in its chemical structure; it forms mainly intrastrand links between two adjacent guanine residues, disrupting DNA replication and transcription (2,3). About 50% of patients with mCRC are resistant to Oxa-based treatment (4), but the mechanism of “de novo” resistance has been only partially explored and remains largely unknown (5).
Following introduction of targeted agents like Cetuximab (Cet) and Panitumumab (Pan) into the clinical practice of treatment of mCRC, KRAS mutation was identified as the main factor predicting a negative outcome and became a prerequisite condition for selecting patients to be treated with anti-EGFR antibodies (6,7,8). In addition to improvement of response rate (RR) and progression-free survival (PFS) with the combination of Cet and FOLFOX in wt KRAS patients, however, a trend toward an improved PFS was unexpectedly observed in KRAS mt patients compared with wt treated with FOLFOX alone (7). This advantage did not reach statistical significant (HR 1.404; p 0.16), but it is nonetheless interesting, given the exploratory nature of the analysis and the large number of patients censored. A similar trend (PFS 8.7 vs 7.8 months) was seen in extended RAS analysis of the PRIME study (9)
This observation induced us to consider the possibility that KRAS mutational status might also select patients with different sensitivity to Oxa. Therefore we retrospectively analyzed our series of patients affected by advanced colorectal cancer and treated with first- or second-line FOLFOX-6, mainly in the years before anti-EGFR antibodies became available (10). RR was significantly higher in mt than in wt KRAS patients (56% vs 26%, HR: 2.148, 95% CI: 1.222-3.781; p 0.008). In the whole population PFS was 10 and 8 months (HR 1.645, 95% CI: 1.161-3.030; p 0.01) and median overall survival (mOS) was 24 and 18 months (HR: 1.64, 95% CI: 1.13-2.89; p 0.01) in mt and wt KRAS patients, respectively (10). Actually, the possibility that KRAS mutation might play a role in the susceptibility to Oxa had been previously asuggested by Vekris et al, who aimed to identify those genes whose expression was related to sensitivity to four platinum compounds, including Oxa, in a panel of 60 cancer cell lines (11). They found that the activity of Oxa was significantly higher in cell lines with a mutation in one of the KRAS genes, but the mechanism underlying this relationship has not been explored.
It is known that the ability of cells to repair platinum-induced DNA lesions or adducts plays an important role in Oxa cytotoxicity. One of the major DNA repair systems in mammalian cells is the nucleotide excision repair (NER), which removes bulky helix distorting adducts produced by Oxa (12). The excision repair cross-complementing group 1 protein (ERCC1) is an important factor in the incision process, the rate-limiting step of the pathway. ERCC1 forms a heterodimer with Xeroderma pigmentosus group F (XPF), and the ERCC1/XPF complex is responsible for the cleavage of the damaged strand (13). A functional ERCC1 is essential for repairing platinum-DNA adducts and it is involved in drug sensitivity “in-vitro” (14,15,16). Based on this evidence, several clinical studies have been conducted with the aim of relating ERCC1 tumor levels with response to Oxa, but results are controversial (17,18,19), and therefore ERCC1 tissue levels cannot be considered a predictive factor of response to Oxa.
Some preclinical studies have revealed a possible link between EGFR pathway and DNA repair (20,21,22). Moreover, ERCC1 expression in response to Oxa was positively regulated by EGFR in wt KRAS cells (23). In these cells Cet may potentiate the effect of Oxa by down regulating NER-related mechanisms involved in resistance and promoting apoptosis. Based on interaction between the pathways of EGFR and ERCC1, the possibility that downstream signal though KRAS might be involved in the activity of Oxa, whose effect might depend on ERCC1, seems plausible. Therefore, the aim of this study was to estabilish if Oxa sensitivity of a panel of colorectal cancer cell lines might be influenced by KRAS mutational status and to evaluate the possible role of ERCC1.
Materials and Methods
Cell lines and culture conditions
Four human colorectal cancer cell lines (HT-29, HCT-8, SW620 and SW 480) were obtained from the American Type Culture Collection (ATCC, Manassas, VA, USA). HT-29 were maintained in McCoy medium supplemented with 10% foetal bovine serum, penicillin (100 IU/ml), streptomycin (100 μg/ ml) and glutamine (4 mM); HCT-8 were cultured in RPMI 1640 medium supplemented with 10 % horse serum, penicillin (100 IU/ml), streptomycin (100 μg /ml) and glutamine (4 mM); SW620 and SW480 were passed on L-15 medium supplemented with 10% fetal bovine serum, penicillin (100 IU/ml), streptomycin (100 μg/ml) and glutamine. Cell cultures were maintained at 37°C in a humidified atmosphere with 5% CO2.
Drugs
Oxa was obtained from Sanofi Aventis (Milan, Italy). The drug was aliquoted at a concentration of 5 mg/ml and kept at 2-5 °C until use.
SN 38 (the active metabolite of irinotecan) was obtained from Sigma-Aldrich. It was diluted to a stock solution of 50 mg/ml in dimethyl sulfoxide (DMSO). The stock solution was kept at -20˚C and diluted at appropriate concentrations with medium before use.
RAS mutations status analysis
DNA was extracted from cell lines pellets using the QIAamp DNA extraction kit (QIAGEN, Milan, Italy), according to manufacturer's instructions. DNA was then quantized by determination of optical density in a spectrophotometer with a wavelength of 260 nm. Mutational analysis of KRAS and NRAS has been carried out for exon 2 (at codons 12 and 13), exon 3 (at codon 61), and exon 4 (at codons 117 and 146). Each reaction provides for the amplification of 100 ng of DNA in 25 μl total mix containing 1x PCR buffer [20 mm TRIS (pH 8.3), 50 mM KCl, 1.5 mM MgCl2], dNTPs (200 mM each), specific primers (20 pM each), 0.25 U Taq polymerase platinum (Invitrogen) and water. For both reactions, after an initial denaturation at 95° for 8 minutes, the DNA was amplified for 40 cycles; for each cycle denaturation at 95 ° C for 40 seconds, annealing at 55 ° C for 40 seconds and extension at 72° C for 50 seconds, with a final step of further extension at 72°C for 4 minutes, were carried out. The amplification products were then visualized by running on a 2.5% agarose gel stained with ethidium bromide. The amplicons were then extracted from the agarose gel using the kit WizardR Gel and PCR Clean-Up Sistem (Promega) according to the procedures described by the supplier, and were then sequenced with ABI Prism 3700 Avant (Applied Biosystems).
RNA extraction and Real-Time PCR
Total RNA was extracted by QIAzol, according to the manufacturer's protocol (QIAGEN). RNA was precipitated in isopropanol, washed in 70% ethanol and then the pellet was allowed to dry in air and was resuspended with RNA-ase free water. The purity and quantity of RNA were assayed by spectrophotometer. RNA samples (2 μg) were reverse transcribed using the enzyme ImProm-II according to the instructions of the supplier company (Promega).
Subsequently 1 microliter of the product of reverse transcription was mixed with the DNA Master Plus SYBR Green I (Roche) and with the specific primers:
ERCC-1:
- For: 5'-GGG AAT TTG GCG ACG TTC TAA-3'
- Rev: 5'-GCG GAG GAG GAA GCT CAG-3 '
KRAS:
- For: 5'-GGG GAG TTT CTT GGC TGT GTA-3'
- Rev: 5'-CTG GTC AGC CTG TGT TTT GTC-3'
18S:
- For 5'-CGGCTACCACATCCAAGGAA-3'
- Rev-5'-GCTGGAATTACCGCGGCT-3'
PCR profile was as follows: 10 min at 95 °C, followed by 45 cycles of 10 s at 95°C, 8 s at 60°C and 20 s at 72°C. Data were analysed using Sequence Detection System Software (Applied Biosystems, Foster City, CA, USA). Parameter CT (threshold cycle) was defined as cycle number at which the fluorescent signal passed a fixed value (threshold) above baseline. Relative mRNA copy numbers were calculated from standard curves generated with 10-fold dilution series of template DNA.
Finally, the expression level for ERCC1 and KRAS of each sample was normalized by subtracting the cycle threshold (CT) of housekeeping gene (18S) from the gene of interest to calculate the ΔCT.
Cytotoxicity assay - MTT Test
3-(4,5-dimethyl-thiazoyl)-2,5-diphenyl-SH-tetrazolium bromide (MTT) assay was performed both on original (HT-29, HCT-8, SW620, SW480) and lentivirus silenced or control cell lines (SW620LC, SW620LV).
Target cells were resuspended in medium at 1x105 cells⁄ml, and 100 μl of 105 cells/ml cell suspension was distributed to each well of 96-well plates (Costar) and allowed to adhere for 24 hours. Wells containing 200 μl medium alone (without cells) and reagents were used as negative control. The cells were exposed to increasing concentrations of Oxa (10, 20, 50, 80, 100 μM) or SN-38 (0.0001, 0.001, 0.01, 0.1, 1 nM). After treatment with incubation doses for 24 h, 20 μl MTT solution (5 mg⁄ ml) was added to each well, and microplates were further incubated at 37 °C for 4 h. Unreacted supernatants in wells were discarded, and 100 μl acidified isopropanol (0.04 N HCl in isopropanol) was added to the cultures and mixed thoroughly to dissolve the dark-blue crystals of formazan. Absorbance values of each well were determined using microplate enzyme-linked immunoassay reader equipped with a 570 nm filter. Negative control well was used for baseline zero absorbance. Results are presented as percentage of viability, determined as (absorbance of experimental well/absorbance of positive control well) x 100. Each experiment was repeated three times.
Stable KRAS silencing and clones selection
The expression of the KRAS in SW620 cells was silenced by lentiviruses containing plasmids with siRNA against human KRAS (MISSION Lentiviral Transduction Particles, Sigma Aldrich). The SW620 were also transfected with lentiviruses containing plasmids without siRNA (MISSION Lentiviral Control) in order to obtain a control line. Both control and siRNA plasmids contained Puromycin resistance gene for mammalian selection. 2x104 SW620 cells were cultured in 96-well plates. After 24 h, cells were incubated at four increasing concentrations of lentiviruses (MOI 2, 5, 10, 15). In order to select transfected clones, after further 24 h, medium containing viral particles was replaced by a Puromycin (2 μg/ml) containing culture medium. The culture medium supplemented with Puromycin (2 μg/ml) was renewed every three days and, thus, after 6 days it was possible to isolate the clones. Subsequently, the clones were expanded and gene silencing was tested by RT-PCR. Among the various clones of KRAS-silenced SW620 has been selected the one with a lower expression of KRAS mRNA (SW620LV). The cell clones with control lentiviruses (SW620LC) showed no differences in mRNA KRAS expression in comparison to SW620 cell line.
Transient ERCC1 silencing
A panel of 6 ERCC1-siRNA (siRNA-S1, siRNA-S2 siRNA-S3 siRNA-S4, siRNA-S5 and control-siRNA) was designed and synthesized by Sigma Aldrich. "N-TER ™ Nanoparticle siRNA Transfection System" kit was used for transfection according to manufacturer's instructions. In order to select the most efficient siRNA, 2.2 x 105 cells for each line (HCT-8, HT-29, SW620, SW480) were seeded in 24-well plates. Four different concentrations were evaluated for each siRNA: 0-10-20-40 μM. Silencing was conducted both in serum and serum-free condition. After 24 hours the transfection medium was replaced with fresh medium and cells were harvested after 48 hours for RNA extraction and evaluation of ERCC1 by RT-PCR. The siRNA-S5 at a concentration of 40 μM showed to reduce mRNA ERCC1 most efficiently and thus was used for the subsequent experiments.
Western blotting
SW620, SW480, HT-29, HCT-8, SW620LC, SW620LV cells were plated on six-well plates in normal culture medium up to 50% confluence. Cells were then rinsed in sterile phosphate-buffered saline (PBS) and cultured for further 24 h in medium with serum, in the presence or absence of different concentrations of Oxa (10-100 μM). Culture medium was discarded and cells were harvested in lysis buffer (20 mmol ⁄ l Tris (pH 7.4), 150 mmol ⁄ l NaCl, 1 mmol ⁄ l EDTA, 1 mmol ⁄ l EGTA, 1% Triton, 2.5 mmol ⁄ l sodium PPi, 1 mmol ⁄ l h-glycerolphosphate, 1 mmol ⁄l Na3VO4, 1 Ag⁄ml leupeptin; Cell Signaling, Beverly, MA, USA) and clarified by centrifugation. After boiling supernatants in reducing SDS sample buffer, equal amounts of protein (80 ng) were loaded per lane and samples were electrophoresed on 15% polyacrylamide SDS gel and transferred to nitrocellulose membranes. Rabbit antibodies for western blot against ERCC1 and KRAS were purchased from Cell Signaling Technology, Inc. (Beverly, MA, USA). Mouse antibodies against β-Actin were obtained from Cell Biolabs, Inc. (San Diego, CA, USA). Protein bands were visualized by chemiluminescence using an enhanced chemiluminescence kit (Amersham Europe GMBH, Cologno Monzese, Italy).
Statistical Analysis
Results were analyzed by the χ2 test for comparing different degrees of toxicity of drugs under study. The Pearson chi-square test was used for testing the relationship of nonparametric data (toxicity's degree and ERCC1 levels); p ≤ 0.05 was accepted as statistically significant. The software Biostats Calculator ver 1.3.1 for iPad was used for data processing.
Results
RAS status in cell lines
As expected, the internal evaluation of the mutational status of RAS showed all RAS wt status for HT-29 and HCT-8, while SW620 and SW480 were KRAS mutated in codon 12 and NRAS wt.
Oxaliplatin causes a statistically significant higher cytotoxicity in mt KRAS cell lines
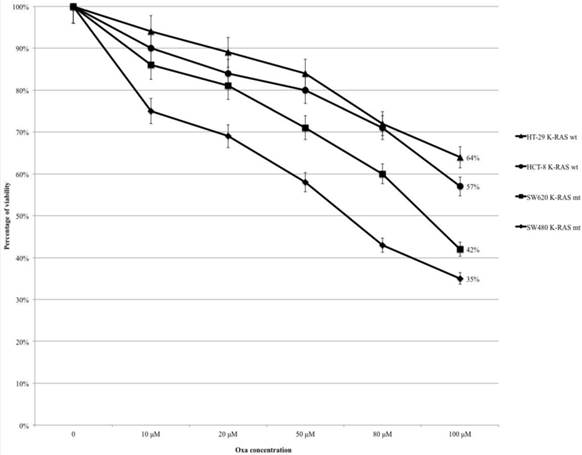
HT-29, HCT-8, SW480 and SW620 cells were treated with Oxa (range 10-100 μM) and the percentage of cytotoxicity was assessed by the MTT assay (Fig. 1). In all cell lines the percentage of viable cells decreased progressively from control to the highest level of treatment (at 100 μM: HT-29 64%, HCT-8 57%, SW620 42% and SW480 35%) (Fig. 1). The mt KRAS cell lines (SW620 and SW480) were more sensitive to Oxa compared with wt KRAS lines, and the odds ratios (OR) were statistically significant (HT-29 vs SW620 OR: 2.68, 95% CI 1.511-4.757 p <0.001; HT-29 vs SW480 OR: 3.6 95% CI 2.012-6.459 p <0.0005; HCT-8 vs SW620 OR:1.831 95% CI 1.045-3.207 p 0.035; HCT-8 vs SW480 OR: 2.43 95% CI 1.371-4.328 p 0.002). Cytotoxicity was not statistically different comparing each other wt KRAS (HT-29 vs HCT-8 OR: 1.464 95% CI 0.826-2.596 p 0.192) and mt KRAS cell lines (SW620 vs SW 480 OR: 0.744 95% CI 0.420-1.317 p 0.310).
MTT-assay: Oxa causes a dose-dependent increased cytotoxicity in KRAS mt cell lines.
SN-38 didn't cause statistically significant different cytotoxicity between mt and wt KRAS cell lines
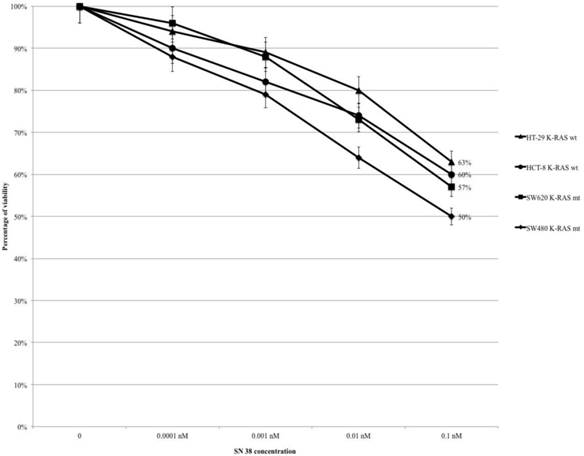
Treating HT-29, HCT-8, SW480 and SW620 cells with SN-38 (range 0.0001-0.1 nM), the percentage of viability gradually decreased in all cell lines from control to the highest level of drug concentration (at 0.1 nM: HT-29 63%, HCT-8 60%, SW620 57% and SW480 50%) (Fig. 2). The mt KRAS cell lines (SW620 and SW480) were more sensitive to SN-38 compared to wt KRAS lines, but the difference is not statistically significant (HT-29 vs SW620 OR: 0.779, 95% CI 0.442-1.373 p 0.387; HT-29 vs SW480 OR: 0.587 95% CI 0.334-1.033 p 0.065; HCT-8 vs SW620 OR: 0.884 95% CI 0.503-1.552 p 0.667; HCT-8 vs SW480 OR: 0.156 95% CI 0.381-1.167 p 0.2). There were no statistically significant differences within wt KRAS lines (HT-29 vs HCT-8 OR: 1.134 95% CI 0.642-2.007 p 0.663) and within mt KRAS lines (SW480 vs SW620 OR: 0.754 95% CI 0.432-1.317 p 0.321).
MTT-assay: SN-38 causes a dose-dependent cytotoxicity but without statistically significant differences.
Silencing KRAS reduces Oxaliplatin's sensitivity in mt KRAS cell lines
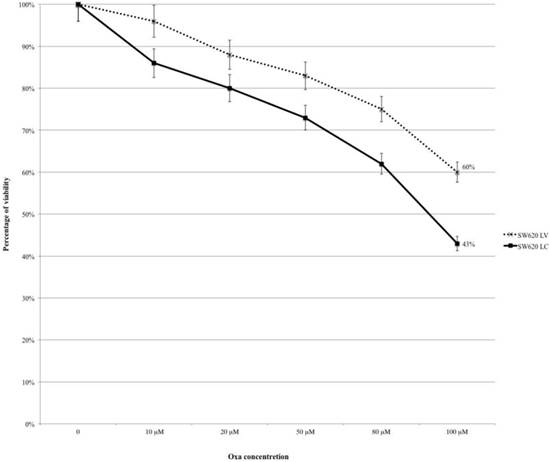
SW620LC and SW620LV were treated with Oxa at the same escalating doses reported in Fig.1 (range 10-100 μM) and cytotoxicity was assessed by the MTT-assay. Cell line treated with control lentivirus (SW620LC) was more sensitive to Oxa than KRAS silenced line (SW620LV) and citotoxicity curves remained diverging at any level of Oxa concentration (at 100 μM: SW620LV 60% and SW620LC 43%; OR: 0.503, 95% CI 0.286-0.883 p 0.017) (Fig. 3).
MTT-assay: Silencing KRAS reduces the sensitivity to Oxa.
Oxaliplatin determines up-regulation of mRNA-ERCC1 only in wt KRAS cells line
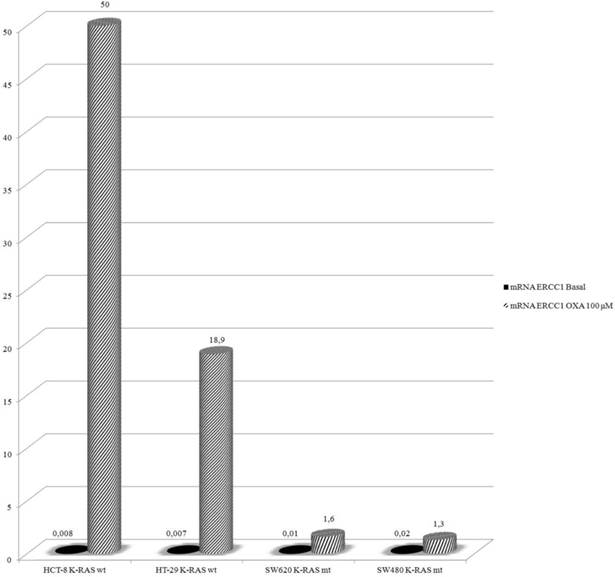
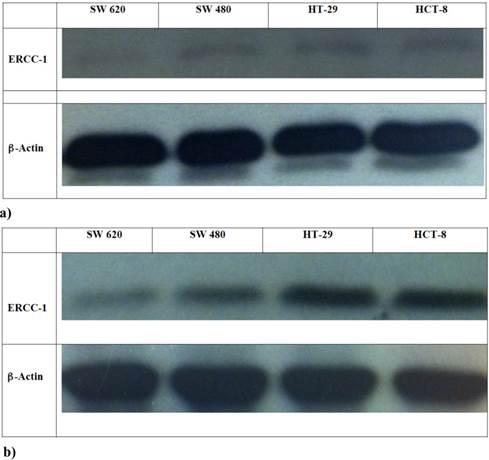
In this experiment HCT-8, HT-29, SW480 and SW620 cells were exposed to Oxa 100 μM for 24 h and then mRNA-ERCC1 was assesed. Basal levels were not significantly different (HT-29 ΔCT: 0.007, HCT-8 ΔCT: 0.008, SW620 ΔCT: 0.01, SW480 ΔCT: 0.02). After exposure to Oxa mRNA-ERCC1 increases in all the cell lines, but in wt KRAS cells the rise was far greater (HCT-8 ΔCT from 0.008 to 50; HT-29 ΔCT from 0.007 to 18.9) than in mt KRAS cells (SW620 ΔCT from 0.01 to 1.6; SW480 ΔCT from 0.02 to 1.3) (Fig. 4). Comparing ERCC1 values after Oxa exposure a large statistically significant difference was found between wt and mt cell lines. This means that only in wt KRAS cells Oxa exposure causes a significant induction of ERCC1, whereas in mt KRAS cell lines ERCC1-dependent repair of Oxa induced damage does not increase significantly (HCT-8 vs SW620 OR: 42.9 95% CI 17.260-106.972 p <0.0005; HCT-8 vs SW480 OR: 89.9 95% CI 39.542-204.520 p < 0.0005; HT-29 vs SW620 OR: 18.56, 95% CI 7.186-47.950 p <0.0005; HT-29 vs SW480 OR: 38.849 95% CI 16.399-92.033 p <0.0005 ). The induction of ERCC1 only in wt KRAS cells line has been confirmed by western blot (Fig. 5a-b).
Real-Time PCR: Oxa determines an increased expression of mRNA-ERCC1 only in KRAS wt cell lines.
a-b Western Blot: ERCC1 expression in cell lines in basal condition (a) and after Oxa exposure (b).
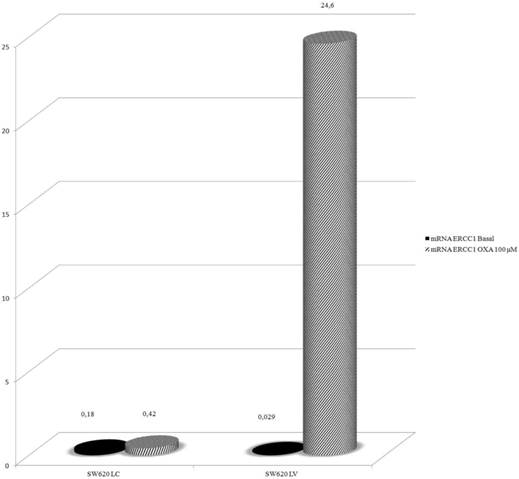
Silencing KRAS restore the ability to induce ERCC1 expression after Oxaliplatin exposure
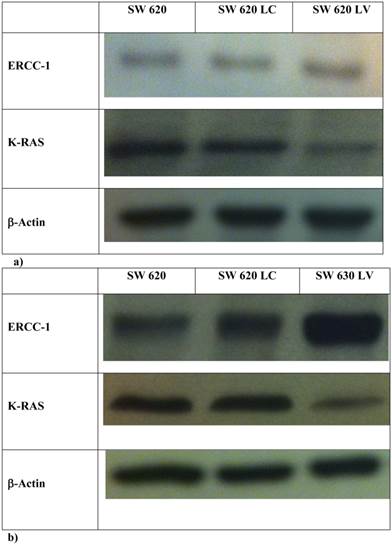
In order to explore further the relationship between KRAS and ERCC1 induction, lentivirus KRAS silenced cells (SW620LV) and their control clones (SW620LC), have been exposed to Oxa 100 μM for 24 h. Basal ERCC1 levels were not significantly different between the two cell lines (SW620LV ΔCT: 0.029 and SW620LC ΔCT: 0.18). However, SW620LV displayed a statistically significant induction of ERCC-1 after exposure to Oxa (ΔCT: from 0.029 to 24.6), whereas SW620LC did not show this potential (ΔCT: from 0.18 to 0.42). The value of mRNA ERCC1 in SW620LV cells after Oxa exposure is significantly higher than in SW620LC cells (OR: 36.5 95% CI 24.2-54.44, p <0.0005) (Fig. 6). Therefore mt KRAS cells acquire the capability of inducting ERCC1, as wt KRAS cell lines do normally, only after silencing KRAS. The induction of ERCC1 after Oxaliplatin exposure in SW620LV has been confirmed by western blot (Fig. 7a-b).
Real-Time PCR: Silencing KRAS restore the ability to induce ERCC1 expression after Oxa exposure.
a-b Western Blot: ERCC1 expression in SW620, SW620LC e SW620LV in basal condition (a) and after Oxa exposure (b).
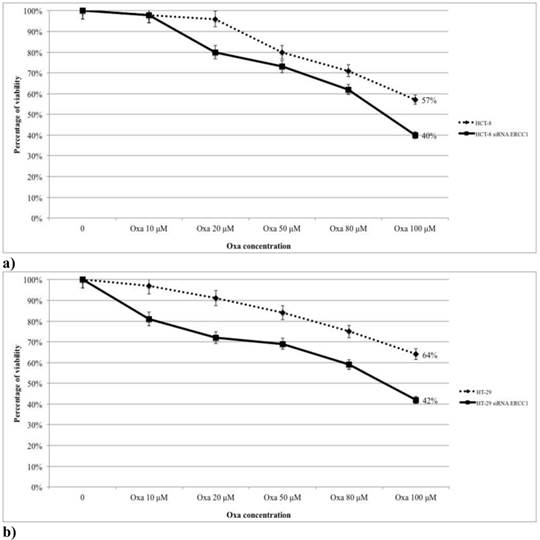
The magnitude of ERCC1 silencing correlates with the variation in sensitivity to Oxaliplatin in SW620LV cell line
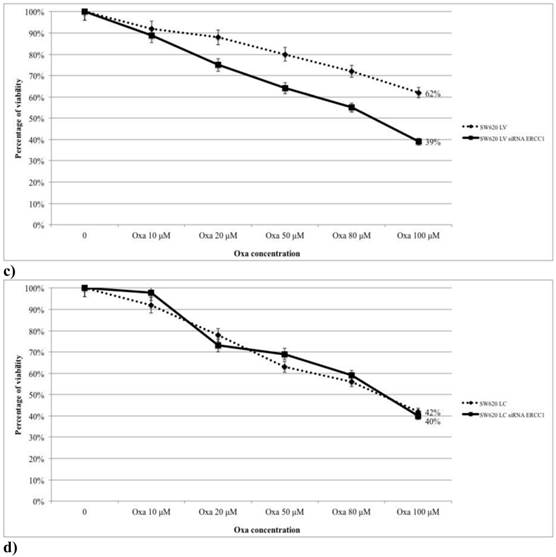
The role of ERCC1 in determining Oxa sensitivity in wt and mt KRAS silenced cell lines has been studied by silencing ERCC1. In both HCT-8 and HT-29 wt KRAS cell lines down-regulation of ERCC1 significantly reduces the percentage of viable cells in the MTT assay at any level of Oxa treatment (p < 0,005 and p <0,005, respectively) (Fig. 8a, 8b). Consistent results have been obtained in SW620LV cell line RNA silenced for ERCC1 in comparison to SW620LV cell line (p< 0,005) (Fig. 8c). In this case there is a statistically significant correlation between the reduction of ERCC1 observed in RT-PCR (Fig. 9) and the extent of variation in Oxa sensitivity (Fig. 8c) (Pearson test, p <0.037). In cell lines not KRAS mRNA silenced (SW620LC), instead, downregulation of ERCC1 (SW620LC siRNA ERCC1) doesn't affect cytotoxicity of escalating Oxa doses (Fig. 8d). Taken together, these results show that ERCC1 plays a crucial role in Oxa cytotoxicity provided that it could not be induced in response to the drug.
a,b,c,d. The magnitude of the silencing of ERCC1 correlates with the variation in sensitivity to Oxa.
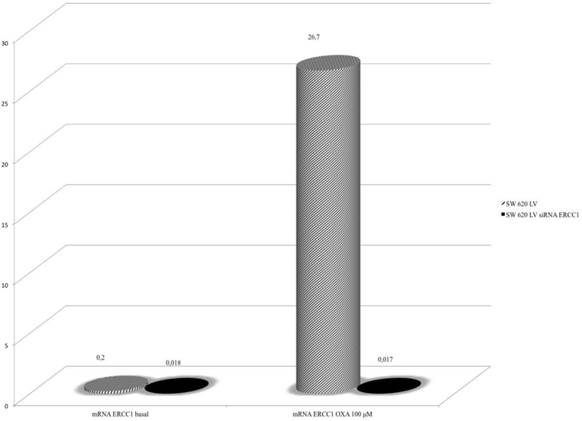
Real-Time PCR: Silencing ERCC1 SW 620LV lose the ability to induce ERCC1 expression after Oxa exposure.
Discussion
The introduction of agents directed to specific molecular targets has stimulated the research of factors predicting response to antineoplastic treatment with a view to limit the exposure to toxic effects of therapy only to patients who have a higher probability to derive benefit from treatment. This pursuit has given also the opportunity to explore in depth the molecular mechanisms involved in tumor development and growth. In CRC the major advance has come from identification of the predictive role of KRAS mutation in response to therapy with anti-EGFR antibodies, alone or in combination with irinotecan- or oxa-based chemotherapy (6, 7, 8, 24, 25, 26). Furthermore, in OPUS and PRIME studies, a clear increase of PFS in mt in comparison to wt KRAS patients, respectively, was unexpectedly observed in the control arm, suggesting the possibility that KRAS mutational status could also affect response to standard chemotherapy. This hypothesis was supported by a retrospective study of our group in which this effect resulted indipendent from basal mRNA ERCC1 (10).
In the present study we aimed at exploring potential mechanisms underlying increased Oxa sensitivity of mt KRAS colorectal cancer cells. Results confirmed a significantly higher cytotoxicity in mt KRAS cells (SW620 and SW480) than in wt lines (HT-29 and HCT-8). Wild type and mutated cell lines did not show statistically significant differences in SN 38 sensitivity, thus confirming the specificity of the synergism between KRAS mutation and Oxa sensitivity. Previous in-vitro studies have suggested that response to Oxa in advanced CRC might be influenced by expression of ERCC1, a pathway also involved in KRAS mutagenesis (21, 22). Moreover, ERCC1 expression in response to Oxa was positively regulated by EGFR in wt KRAS cells (23). Based on these observations, we assumed that KRAS mutational status could influence ERCC1 expression and therefore we determined ERCC1 levels at baseline and after exposure to Oxa in our cell lines. We found no significant difference in term of ERCC1 basal levels between mt and wt cell lines and may conclude that basal levels of ERCC1 do not correlate with response to Oxa in agreement with our previous retrospective clinical study (10). However, after exposure to Oxa, only wt KRAS cells, more resistant to the drug, were able to induce ERCC1. This result allows us to suppose a link between ERCC1 induction and KRAS mutation; therefore, we tested this hypothesis by silencing mutated KRAS in SW620 cell line. In the resulting SW620LV a statistically significant decrease in Oxa sensitivity was actually observed, thus confirming a direct correlation between KRAS and Oxa activity. Interestingly, silenced mutated KRAS SW620 acquired not only a more resistant phenotype but even the ability to induce ERCC1 after Oxa exposure, as it occurs “de novo” in wt KRAS cells. On the contrary, by silencing mRNA ERCC1, Oxa cytotoxicity increases in wt KRAS cell lines as well in SW620LV cells, demonstrating the role of this enzyme in resistance to Oxa.
A positive regulation of ERCC1 expression by EGFR pathway has been already seen in wt KRAS cell lines resulting, however, in a synergistic interaction between Oxa and Cet (23). This is in agreement with ERCC1 induction that we have observed in wt KRAS cell lines, but suggests that cytotoxicity induced by Cet might overcome the relative Oxa-resistance due to ERCC1 induction. Further indirect evidence of Oxa-induced ERCC1 expression comes from the observation that Oxa resistant cell lines showed a lesser ability to repair DNA than their parental cell lines when exposed to Cet (28). Taken together with our results, these findings suggest that ERCC1 downregulation after exposure to Oxa in mt KRAS cells would be considered as an evolutionary advantage triggered by the abnormal proliferative stimulus represented by KRAS mutation.
How KRAS mutation drives ERCC1 downregulation remains to be understood, but recently it has been shown that in breast cancer cells the expression of ERCC1 is regulated by H-RAS, one of the three proteins of the RAS family, through AP-1 (29). Contrary to what was observed in the past, it has also been shown that the three protein of RAS family (N-RAS, KRAS and H-RAS), have different activities and each may affect the synthesis and function of the others (30-31). Therefore it is conceivable that mutational status of KRAS can influence the activity of H-RAS, and consequently the expression of ERCC1. Our preliminary data (not shown), indeed, suggest that downregualtion of ERCC1 is related to decreased levels both of H-RAS and AP-1 only in mt KRAS cell lines.
Our results show that ERCC1 gene expression is inducible, contributes to Oxa resistance and seems to be regulated by KRAS mutational status, thereby explaining the different Oxa sensitivity of mt in comparison to wt KRAS cell lines. The demonstration that ERCC1 is an enzyme potentially inducible by Oxa may contribute to explain the lack of correlation, observed in numerous studies, between basal ERCC1 tissue levels and response to Oxa therapy (17-19) Other in-vitro studies have associated the inducible up-regulation of ERCC1 to cellular resistance to Oxa (32). In agreement with “in-vitro” data, Merla et al have recently shown that ERCC-1 gene expression may be induced by oxaliplatin-based chemotherapy also in-vivo and that the enzyme level is related to clinical outcome (33). By serial sampling of ERCC-1 levels in mononuclear blood cells of mCRC patients during any cycle of FOLFOX-6 therapy, they obtained median PFS of 7.25 months in patients with ERCC1 induction significantly shorter than 13 months in patients with stable or reduced ERCC1, whereas in agreement with other studies, they didn't find any significant difference in PFS when baseline expression of ERCC-1 was considered. They concluded that gene induction is a potential marker of resistance to chemotherapy, however, ERCC1 gene induction may not be considered a valid biomarker because it lacks of a real predictive value, not allowing selecting patients before starting treatment. Conversely, based on our results, the cellular ability to modulate ERCC1 is actually surrogated by KRAS mutational status, that might be considered a valid predictor of response to Oxa.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Jemal A, Siegel R, Yu J. et al. Cancer statistics 2010. CA: A Cancer Journal for Clinicians. 2010;60:277-300
2. Raymond E, Faivre S, Woynarowski JM. et al. Oxaliplatin: mechanism of action and antineoplastic activity. SeminOncol. 1998(Suppl 5):4-12
3. Woynarowski JM, Chapman WG, Napier C. et al. Sequence and region-specificity of oxaliplatin adducts in naked and cellular DNA. Mol Pharmacol. 1998Nov;54:770-7
4. de Gramont A, Figer A, Seymour M. et al. Leucovorin and fluorouracil with or without oxaliplatin as first line treatment in advanced colorectal cancer. J Clin Oncol. 2000;18(16):2938-2947
5. Mamenta EL, Poma EE, Kaufmann WK. et al. Enhanced replicative bypass of platinum-DNA adducts in cisplatin-resistant human ovarian cancer cell lines. Cancer Res. 1994;54:3500-5
6. E. Van Cutsem, C.H. Kohne, I. Lang et al. Cetuximab plus irinotecan, fluorouracil, and leucovorin as first-line treatment for metastatic colorectal cancer: updated analysis of overall survival according to tumor KRAS and BRAF mutation status. J Clin Oncol. 2011;29:2011-2019
7. Bokemeyer C, Bondarenko I, Makhson A. et al. Fluorouracil, leucovorin, and oxaliplatin with and without cetuximab in the first-line treatment of metastatic colorectal cancer. J Clin Oncol. 2008;27:663-671
8. Douillard JY, Siena S, Cassidy J, Tabernero J, Burkes R, Barugel M, Humblet Y, Bodoky G, Cunningham D, Jassem J, Rivera F, Kocákova I, Ruff P, Błasińska-Morawiec M, Šmakal M, Canon JL, Rother M, Oliner KS, Wolf M, Gansert J. Randomized, phase III trial of panitumumab with infusional fluorouracil, leucovorin, and oxaliplatin (FOLFOX4) versus FOLFOX4 alone as first-line treatment in patients with previously untreated metastatic colorectal cancer: the PRIME study. J Clin Oncol. 2010Nov1;28(31):4697-705
9. Douillard JY1, Oliner KS, Siena S, Tabernero J, Burkes R, Barugel M, Humblet Y, Bodoky G, Cunningham D, Jassem J, Rivera F, Kocákova I, Ruff P, Błasińska-Morawiec M, Šmakal M, Canon JL, Rother M, Williams R, Rong A, Wiezorek J, Sidhu R, Patterson SD. Panitumumab-FOLFOX4 treatment and RAS mutations in colorectal cancer. N Engl J Med. 2013Sep12;369(11):1023-34
10. Basso M, Strippoli A, Orlandi A, Di Salvatore M, Schinzari G, Martini M, Calegari A, Cassano A, Barone C. KRAS mutational status affects oxaliplatin-based chemotherapy independently from basal mRNA ERCC1 expression in metastatic colorectal cancer patients. Br J Cancer. 2013;108(1):115-20
11. Vekris A, Meynard D, Haaz MC, Bayssas M, Bonnet J, Robert J. Molecular determinants of the cytotoxicity of platinum compounds: the contribution of in silico research. Cancer Res. 2004Jan1;64(1):356-62
12. Graf N, Ang WH, Zhu G, Myint M, Lippard SJ. Role of endonucleases XPF and XPG in nucleotide excision repair of platinated DNA and cisplatin/oxaliplatin cytotoxicity. Chembiochem. 2011;12(7):1115-23
13. Tsodikov OV, Enzlin JH, Schärer OD. et al. Crystal structure and DNA binding functions of ERCC1, a subunit of the DNA structure-specific endonuclease XPF-ERCC1. Proc Natl Acad Sci USA. 2005;102:11236-41
14. Arnould S, Hennebelle I, Canal P. et al. Cellular determinants of oxaliplatin sensitivity in colon cancer cell lines. Eur J Cancer. 2003;39:112-9
15. Yu JJ, Lee KB, Mu C. et al. Comparison of two human ovarian carcinoma cell lines (A2780/CP70 and MCAS) that are equally resistant to platinum, but differ at codon 118 of the ERCC1 gene. Int J Oncol. 2000;16:555-60
16. Park DJ, Stoehlmacher J, Zhang W. et al. ERCC1 polymorphism is associated with differential ERCC1 gene expression. Proc Am Assoc Cancer Res. 2002;43:321 (abstract 1591)
17. Shirota Y, Stoehlmacher J, Brabender J. et al. ERCC1 and thymidylate synthase mRNA levels predict survival for colorectal cancer patients receiving combination oxaliplatin and fluorouracil chemotherapy. J Clin Oncol. 2001;19:4298-304
18. Lenz HJ, Zhang W, Shi MM. et al. ERCC-1 gene expression levels and outcome to FOLFOX chemotherapy in patients enrolled in CONFIRM1 and CONFIRM2. J Clin Oncol. 2008:abstract4131
19. Kim SH, Kwon HC, Oh SY. et al. Prognostic value of ERCC1, thymidylate synthase, and glutathione S-transferase pi for 5-FU/oxaliplatin chemotherapy in advanced colorectal cancer. Am J Clin Oncol. 2009;32:38-43
20. Dickstein BM, Wosikowski K, Bates SE. Increased resistance to cytotoxic agents in ZR75B human breast cancer cells transfected with epidermal growth factor receptor. Mol Cell Endocrinol. 1995;110:205-211
21. Yacoub A, McKinstry R, Hinman D, Chung T, Dent P, Hagan MP. Epidermal growth factor and ionizing radiation up-regulate the DNA repair genes XRCC1 and ERCC1 in DU145 and LNCaP prostate carcinoma through MAPK signaling. Radiat Res. 2003;159:439-452
22. Yang G, Curley D, Bosenberg MW, Tsao H. Loss of Xeroderma Pigmentosum C (Xpc) enhances melanoma photocarcinogenesis in Ink4a-Arf-Deficient mice. Cancer Res. 2007;67(12):5649-5657
23. Balin-Gauthier D, Delord JP, Pillaire MJ, Rochaix P, Hoffman JS, Bugat R, Cazaux C, Canal P, Allal BC. Cetuximab potentiates oxaliplatin cytotoxic effect through a defect in NER and DNA replication initiation. Br J Cancer. 2008Jan15;98(1):120-8
24. Saltz LB, Meropol NJ, Loehrer PJ Sr, Needle MN, Kopit J, Mayer RJ. et al. Phase II trial of cetuximab in patients with refractory colorectal cancer that expresses the epidermal growth factor receptor. J Clin Oncol. 2004;22:1201-8
25. Cunningham D, Humblet Y, Siena S, Khayat D, Bleiberg H, Santoro A, Bets D, Mueser M, Harstrick A, Verslype C, Chau I, Van Cutsem E. Cetuximab monotherapy and cetuximab plus irinotecan in irinotecan-refractory metastatic colorectal cancer. N Engl J Med. 2004Jul22;351(4):337-45
26. Van Cutsem E, Siena S, Humblet Y, Canon JL, Maurel J, Bajetta E, Neyns B, Kotasek D, Santoro A, Scheithauer W, Spadafora S, Amado RG, Hogan N, Peeters M. An open-label, single-arm study assessing safety and efficacy of panitumumab in patients with metastatic colorectal cancer refractory to standard chemotherapy. Ann Oncol. 2008Jan;19(1):92-8
27. Tveit KM, Guren T, Glimelius B, Pfeiffer P, Sorbye H, Pyrhonen S, Sigurdsson F, Kure E, Ikdahl T, Skovlund E, Fokstuen T, Hansen F, Hofsli E, Birkemeyer E, Johnsson A, Starkhammar H, Yilmaz MK, Keldsen N, Erdal AB, Dajani O, Dahl O, Christoffersen T. Phase III trial of cetuximab with continuous or intermittent fluorouracil, leucovorin, and oxaliplatin (Nordic FLOX) versus FLOX alone in first-line treatment of metastatic colorectal cancer: the NORDIC-VII study. J Clin Oncol. 2012;30(15):1755-62
28. Prewett M, Deevi DS, Bassi R, Fan F, Ellis LM, Hicklin DJ, Tonra JR. Tumors established with cell lines selected for oxaliplatin resistance respond to oxaliplatin if combined with cetuximab. Clin Cancer Res. 2007Dec15;13(24):7432-40
29. Youn CK, Kim MH, Cho HJ, Kim HB, Chang IY, Chung MH, You HJ. Oncogenic H-Ras up-regulates expression of ERCC1 to protect cells from platinum-based anticancer agents. Cancer Res. 2004 Jul 15;64(14):4849-57. Erratum in: Cancer Res. 2004Nov15;64(22):8484
30. Ferreira L, Fuentes-Calvo I, Muñoz-Félix JM, Muñiz-Martín C, Sánchez-Juanes F, Raposo C, González-Buitrago JM, López-Novoa JM, Martínez-Salgado C. Functional specific roles of H-ras and N-ras. A proteomic approach using knockout cell lines. Electrophoresis. 2012May;33:1385-96
31. Castellano E, Santos E. Functional specificity of ras isoforms: so similar but so different. Genes Cancer. 2011Mar;2(3):216-31
32. Seetharam RN, Sood A, Basu-Mallick A, Augenlicht LH, Mariadason JM, Goel S. Oxaliplatin resistance induced by ERCC1 up-regulation is abrogated by siRNA-mediated gene silencing in human colorectal cancer cells. Anticancer Res. 2010Jul;30(7):2531-8
33. Merla A. et al. The kinetics of the excision repair cross-complementing group-1 (ERCC1) gene in patients with colorectal cancer. J Clin Oncol. 2012;30(suppl):abstr10574
Author contact
![]() Corresponding author: Prof. Carlo Barone, Medical Oncology, Università Cattolica del Sacro Cuore, Largo Agostino Gemelli, n° 8, 00168 Rome, Italy. Tel: +390630154844 Fax: + 39063051343 e-mail: carlobaroneunicatt.it.
Corresponding author: Prof. Carlo Barone, Medical Oncology, Università Cattolica del Sacro Cuore, Largo Agostino Gemelli, n° 8, 00168 Rome, Italy. Tel: +390630154844 Fax: + 39063051343 e-mail: carlobaroneunicatt.it.