Impact Factor ISSN: 1837-9664

 Global reach, higher impact
Global reach, higher impactJ Cancer 2015; 6(3):203-217. doi:10.7150/jca.10640 This issue Cite
Review
Immune Suppression during Oncolytic Virotherapy for High-Grade Glioma; Yes or No?
Carolien A.E. Koks1 ![]() , Steven De Vleeschouwer2,3, Norbert Graf4, Stefaan W. Van Gool1,5
, Steven De Vleeschouwer2,3, Norbert Graf4, Stefaan W. Van Gool1,5
1. Pediatric Immunology, Department of Microbiology and Immunology, KU Leuven, Belgium
2. Department of Neurosciences, KU Leuven, Belgium
3. Neurosurgery, University Hospitals Leuven, Belgium
4. Department for Pediatric Oncology, University of Saarland Medical School, Germany
5. Pediatric Neuro-oncology, University Hospitals Leuven, Belgium
Received 2014-9-25; Accepted 2014-11-14; Published 2015-1-15
Abstract
Oncolytic viruses have been seriously considered for glioma therapy over the last 20 years. The oncolytic activity of several oncolytic strains has been demonstrated against human glioma cell lines and in in vivo xenotransplant models. So far, four of these stains have additionally completed the first phase I/II trials in relapsed glioma patients. Though safety and feasibility have been demonstrated, therapeutic efficacy in these initial trials, when described, was only minor. The role of the immune system in oncolytic virotherapy for glioma remained much less studied until recent years. When investigated, the immune system, adept at controlling viral infections, is often hypothesized to be a strong hurdle to successful oncolytic virotherapy. Several preclinical studies have therefore aimed to improve oncolytic virotherapy efficacy by combining it with immune suppression or evasion strategies. More recently however, a new paradigm has developed in the oncolytic virotherapy field stating that oncolytic virus-mediated tumor cell death can be accompanied by elicitation of potent activation of innate and adaptive anti-tumor immunity that greatly improves the efficacy of certain oncolytic strains. Therefore, it seems the three-way interaction between oncolytic virus, tumor and immune system is critical to the outcome of antitumor therapy. In this review we discuss the studies which have investigated how the immune system and oncolytic viruses interact in models of glioma. The novel insights generated here hold important implications for future research and should be incorporated into the design of novel clinical trials.
Keywords: glioblastoma, oncolytic virotherapy, antitumor immunity
Introduction
In 1904 a report was published describing complete remission in a chronic leukemia patient, following what was thought to be an influenza infection (1). Interestingly, this was described some 30 years before it was determined that influenza is caused by a viral infection, not a bacterium (2). Initial case reports like this sparked the first interest in using viruses to treat human cancers and form the earliest basis of the now quickly evolving oncolytic virotherapy (OVT) field.
In the last two decades, advances in our understanding of tumor biology and virology have only increased the interest in using oncolytic viruses (OV) for cancer therapy. To date, the oncolytic activity of over 20 viruses has been characterized, and new oncolytic candidates continue to emerge. Within the last decade, large-scale and well-documented clinical application was reached. Hundreds of cancer patients have been treated, in clinical trials evaluating over ten different oncolytic strains in malignancies such as melanoma, glioma, hepatocellular carcinoma, and ovarian cancer (3-7). Virotherapeutics such as Herpes simplex virus (HSV), Newcastle disease virus (NDV), and vaccinia virus have demonstrated systemic safety and efficacy in clinical practice and OVs have been indicated as a safer alternative than other cytotoxic agents used in phase I oncology studies (8).
In November 2005 the first OV (Oncorine; a genetically modified adenovirus) was approved for commercialization and routine application in the treatment of nasopharyngeal carcinoma, by the Chinese State Food and Drug Administration (FDA) (9). The first OV to generate positive results in a phase III clinical trial is the drug called talimogene laherparepvec (Tvec; a genetically modified HSV provided by Amgen), making it a likely first candidate for routine use FDA approval (in the treatment of advanced melanoma) (10).
A tumor entity to quickly become a target for OVT is Glioblastoma Multiforme (GBM). GBM is classified as a grade IV neoplasm by the World Health Organization and is the most frequent primary brain tumor in adults (11). The interest in testing novel approaches for the treatment of this disease stems from the dismal prognosis of affected patients (12;13). Despite major improvements in the fields of conventional therapies; surgery, radio- and chemotherapy, overall survival remains extremely low, residing at 14.6 months with mortality rates reported to be around 88% within three years (12). At time of relapse - which remains universal - the prognosis is particularly bleak, with a median survival of only nine months in the most successful salvage strategies (14). With the realization that further advances in conventional therapies alone would not produce any long-term solutions for GBM patients, a strong need for more effective long-term treatments that are tumor-specific and able to kill all (residual) tumor cells, emerged. OVT represents such an approach, which can match the heterogeneity of the tumor and utilize the same activated pathways that drive tumor cell growth.
So far, 15 different OV have been tested preclinically in models of GBM (Table 1) (15;16). Four oncolytic strains have additionally completed the first phase I clinical trials in GBM patients. In total, 120 patients with GBM (occasionally anaplastic astrocytoma), mainly relapsed cases, were treated with OV via intratumoral administrations or intravenous injections. As these trials were designed primarily to evaluate safety and feasibility, the outcome has been clearly positive. There was a near complete absence of serious adverse events and no maximally tolerated dose (MTD) was reached in any trial. These results compare favorably to phase I trials conducted with non-biological cytotoxic drugs, where a MTD is virtually always reached and some toxicity is to be expected. Though dosing and application regimens were conservative, efficacy was described in the form of tumor shrinkage, long-term survival, and even complete responses. Results compare favorably to the multicenter studies with current gold standard Temozolomide in GBM at first relapse, which describe complete responses to treatment in only 1 percent of Temozolomide-pretreated and 2 percent of Temozolomide-naïve patients, respectively (17). In this respect, OVT warrants further investigation as a valuable novel approach for the treatment of GBM. At the same time however, the limited efficacy seen in initial clinical application does not nearly match the significant therapeutic effects that were demonstrated in preclinical work. This suggests that new studies are urgently needed to investigate the reasons behind this discrepancy and ways of improving clinical efficacy.
Oncolytic virus candidates for glioma therapy
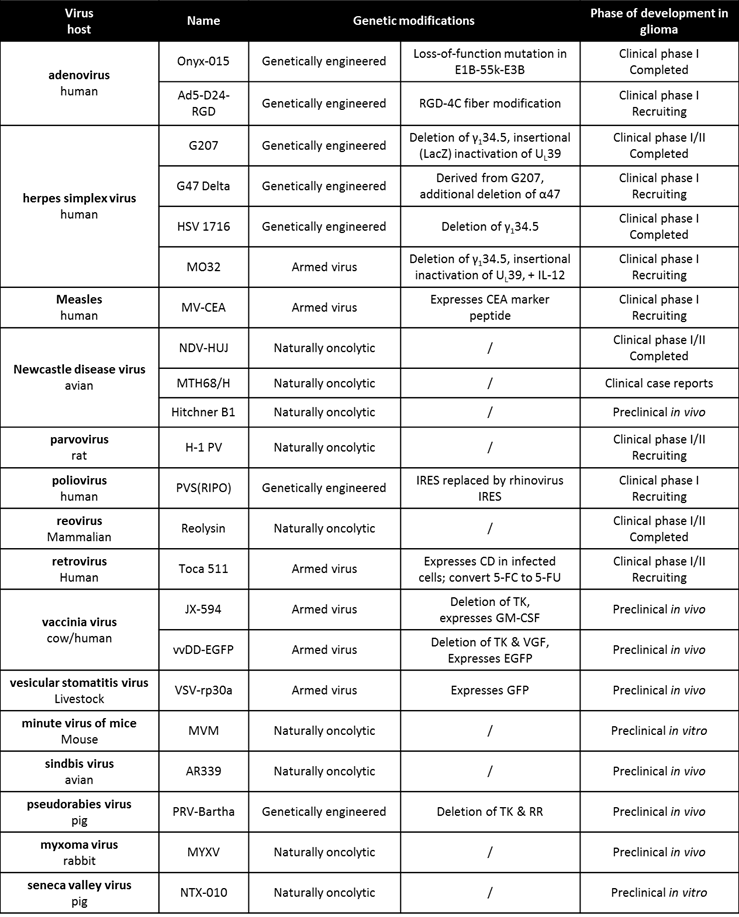
Data last verified on October 20, 2014 (www.clinicaltrials.gov)
The setup of initial clinical trials was based on results obtained in preclinical in vitro systems and xenotransplant models, where OVT was shown to be highly efficient. Based on these preclinical models, the efficacy of OVT was hypothesized to be dependent on the replication capacity of the virus and the extent of virus-mediated tumor cell death. The role of the immune system as mode of action during OVT was not often considered in initial investigations, nor in the setup of initial clinical trials. However, in hindsight, when using a highly immunogenic therapeutic agent such as a virus, it seems highly unlikely that the results from immunodeficient animals could be directly translated into a clinical, patient setting.
In this review we will investigate the complex relationship between the immune system and OVT for glioma. Due to the highly immunogenic nature of viruses, many groups initially hypothesized the immune system to be a strong hurdle for successful OVT and several have aimed to improve therapeutic outcome by suppressing or circumventing innate immunity. Several remarks concerning a unilateral view point on this matter can be raised, however, and we will discuss data obtained in immunocompetent animals which now change the OVT paradigm. Finally, we will discuss ways in which OV can induce both innate and adaptive antitumor immunity, and which are only recently being investigated in models of GBM. From this overview, we will highlight areas for future research in this field and we aim to answer the question raised in the title of this work. Should we keep/put GBM patients on immunosuppressive regimens to allow for successful OVT? Or might the co-administration of immunosuppressive agents rather interfere with the therapeutic benefit of OV?
Early preclinical studies
Some 20 years ago, oncolytic HSV was demonstrated to directly replicate in and kill a whole range of both murine and human glioma cells (18). When tested in immunodeficient SCID mice bearing human glioma xenotransplants, HSV therapy significantly prolonged survival of treated animals as compared to untreated controls (18). Following this initial report, several HSV mutants were developed, demonstrating cytopathic effects on human glioma cells in vitro and in vivo; in subcutaneous and intracranial xenotransplant models (19;20). Around the same time, a second group of genetically engineered viruses was developed for study in glioma therapy; adenoviruses. A tumor-selective adenovirus, Delta24, was shown to replicate in and lyse human glioma cells with great efficiency (21). In vivo, multiple intratumoral injections of Delta24 induced inhibition of tumor growth of subcutaneously injected human glioma cells. Amongst the first naturally occurring oncolytic strains to be investigated in the treatment of preclinical glioma was reovirus (22). Reovirus was demonstrated to kill both established malignant glioma cell lines and primary patient cultures and to cause dramatic (often complete) tumor regression in both subcutaneous and intracerebral human malignant glioma mouse models. More recently, the strong cytotoxic potential of NDV towards human glioma cells has also been demonstrated in vitro and in vivo (23).
Translation into clinical trials
The initial preclinical findings indicating direct human glioma cell killing by OV, together with extensive safety assessments in rodents and non-human primates, resulted in the initiation of the first clinical trials in relapsed glioma patients. To date, HSV, adenovirus, reovirus and NDV have completed the first round of clinical application (Table 2).
Immunological aspects of oncolytic virotherapy in glioma patients

The possibility that the immune status of the patient might influence the therapeutic potential of OVT was not taken into consideration in the setup of these initial trials. Immunological endpoints were limited to measuring cytokine concentrations and antiviral neutralizing antibody titers over time following treatment. Immune status and antiviral immunity of patients were often reported. The immunological aspects investigated in these trials are outlined in Table 2.
In trials employing HSV mutant 1716, patients were reported to remain on highly immunosuppressive dexamethasone regimens during and following intracranial OVT (24-26). In cases where dexamethasone dosing was aimed to be reduced gradually, complete withdrawal was only achieved several weeks after the start of OVT (26). Other patients received valproic acid (VPA) to manage seizures or concomitant and/or subsequent chemo- and/or radiotherapy (25;26). When looking at the immune status of these patients at the time of HSV injection, lymphopenia and low T cell counts were detected in all patients, while total white cell counts remained normal (24-26). Cellular proliferative responses were also reduced in most patients, when compared with normal healthy controls. In line with what is generally accepted, these data indicated a reduced degree of immunocompetence in GBM patients. Despite the immunosuppressive effects of malignant glioma and of dexamethasone treatment, changes in IgG and IgM serum levels in treated patients demonstrated the induction of an immunological response to HSV1716 following the intratumoral administration of the virus (25). The presence of anti-HSV antibodies did, however, not block successful HSV1716 replication at the tumor site in these seropositive patients (25). High, continued administration of immunosuppressive dexamethasone was linked to lack of generating anti-HSV immune responses in these cases (26).
In clinical studies with the HSV G207 strain, patients seronegative for HSV-1 antibody prior to OV inoculation showed seroconversion, despite chronic dexamethasone treatment, if the administered virus dose was high enough (27). All previously seronegative patients seroconverted following HSV therapy and some patients had increasing antibody titers over time during the course of study (3). Patients included in these trials demonstrated a similar reduction in immunocompetence, marked by low CD4+ and CD8+ T cell counts, both pre- and post-G207 (3). However, though only minimal immune cell infiltrates could be detected in tumor tissue sections before G207 inoculation, significant positive staining with anti-CD3 and anti-CD8 was seen in some patients, post-OV administration. Simultaneously, infiltrating monocytes, macrophages and microglia were also detected in these sections.
The adenovirus mutant ONYX-015 has also been investigated in a phase I trial using the intratumoral injection approach (28). Out of 22 seronegative patients treated with ONYX-015, only 2 seroconverted from negative to positive for adenovirus antibodies (28). These patients received the highest OV dosages (109 - 1010) in a dose-escalating trial. This low number was attributed to the relatively immunocompromised state of the patients, who were on steroid medication and had previously been treated with chemotherapy and radiation.
Wild type reovirus has been investigated in a phase I trial for relapsed glioma patients, though immunological aspects were not discussed (29).
NDV remains the only viral agent which has been tested in glioma patients following intravenous administration. So far, one formal phase I study using the lentogenic HUJ strain and three case reports/series using the mesogenic MTH-68/H strain have been published (30-33). All enrolled patients were negative for anti-NDV antibodies at baseline (30). All patients however seroconverted during the course of the trial, though antibody titers remained low throughout repetitive dosing. In patients receiving long-term therapy, antibodies either plateaued or started to decrease after several weeks.
Though immunosuppressive mechanisms are at play at the tumor site and their immunity is often compromised, treated GBM patients were able to mount antiviral immune responses to the administered OV, as measured by neutralizing antibody titers. Additionally, two studies have shown the potential of OV to kill glioma cells in vitro and in xenotransplant models, but failing to do so in immunocompetent tumor-bearing animals (34;35). Based on this understanding the view emerged that the immune system might be heavily inhibitory to OVT and that direct translation of results obtained in immunodeficient animals to a relatively immunocompetent patient might prove problematic. Therefore, in more recent studies, several groups have started to investigate the hypothesis that, besides improving the potency of the oncolytic agent employed, the maintenance or induction of immunosuppression might also be critical to improving the efficacy of OVT in clinical applications. Immunosuppressive co-treatments could avoid rapid immune-mediated viral clearance and thus prolong viral persistence in the tumor environment.
Suppression of antiviral immunity using immunosuppressive agents
Cyclophosphamide
A first immunosuppressive agent investigated in combination with OVT is cyclophosphamide (CPM). CPM is an alkylating agent used in chemotherapeutic strategies for the treatment of several types of malignancies (e.g. breast carcinoma, (non-)Hodgkin's lymphoma, certain types of leukemia). It has been described to reduce vascular permeabilisation and, given its immunosuppressive actions at high dosages, it is also used in the treatment of severe autoimmune disease.
In an immunocompetent rat glioma model using intratumoral treatment with HSV, CPM pretreatment increased the replication of the virus in the injected tumors by suppressing immune activity (36). Here, increased survival of HSV within the infected tumor microenvironment led to increased propagation in the tumor cells. It was demonstrated that this was not due to a direct enhancement of viral replication in tumor cells, but coincided with an impaired mRNA production of antiviral cytokines in peripheral blood mononuclear cells. Further work demonstrated a marked increase in intratumoral natural killer (NK) cells and phagocytes early after viral infection, as well as increased macrophage influx at later time points (37). CPM pretreatment was able to inhibit the intratumoral infiltration of phagocytes, as well as the interferon (IFN-)γ production by NK cells. The same increase in OV titers in the tumor microenvironment of treated animals could also be obtained by direct depletion of macrophages and microglia via clodronate liposomes (35). The effect of CPM pretreatment on the recruitment of CD4+ and CD8+ T cells at later time points in this model was not shown (37). CPM likewise prolonged viral-mediated gene expression in the brain of glioma-bearing mice treated with oncolytic adenovirus (38).
Rapamycin
More recently, Rapamycin (Sirolimus) was also combined with OVT as a means of improving viral spreading in vivo. Rapamycin is used in the prevention of (kidney) transplant rejection and is a potent immunosuppressant that inhibits the development and/or activation of both innate and adaptive immune responses. It has been demonstrated to have direct activity against glioma (39;40) and was thought to be a safer clinical alternative than CPM, which, at the high dosages required for the immunosuppressive function, is often toxic.
Whereas Myxoma virus was initially demonstrated to prolong survival of immunocompromised hosts carrying human gliomas, it failed to do so in an immunocompetent syngeneic racine glioma model (34). Co-administration of Rapamycin in this model however significantly improved the survival of glioma-bearing rats. This finding strongly supports the concept that the antiviral immune response is a major barrier to effective OVT. The mechanism behind this was shown to be the inhibition of intratumoral infiltration of microglia and peripheral macrophages after viral treatment. This coincided with higher viral titers in the tumor microenvironment in the days following OV administration. Furthermore, Rapamycin suppressed the production of type I IFN, as well as inhibiting the ability of glioma cells to respond to IFN-α/β.
Valproic acid
The histone deacetylase inhibitor VPA has also been investigated in this respect, albeit in an immunocompromised setting using nude mice (41). Similar to CPM and Rapamycin, VPA was shown to decrease the recruitment of macrophages and NK cells into the tumor microenvironment at early time points after viral infection. At later time points however, strong recruitment of both cell populations was noted demonstrating the transient nature of the immunosuppressive environment following administration of a single dose of VPA. Like Rapamycine, VPA was demonstrated to directly inhibit type I IFN, by preventing the transcription of IFN-stimulated genes. It's important to note that any approach which induces direct type I IFN inhibition is potentially dangerous, as it could cause severe toxicity towards healthy cells and thereby risk compromising the tumor-specificity of OVT. In vitro, VPA impaired NK cell function through the inhibition of granzyme B and perforin expression.
Evading antiviral immunity using carrier cells
In order to tackle not only the hurdle of the host antiviral immune response but also the issue of inefficient viral distribution within the tumor and to distant tumor sites, a new approach using carrier cells was investigated. This approach comprises the use of cells with natural tropism for tumor sites as OV carriers, thereby hiding the virus from immune detection. It was hypothesized that this type of approach would enable systemic application, resulting in improved viral spreading throughout the tumor and to multiple tumor sites. Additionally, these virus-infected cells have the potential to act as in situ virus-producing factories, which generate OV progeny at the tumor beds. The ideal carrier therefore should be easily infected with the therapeutic virus, produce high levels of progeny that can infect target tumor cells and be relatively resistant to OV-mediated toxicity. Stem cells possess an additional desirable characteristic, as their immunosuppressive properties have been well documented in literature (42;43). These qualities would not only allow therapeutic viruses to be hidden from host immunosurveillance, but may also suppress local inflammation during virotherapy, thus allowing the virus to replicate and kill tumor cells without immune restriction.
Mesenchymal stem cells
Hai et al. investigated the possibility of using mesenchymal stem cells (MSC) as carriers for adenovirus delivery in an immunocompetent orthotopic glioma model (44). MSC are derived from the bone marrow, are multipotent cells and can differentiate into a variety of mesenchymal tissue cells; osteoblasts, chondrocytes, and adipocytes.
MSC had previously been shown to possess inherent tumor tropism in this model, as well as in glioma xenotransplant models, where they could effectively deliver Delta24-RGD to the glioma site (45;46). The mechanism behind this tumor tropism is still largely unknown, but was linked decades ago to the inflammatory signaling within tumors, which resembles that of unresolved wounds (47). In glioma, this has been shown to be specifically associated with chemotaxis induced by platelet-derived growth factor (PDGF), epidermal growth factor (EGF) and stromal-derived factor-1 (SDF-1) in the tumor tissue (48). PDGF for example, was shown to increase the attraction of human MSC in vitro and in vivo and this tropism is mediated via PDGF-beta receptors on human MSC (48). It has also been suggested that neovascularization might be critical for MSC to localize to and gain entry into gliomas following intravascular delivery (45). Indeed, the hypoxia-induced pro-angiogenic molecule vascular endothelial cell growth factor (VEGF) has been shown to induce the homing of MSC to tumor sites in murine glioma (49).
Following intra-arterial injection in glioma-bearing mice, MSC were detected throughout the tumors, but not in non-tumoral areas of the brain (44). In immunocompetent animals, MSC had the capacity of migrating to the glioma site after having been implanted into the contralateral hemisphere. When MSC were infected with adenovirus (MSC-Ad35), they successfully replicated the virus without the process negatively affecting their own viability. In vivo it was shown that treatment with MSC-Ad35 was able to significantly reduce tumor growth and prolong survival of glioma-bearing mice, where Ad-35 alone could not. In these studies the addition of MSC as virus carriers was crucial to the antiglioma activity of adenovirus, both in immunocompetent and immunodeficient animals. Free adenovirus particles were not able to reach the glioma site or induce therapeutic activity following intra-arterial or intracerebral injection. This implied that MSC likely played a role in improving the tumor homing and spread of the virus, additionally to the capacity to shield the virus from the host immune system.
Neural stem cells
Another cell type investigated in models of glioma as a potential virus carrier is the neural stem cell (NSC) (50). NSC are derived from fetal, neonatal or postnatal tissues and are multipotent, capable of differentiating into three major types of central nervous system cells: neurons, astrocytes, and oligodendrocytes.
The NSC tropism for gliomas is well-known and seems closely linked to hypoxia within the tumor (51). In NSC, hypoxia induces up-regulation of CXCR4, urokinase-type plasminogen activator receptor (uPAR), VEGF receptor 2 and c-Met receptors. Inhibiting the function of these receptors inhibits NSC migration, as does knockdown of hypoxia-inducible factor-1alpha (HIF1α) in the glioma cells, which decreases the expression of receptor ligands SDF-1, uPAR and VEGF.
Acting as virus-producing cellular factories, NSC could successfully replicate and release adenovirus progeny to glioma cells (50). The migratory capacity of NSC was not diminished following loading with adenovirus, rather the OV infection up-regulated chemoattractant receptors and significantly enhanced migration of NSC, both in vitro and in vivo (52). At the same time, delivery via NSC carriers decreased the amount of non-specific distributing within the brain. Of note, when free virus particles were injected intratumorally, migration into the contralateral hemisphere was seen in all animals treated. This contradicts other reports demonstrating the incapability of injected OV to migrate from the injection site (44;53). In an earlier report, neural precursor cells were also found capable of delivering an HSV-1 mutant to established intracerebral gliomas in nude mice, allowing for extensive spread throughout the tumor and into the surrounding parenchyma (54). However, in this instance the HSV-1 mutant rRp450 was demonstrated to kill the carrier cells, requiring replication block in the precursors by G1 growth arrest induction. This indicates the importance of optimal OV - carrier matching.
Based on these reports and others, all demonstrating the potential of NSC to carry a therapeutic payload to glioma sites for antitumoral activity, the FDA has recently approved the HB1.F3-CD immortalized NSC line for clinical trial (50).
Limits to clinical application
In a preclinical study directly comparing the two cell lines for their ability to deliver oncolytic adenovirus to intracranial human glioma xenotransplants, both carrier systems supported intracellular adenoviral replication and increased virus distribution to the tumor site (55). However, the amount of virus released from NSC was a log higher than that released by MSC in this system. The molecular mechanism behind this enhanced ability of NSC to support adenoviral replication has not been investigated. It might be related to the immortalized nature of the NSC line used, which provides for a greater rate of doubling and improved cellular stability. Moreover, only adenovirus-loaded NSC significantly prolonged the survival of tumor-bearing mice in this orthotopic human glioma model, despite the comparable migratory capacity of NSC and MSC (55).
NSC might therefore be more suitable as therapeutic delivery vehicles for brain tumors. Due to their developmental origin they have the inherent capacity to migrate into the host brain without disrupting the normal functions of the target organ. However, the therapeutic potential of OV-loaded NSC so far has not been investigated in immunocompetent glioma models. Furthermore, the clinical application of NSC is limited due issues associated with their potential immunogenicity and the risk of secondary malignancies associated with the use of an immortalized cell line. As a potential benefit to the use of MSC, it has been demonstrated in both immunocompetent and immunodeficient tumor-bearing mice that MSC viability declines over time (46). This may be an advantage of MSC as a delivery vehicle because they may deliver a therapeutic agent and then gradually disappear, independent of the immune status of the host. The feasibility of using autologous MSC as cell carriers for OVT is very attractive, in contrast to NSC. However, it is harder to predict MSC behavior as the cell type is much more heterogeneous. Simultaneously, for some patients suffering from brain tumors it might be unlikely to be able to obtain sufficient healthy bone marrow to allow for effective therapy.
Adipose-derived stem cells
Given the logistic and/or ethical issues associated with the expansion, propagation, and manipulation of functional adult NSC and MSC, adipose-derived stem cells (ADSC) have also been investigated as a potential OV carrier for the treatment of GBM (53). Adipose tissue is ubiquitous and uniquely expandable and most patients possess excess fat that can be harvested.
ADSC were first described in 2001 and have since become one of the most popular adult stem cell populations in the field of regenerative medicine, where they are investigated for their potential to mediate inflammation and vascularization during tissue regeneration (56). Given their mesodermal, ectodermal, and endodermal differentiation potential, they are also explored with regards to reprogramming into induced pluripotent cells.
ADSC were found to be permissive for myxoma virus (vMyxgfp) replication, supporting multiple rounds of replication leading to productive infection (53). The viral infection had no negative impact on ADSC viability. In vitro, co-culture of human GBM cells and myxoma virus-infected ADSC showed cross infection and concomitant cell death exclusively in GBM cells. In vivo, intracranial injection with myxoma virus-infected ADSC led to successful delivery of the OV to the tumor, resulting in a significant survival increase in a human GBM xenotransplant model in nude mice.
Issues regarding immunosuppression
While the data described above support the hypothesis that innate immunosuppression will benefit OVT, it is important to keep alternative and/or additional interpretations in mind as well.
CPM has been used as an anticancer therapy since 1959 and it is well understood that pleiotropic immunomodulatory effects can be obtained by CPM treatment, depending on the dosing schedule applied. As high doses of CPM result in potent cytotoxicity and lymphoablation, the routine use of CPM in glioma patients is limited to the administration of continuous low (metronomic) doses. Metronomic CPM has been shown to have immunostimulatory effects that include expansion of antigen-specific tumor-reactive T cells, transient depletion of regulatory T cells (Treg), restoration of dendritic cell (DC) homeostasis and induction of several cytokines (57-59). By stimulating the effector arm of the immune response, while simultaneously inhibiting immunosuppression, low dosages of CPM are able to result in antitumor immune responses and enhance immune-based tumor rejection regimens (57;58). In addition to these immunostimulatory properties, low doses of CPM have further been shown to possess antiangiogenic effects as well (60). Irrelevant to the dosage, CPM is known to exert intrinsic pro-immunogenic activities on tumor cells and has been shown to induce hallmarks of immunogenic cell death (ICD) on a variety of tumor types (60). Treating lymphoma cells with CPM for example, induced surface exposure of Calreticulin (ectoCRT) and extracellular release of high-mobility group protein 1 (HMGB1) by the dying tumor cells (61).
Although most studies evaluating CPM as an immunosuppressive agent to combine with OVT use higher doses of the drug, it becomes interesting to speculate that at least some of the reported activities of CPM in combination with OV in glioma and other cancer models might be attributable to these immunostimulatory properties. It has indeed been shown that using low dosages of CPM, which have no effect on the antiviral immunity or on the neutralizing antibody titers, has a beneficial effect on adenovirus therapy due to the depletion of Treg, thus taking advantage of the immunostimulatory actions of CPM (62). Furthermore, a recent paper employing the U251 glioma xenotransplant model has demonstrated that both metronomic doses of CPM, as well as MTD doses, can activate antitumor immunity, associated with brain tumor recruitment of NK cells, macrophages, and DC. In this report, low doses induced potent immune activation while the MTD rather induced a transient, weak innate immune response (63).
The described benefits of using immunomodulating agents (i.e. CPM, Rapamycin, VPA) were largely attributed to the decreased brain influx and/or function of macrophages and NK cells, which these drugs can induce. The role of these cell populations in OVT for (brain) tumors remains controversial, however. IFN type I-mediated activation of NK cells renders them highly capable of killing virus-infected cells and of priming the adaptive antiviral immune response. Therefore, the role of NK cells in OVT has often been viewed as detrimental to successful therapy. However, NK cells have also been described to possess strong antitumoral activities and the NK cell-mediated lysis of glioma cells could in fact be further improved by oncolytic Myxoma virus infection (64). The mechanism behind this was related to Myxoma virus-induced down-regulation of class I Major Histocompatibility Complex surface expression on infected target cells, which favors NK cell activation. Several other OV strains (i.e. influenza A virus, parvovirus) have also been demonstrated to promote NK cell activity against tumor targets by up-regulating or down-regulating NK cell activating or inhibitory ligands, respectively, on the surface of infected tumor cells (64).
Both reovirus and measles virus have been shown to stimulate innate antitumor immunity (65;66). Following OV infection, innate immune cells exerted cytotoxicity towards several tumor cell lines. NK cells became activated and functional following OV treatment, as demonstrated by increased expression of the activation marker CD69 and active degranulation. In a subcutaneous model of non-small cell lung carcinoma, intratumoral coxsackievirus B3 administration recruited significantly greater numbers of NK cells, granulocytes, macrophages, and DC into the tumor bed (67). Degranulation experiments demonstrated significantly more NK cell and granulocyte activation in treated animals as compared to controls. Depletion of either cell fraction significantly abrogated the therapeutic effect of the coxsackie virotherapy, illustrating their substantial contribution to virus-induced antitumor immunity. In this setting, even without successful viral replication, the OV infection of gliomas would improve NK cell-mediated clearance of infected tumor cells. Furthermore, these immunostimulatory properties of OV on innate immunity may subsequently prime effective generation of adaptive immunity.
Successful oncolytic virotherapy in immune competent animals
The hypothesis that immune interactions are detrimental to OVT is mostly challenged by recent studies that have demonstrated successful OVT in immunocompetent glioma models without immunosuppressive co-treatments.
Parvovirus H-1 (H-1PV)-based virotherapy was tested for rat and human gliomas, in parallel, using immunocompetent and immunodeficient rat models, respectively (68). Both the rat RG-2 as the human U87 glioma cell lines employed in this study were demonstrated to be highly susceptible to cytotoxic killing by H-1PV in vitro. In both models, large orthotopic tumors were treated with a single stereotactic injection and/or multiple intravenous H-1PV injections. Systemic application was attempted based on initial findings indicating the H-1PV is able to cross the blood-brain barrier in healthy animals. To increase the viral load to the tumor area following intravenous therapy, the virus dose was 50-fold higher than for stereotactic treatment. In a follow up study, efficient glioma regression, resulting in significant prolongation of glioma-bearing rat survival was demonstrated after a single intranasal instillation of H-1PV as well (69). Successful delivery to the brain via intranasal route represents an interesting approach with regards to clinical translation as it is practical, painless, noninvasive and it bypasses the blood-brain barrier. Several types of therapeutic agents, including growth factors, proteins, and anticancer agents have been delivered successfully to the brain of animals and humans through this route (70-74). H-1PV therapy resulted in significantly improved survival in both glioma models, however; only in immunocompetent glioma-bearing rats could single stereotactic treatment or multiple systemic application of the virus also induce full remission of advanced and even symptomatic intracranial gliomas (68). Successful viral replication was measured specifically in the tumors and indicated the contribution of secondary infection by progeny virus to the efficiency of oncolysis. No therapy-related damage to the surrounding healthy brain tissue, or to other organs, was found and therapy induced only minor inflammation. Of note, both intravenous and intracranial injection resulted in the appearance of H-1PV-neutralizing serum antibodies five to seven days after the start of therapy. No obvious infiltration of tumors with immune cells following OVT was noticed and the number of CD3+ cells in the tumor area did not increased. However, tumor re-induction in cured animals, one year after successful H-1PV therapy, failed to result in the development of RG-2 gliomas, even when cells were injected in 30-fold higher numbers into the contralateral hemisphere. This strongly indicates the induction of immunological antitumor memory by H-1PV therapy. Based on these data it was hypothesized that H-1PV, and possibly other OV, might serve, at least in part, as an adjuvant to promote anticancer vaccination through the release of tumor-associated antigens and additional immunostimulatory activities.
Likewise, repeated intratumoral administration of a recombinant, murine granulocyte macrophage colony-stimulating factor (GM-CSF) expressing vaccinia virus (JX-594m) was able to significantly improve median survival in both the rat RG-2 and the mouse GL261 immunocompetent, orthotopic glioma models (75). In vitro, JX-594m killed all five malignant glioma cell lines tested, as well as ex vivo grown brain tumor-initiating cells (BTIC) from patient samples. The ability of OV to infect and kill BTIC indicates their potential to overcome treatment resistance to chemo- and radiotherapy, given that resistance is based on the persistence of these cell types. The therapeutic responses measured in vivo were similar in both glioma models, despite the RG-2 cell line being much more sensitive to JX-594m-induced cytotoxicity in vitro, as compared to the GL261 cells. This finding indicates a discrepancy between the direct cytopathic effects of an oncolytic agent, as measured in vitro, and the therapeutic potential - a discrepancy that has recently been discussed by many groups (76;77). Further safety/toxicity studies in none-tumor-bearing rodents treated with supratherapeutic doses of JX-594m demonstrated predictable GM-CSF-dependent inflammation and necrosis. Experiments in tumor-bearing animals demonstrated that the addition of the cytokine is however not necessary for the survival benefit of vaccinia virus-based virotherapy in this model and the intracranial administration of JX-594 (expressing human GM-CSF) was well tolerated.
The replication-competent HSV mutant R-LM113, fully retargeted to the human epidermal growth factor receptor 2 (HER2), significantly prolonged survival of glioma-bearing BALB/c mice carrying HER2-expressing high-grade gliomas, both when administered at time of tumor inoculation, as well as at time of established tumor (78).
The studies indicate that the host immune defenses do not curtail the oncolytic antitumor effect of replication competent OV. Several reasonable explanations might be offered for this finding. The antiviral activities of recruited NK cells and macrophages might be inefficient in the context of the glioma microenvironment, thereby allowing viral replication. Indeed, macrophage/microglia function might be suppressed when infiltrating into the GBM microenvironment. Glioma cancer stem cells have been shown to produce soluble colony-stimulating factor (sCSF), transforming growth factor (TGF)-β1 and macrophage inhibitory cytokine-1 (MIC-1), which inhibited the phagocytic function of the macrophages/microglia present (79). Alternatively, viral replication might simply proceed faster than immune-mediated clearance of virus particles.
In the last two to three years a third option; the idea that virotherapy is - at least partly - immunotherapy has been uncovered. In the initial OVT concept extensive propagation of the OV inside infected tumor cells was deemed crucial to successful therapy. The productive generation of progeny OV could spread the infection to nearby tumor cells that escaped the initial round of oncolysis. Therefore, highly cytotoxic agents and a maximum potential for intratumoral spread were essential. In recent times however, an extensive body of evidence is emerging that supports the notion that, rather than utilizing OV solely for tumor eradication, OVT can generate strong innate and adaptive antitumor immunity (16). The immune effects might increase the benefit of OVT and form an integrated part of the therapy (80). Evidence from several cancer models has indicated that OV might even trigger this antitumor immunity relative independent of viral replication or killing. In this new paradigm, increasing therapeutic efficacy does not have to equal enhancing viral replication and spread, but rather enhancing antitumor immunity. In this case, immunosuppression might actually reduce tumor therapy.
Oncolytic virus-induced antitumor immunity
One way in which an OV may fulfill this new 'oncolytic paradigm' is by inducing ICD (81;82). Most anticancer therapies induce non-immunogenic cell death, which induces tolerance towards tumor cells (83). However, in recent times it has emerged that certain therapies can induce a cancer cell death sub mechanism that is actively immunostimulatory because it is associated with the emission of potent danger signals, thereby leading to effective activation of antitumor immunity (82). The concept of ICD has been expertly reviewed by Kroemer and Krysko in recent years (81;82).
Several viral strains have been demonstrated to induce immunogenic signals in cancer cells (Table 3). Oncolytic H-1PV infection was shown to induce the release of heat-shock protein (HSP)72 during virus-induced apoptosis in susceptible human melanoma cells (84). The HSP72 release was even higher and of a longer duration than following conventional heat-shock treatments. Until this point, as a standalone therapy, three naturally occurring OV (i.e. measles virus, coxsackievirus B3, NDV) have been shown to induce the molecular signatures of ICD in vitro and to cause stimulation of immune cells (66;67;85).
Immunogenic cell death features induced by oncolytic viruses

Besides inducing the release of type I and type III IFN, inflammatory cytokines IL-6 and IL-8 and chemokine RANTES, measles virus provoked passive release of HMGB1 in human melanoma cells after infection (66). HMGB1 is known to act upon DC through toll-like receptor 4 (86) and indeed the conditioned media from measles-infected melanoma cells up-regulated CD80/CD86 expression on DC, phenotypically activating the cells for potential support of priming of adaptive antitumor immunity. In an in vitro system, measles virus-mediated melanoma cell death was capable of stimulating a melanoma-specific adaptive immune response. CD8+ cytotoxic T lymphocytes (CTL) co-cultured with measles virus-loaded DC infected melanoma cells degranulated specifically on recognition of melanomal targets. The same population was also IFN-γ+, indicating a Th1 cytokine response. In a functional killing assay, CTL primed by DC loaded with virus-infected tumor cells had more activity against uninfected melanoma targets than those primed by DC loaded with uninfected tumor cells. These data collectively indicated that virus-infected melanoma cells are more effective than uninfected cells as an antigen source for loading of DC for priming of a specific anti-melanoma immune response.
Likewise, coxsackievirus B3 infection in human non-small cell lung cancer led to abundant ectoCRT expression, ATP secretion, and HMGB1 release (67). Moreover, intratumoral administration markedly recruited NK cells, granulocytes, macrophages, and mature DC into tumor tissues. Recruited NK cells were shown to contribute to the antitumor effect of OVT, as discussed above. Tumor-infiltrating DC expressed significantly higher levels of co-stimulatory molecules CD80 and CD86, as well as maturation marker CCR7.
In glioma research, recent work by our group has demonstrated the induction of ICD following NDV therapy in an orthotopic, syngeneic murine GBM model (85). Without treatment, immunocompetent animals survive significantly longer in this model than their immunodeficient counterparts, demonstrating that the GL261 tumor is immunogenic and capable of eliciting a limited endogenous antitumor immune response, which prolongs survival of glioma-bearing mice but is insufficient to induce cure (87). NDV treatment cured 50% of immunocompetent animals of established glioma (85). On the contrary, in immunodeficient animals no long-term survival was induced, although NDV treatment significantly prolonged overall survival. In vitro, NDV-treated GL261 glioma cells underwent necroptotic cell death accompanied by ectoCRT and passive release of HMGB1. NDV treatment also induced up-regulation of glioma-associated antigen expression on the surface of infected cells. Elevated infiltration of IFN-γ+ CD4+ and CD8+ T cell populations along with reduced accumulation of immunosuppressive myeloid derived suppressor cells in the brain of NDV-treated glioma-bearing mice indicated significant modification of the immunological tumor microenvironment after therapy. Ex vivo T cell restimulation experiments demonstrated the presence of an activated T cell population specifically recognizing GL261 tumors, which was not present in untreated tumor-bearing animals. In vivo depletion of CD8+ T cells abolished the therapeutic effect almost completely, indicating these cells as the principle mediators of antitumor activity in this model. We could further show the induction of a long-term, tumor-specific immunological memory response following NDV therapy in this model. The induction of tumor-rejecting anticancer immunity in an immunocompetent syngeneic animal model is an absolute prerequisite for characterizing the existence of bone fide ICD (82). In our model long-term surviving mice, cured of their initial gliomas through NDV therapy, resisted secondary glioma induction, but not secondary growth of a different tumor type (85). It was therefore clear that in this model, though GL261 cells were sensitive to NDV-induced cytotoxicity in vitro, the therapeutic effect of the therapy indeed relied mainly on the induction of ICD in the tumor cells, which primed adaptive antitumor immunity.
The induction of tumor-rejecting anticancer immunity by OVT has also been described for an oncolytic parvovirus (Minute Virus of Mice) in a subcutaneous glioma model using GL261 cells ex vivo pretreated with virus (88). Whereas direct infection of DC and microglia with parvovirus - leading to successful viral entry, but not replication - had no significant impact on the activation of naïve DC or microglia, parvovirus-infected GL261 murine glioma cells did activate both populations, as demonstrated by up-regulation of CD80 and CD86 activation marker expression and increased release of tumor necrosis factor (TNF)-α and IL-6 (88). DC activation was limited following exposure to intact or lysed (through repeated freeze/thaw cycles) uninfected GL261 cells, indicating that parvovirus enhanced the capacity of glioma cells for DC maturation though a mechanism that is not limited to mere lysis of the cells. To investigate whether parvovirus also acted as an adjuvant under in vivo conditions and could endow infected GL261 glioma cells with an enhanced capacity for priming a tumor-specific adaptive immune response, GL261 cells were infected in vitro with parvovirus and injected subcutaneously into either immunocompetent or B and T cell deficient Rag2-/- mice. GL261 cells ex vivo loaded with parvovirus did not have the ability to form tumors in immunocompetent mice. However, in immunodeficient animals, 80 percent of animals developed tumors. This finding indicates the importance of an additional immune component to allow full suppression of tumor outgrowth. Parvovirus infection of GL261 cells could prolong the repression of tumor growth in
Rag2-/- mice in a MOI-dependent manner, though. Tumor rechallenge experiments, in which long-term survivors of parvovirus-infected tumor implantation received a second injection with uninfected GL261 cells, demonstrated the induction of long lasting T cell memory in these animals, as all remained tumor free.
Finally, the efficacy of oncolytic adenovirus therapy was also demonstrated to be highly dependent on the function of the host immune system in orthotopic glioma (89), though ICD was not investigated in this model. Delta24-RGD treatment enhanced the infiltration of CD4+ and CD8+ T cells and of macrophages. They further demonstrated the presence of protective immunological memory in treated animals and the therapeutic effect of OVT was completely lost upon co-treatment with the immunosuppressive agent dexamethasone.
In these models, it thus seems that OV and the immune system act synergistically to eliminate the tumor cells under conditions in which each component alone is inefficient. This novel data is in line with recent reports from other cancer fields, also demonstrating T cell activation and enhanced proliferation and effector function, activation of DC, and stimulation of innate antitumor activity following OVT (90-92). The immune responses against tumor targets, triggered by inflammatory responses to OV, have been demonstrated to be a vital component of successful treatment, capable of overcoming immunosuppressive tumor microenvironments and clearing metastatic disease (77).
Conclusion
OVT represents a rapidly evolving and highly exciting new field within cancer research. To date, the field has mainly concentrated on developing viruses that replicate robustly and extensively in tumors, but with only moderate effect. In retrospect, expecting extensive viral replication in an immunocompetent individual might be unlikely, despite the immunosuppressive mechanisms at play at the tumor site. The immune system was therefore expected to be a strong inhibitory factor in OVT for glioma, as it may limit viral replication and spread within the tumor. However, new evidence is mounting that OV may enhance both innate and adaptive antitumor immune responses and that the net effect may be a benefit to therapy. This new concepts has introduced a real shift within the OVT domain within the last two to three years.
As discussed, OV-induced tumor cell death may of an immunogenic nature and create an appropriate inflammatory tumor microenvironment to allow for effective priming of an adaptive antitumor immune response. Indeed, in several preclinical models the bulk of the therapeutic effect of OVT was demonstrated to be immune-mediated, rather than induced by direct viral oncolysis. In our own work viral presence within the glioma microenvironment induced influx and activation of T cells, as well as a decreased presence of immunosuppressive immune cells. Whereas direct cytotoxicity translated into only a minor improvement in overall survival in immunodeficient mice, ND virotherapy in immunocompetent glioma-bearing animals resulted in relevant improvements in median survival as well as a percentage of long-term surviving animals (85).
The role of the immune system in OVT for glioma patients is not straightforward and the range of immune interactions involved may be beneficial or detrimental in nature. In balancing the therapeutic and damaging effects some groups have suggested the possibility of early, transient immunosuppression to enhance viral replication, followed by a restoration of immune activity to harness the immunotherapeutic potential of OVT. However, the early, innate immune reactivity induced by OVT has also demonstrated to partake in the therapeutic benefit of the therapeutic approach, and could be important in the priming of further adaptive antitumor reactivity (92;93).
Furthermore, it should be taken into consideration that employing immunomodulating agents in the concentrations necessary to induce immunosuppression can be highly toxic and induce severe side effects. Using agents such as Rapamycin and VPA, which directly block the function of type I IFN, introduces an additional risk of systemic virus toxicity.
Factors such as the baseline immune status and prior antiviral immunity of the patient are likely to play a role as well. The use of oncolytic agents for which the prevalence of preexisting immunity in the population is low, therefore might be advantageous. Non-human viruses (e.g. NDV) might therefore be good candidates. These naturally oncolytic agents have several other benefits as compared to genetically engineered strains. They often show antitumor activity against a broad spectrum of tumor cells, and possible treatment-related toxicities are relatively easily predicted and addressed. Contrary to genetically engineered strains, these viruses are not human pathogens, and safety databases concerning their use in humans are available (94).
Studies are urgently needed to investigate optimal schedules for combining OVT with immunosuppressive therapeutics such as corticosteroids, Temozolomide, and radiation therapy. These modalities form the present standard of care for GBM patients and were often co-administered with oncolytic agents in clinical trial. Indeed, a recent preclinical study has indicated the loss of therapeutic efficacy upon co-treatment with dexamethasone (89). The effect of co-administration with other immmunomodulators during the course of OVT should therefore no longer be overlooked in clinical applications. Additionally, future clinical trials should aim to include more relevant immunological end-points, besides the conventional measures of safety and efficacy. The amount of immunological information available from clinical trials performed thus far is limited. Data obtained from treated GBM patients can however crucially direct preclinical investigations and aid in optimizing the therapeutic approach. In line with this, preclinical work should focus on further unraveling the immune mechanisms at play and on studying combination therapies, as highly aggressive malignancies like GBM are likely to require a multimodal approach including several rationally combined therapies. Incorporating these mounting preclinical and clinical findings into novel strategies will be the surest way to maximizing the potential impact of viral agents and the benefit to patients.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Dock G. The influence of complicating diseases upon leukemia. Am J Med Sci. 1904(127):563-92
2. Van Epps HL. Influenza: exposing the true killer. J Exp Med. 2006Apr17;203(4):803
3. Markert JM, Liechty PG, Wang W, Gaston S, Braz E, Karrasch M, Nabors LB, Markiewicz M, Lakeman AD, Palmer CA, Parker JN, Whitley RJ. et al. Phase Ib trial of mutant herpes simplex virus G207 inoculated pre-and post-tumor resection for recurrent GBM. Mol Ther. 2009Jan;17(1):199-207
4. Amgen press release. Amgen announces top-line results of phase 3 talimogene laherparepvec trial in melanoma. 2013.
5. Heo J, Reid T, Ruo L, Breitbach CJ, Rose S, Bloomston M, Cho M, Lim HY, Chung HC, Kim CW, Burke J, Lencioni R. et al. Randomized dose-finding clinical trial of oncolytic immunotherapeutic vaccinia JX-594 in liver cancer. Nat Med. 2013Mar;19(3):329-36
6. Burke JM, Lamm DL, Meng MV, Nemunaitis JJ, Stephenson JJ, Arseneau JC, Aimi J, Lerner S, Yeung AW, Kazarian T, Maslyar DJ, McKiernan JM. A first in human phase 1 study of CG0070, a GM-CSF expressing oncolytic adenovirus, for the treatment of nonmuscle invasive bladder cancer. J Urol. 2012Dec;188(6):2391-7
7. Liu TC, Galanis E, Kirn D. Clinical trial results with oncolytic virotherapy: a century of promise, a decade of progress. Nat Clin Pract Oncol. 2007Feb;4(2):101-17
8. Horstmann E, McCabe MS, Grochow L, Yamamoto S, Rubinstein L, Budd T, Shoemaker D, Emanuel EJ, Grady C. Risks and benefits of phase 1 oncology trials, 1991 through 2002. N Engl J Med. 2005Mar3;352(9):895-904
9. Liang M. Clinical development of oncolytic viruses in China. Curr Pharm Biotechnol. 2012Jul;13(9):1852-7
10. Galluzzi L, Lugli E. Cancer immunotherapy turns viral. Oncoimmunology. 2013Apr1;2(4):e24802
11. Louis DN, Ohgaki H, Wiestler OD, Cavenee WK, Burger PC, Jouvet A, Scheithauer BW, Kleihues P. The 2007 WHO classification of tumours of the central nervous system. Acta Neuropathol. 2007Aug;114(2):97-109
12. Stupp R, Hegi ME, Mason WP, van den Bent MJ, Taphoorn MJ, Janzer RC, Ludwin SK, Allgeier A, Fisher B, Belanger K, Hau P, Brandes AA. et al. Effects of radiotherapy with concomitant and adjuvant temozolomide versus radiotherapy alone on survival in glioblastoma in a randomised phase III study: 5-year analysis of the EORTC-NCIC trial. Lancet Oncol. 2009May;10(5):459-66
13. Stummer W, Pichlmeier U, Meinel T, Wiestler OD, Zanella F, Reulen HJ. Fluorescence-guided surgery with 5-aminolevulinic acid for resection of malignant glioma: a randomised controlled multicentre phase III trial. Lancet Oncol. 2006May;7(5):392-401
14. Greenspoon JN, Sharieff W, Hirte H, Overholt A, Devillers R, Gunnarsson T, Whitton A. Fractionated stereotactic radiosurgery with concurrent temozolomide chemotherapy for locally recurrent glioblastoma multiforme: a prospective cohort study. Onco Targets Ther. 2014;7:485-90
15. Wollmann G, Ozduman K, van den Pol AN. Oncolytic virus therapy for glioblastoma multiforme: concepts and candidates. Cancer J. 2012Jan;18(1):69-81
16. Russell SJ, Peng KW, Bell JC. Oncolytic virotherapy. Nat Biotechnol. 2012Jul;30(7):658-70
17. Weller M, Cloughesy T, Perry JR, Wick W. Standards of care for treatment of recurrent glioblastoma--are we there yet? Neuro Oncol. 2013Jan;15(1):4-27
18. Chambers R, Gillespie GY, Soroceanu L, Andreansky S, Chatterjee S, Chou J, Roizman B, Whitley RJ. Comparison of genetically engineered herpes simplex viruses for the treatment of brain tumors in a scid mouse model of human malignant glioma. Proc Natl Acad Sci U S A. 1995Feb28;92(5):1411-5
19. Andreansky S, Soroceanu L, Flotte ER, Chou J, Markert JM, Gillespie GY, Roizman B, Whitley RJ. Evaluation of genetically engineered herpes simplex viruses as oncolytic agents for human malignant brain tumors. Cancer Res. 1997Apr15;57(8):1502-9
20. Samoto K, Ehtesham M, Perng GC, Hashizume K, Wechsler SL, Nesburn AB, Black KL, Yu JS. A herpes simplex virus type 1 mutant with gamma 34.5 and LAT deletions effectively oncolyses human U87 glioblastomas in nude mice. Neurosurgery. 2002Mar;50(3):599-605
21. Fueyo J, Gomez-Manzano C, Alemany R, Lee PS, McDonnell TJ, Mitlianga P, Shi YX, Levin VA, Yung WK, Kyritsis AP. A mutant oncolytic adenovirus targeting the Rb pathway produces anti-glioma effect in vivo. Oncogene. 2000Jan6;19(1):2-12
22. Wilcox ME, Yang W, Senger D, Rewcastle NB, Morris DG, Brasher PM, Shi ZQ, Johnston RN, Nishikawa S, Lee PW, Forsyth PA. Reovirus as an oncolytic agent against experimental human malignant gliomas. J Natl Cancer Inst. 2001Jun20;93(12):903-12
23. Alkassar M, Gartner B, Roemer K, Graesser F, Rommelaere J, Kaestner L, Haeckel I, Graf N. The combined effects of oncolytic reovirus plus Newcastle disease virus and reovirus plus parvovirus on U87 and U373 cells in vitro and in vivo. J Neurooncol. 2011Sep;104(3):715-27
24. Rampling R, Cruickshank G, Papanastassiou V, Nicoll J, Hadley D, Brennan D, Petty R, MacLean A, Harland J, McKie E, Mabbs R, Brown M. Toxicity evaluation of replication-competent herpes simplex virus (ICP 34.5 null mutant 1716) in patients with recurrent malignant glioma. Gene Ther. 2000May;7(10):859-66
25. Papanastassiou V, Rampling R, Fraser M, Petty R, Hadley D, Nicoll J, Harland J, Mabbs R, Brown M. The potential for efficacy of the modified (ICP 34.5(-)) herpes simplex virus HSV1716 following intratumoural injection into human malignant glioma: a proof of principle study. Gene Ther. 2002Mar;9(6):398-406
26. Harrow S, Papanastassiou V, Harland J, Mabbs R, Petty R, Fraser M, Hadley D, Patterson J, Brown SM, Rampling R. HSV1716 injection into the brain adjacent to tumour following surgical resection of high-grade glioma: safety data and long-term survival. Gene Ther. 2004Nov;11(22):1648-58
27. Markert JM, Medlock MD, Rabkin SD, Gillespie GY, Todo T, Hunter WD, Palmer CA, Feigenbaum F, Tornatore C, Tufaro F, Martuza RL. Conditionally replicating herpes simplex virus mutant, G207 for the treatment of malignant glioma: results of a phase I trial. Gene Ther. 2000May;7(10):867-74
28. Chiocca EA, Abbed KM, Tatter S, Louis DN, Hochberg FH, Barker F, Kracher J, Grossman SA, Fisher JD, Carson K, Rosenblum M, Mikkelsen T. et al. A phase I open-label, dose-escalation, multi-institutional trial of injection with an E1B-Attenuated adenovirus, ONYX-015, into the peritumoral region of recurrent malignant gliomas, in the adjuvant setting. Mol Ther. 2004Nov;10(5):958-66
29. Forsyth P, Roldan G, George D, Wallace C, Palmer CA, Morris D, Cairncross G, Matthews MV, Markert J, Gillespie Y, Coffey M, Thompson B. et al. A phase I trial of intratumoral administration of reovirus in patients with histologically confirmed recurrent malignant gliomas. Mol Ther. 2008Mar;16(3):627-32
30. Freeman AI, Zakay-Rones Z, Gomori JM, Linetsky E, Rasooly L, Greenbaum E, Rozenman-Yair S, Panet A, Libson E, Irving CS, Galun E, Siegal T. Phase I/II trial of intravenous NDV-HUJ oncolytic virus in recurrent glioblastoma multiforme. Mol Ther. 2006Jan;13(1):221-8
31. Csatary LK, Gosztonyi G, Szeberenyi J, Fabian Z, Liszka V, Bodey B, Csatary CM. MTH-68/H oncolytic viral treatment in human high-grade gliomas. J Neurooncol. 2004Mar;67(1-2):83-93
32. Csatary LK, Bakacs T. Use of Newcastle disease virus vaccine (MTH-68/H) in a patient with high-grade glioblastoma. JAMA. 1999May5;281(17):1588-9
33. Wagner S, Csatary CM, Gosztonyi G, Koch HC, Hartmann C, Peters O, Hernaiz-Driever P, Theallier-Janko A, Zintl F, Langler A, Wolff JE, Csatary LK. Combined treatment of pediatric high-grade glioma with the oncolytic viral strain MTH-68/H and oral valproic acid. APMIS. 2006Oct;114(10):731-43
34. Lun X, Alain T, Zemp FJ, Zhou H, Rahman MM, Hamilton MG, McFadden G, Bell J, Senger DL, Forsyth PA. Myxoma virus virotherapy for glioma in immunocompetent animal models: optimizing administration routes and synergy with rapamycin. Cancer Res. 2010Jan15;70(2):598-608
35. Fulci G, Dmitrieva N, Gianni D, Fontana EJ, Pan X, Lu Y, Kaufman CS, Kaur B, Lawler SE, Lee RJ, Marsh CB, Brat DJ. et al. Depletion of peripheral macrophages and brain microglia increases brain tumor titers of oncolytic viruses. Cancer Res. 2007Oct1;67(19):9398-406
36. Wakimoto H, Fulci G, Tyminski E, Chiocca EA. Altered expression of antiviral cytokine mRNAs associated with cyclophosphamide's enhancement of viral oncolysis. Gene Ther. 2004Jan;11(2):214-23
37. Fulci G, Breymann L, Gianni D, Kurozomi K, Rhee SS, Yu J, Kaur B, Louis DN, Weissleder R, Caligiuri MA, Chiocca EA. Cyclophosphamide enhances glioma virotherapy by inhibiting innate immune responses. Proc Natl Acad Sci U S A. 2006Aug22;103(34):12873-8
38. Lamfers ML, Fulci G, Gianni D, Tang Y, Kurozumi K, Kaur B, Moeniralm S, Saeki Y, Carette JE, Weissleder R, Vandertop WP, van Beusechem VW. et al. Cyclophosphamide increases transgene expression mediated by an oncolytic adenovirus in glioma-bearing mice monitored by bioluminescence imaging. Mol Ther. 2006Dec;14(6):779-88
39. Cloughesy TF, Yoshimoto K, Nghiemphu P, Brown K, Dang J, Zhu S, Hsueh T, Chen Y, Wang W, Youngkin D, Liau L, Martin N. et al. Antitumor activity of rapamycin in a Phase I trial for patients with recurrent PTEN-deficient glioblastoma. PLoS Med. 2008Jan22;5(1):e8
40. Galanis E, Buckner JC, Maurer MJ, Kreisberg JI, Ballman K, Boni J, Peralba JM, Jenkins RB, Dakhil SR, Morton RF, Jaeckle KA, Scheithauer BW. et al. Phase II trial of temsirolimus (CCI-779) in recurrent glioblastoma multiforme: a North Central Cancer Treatment Group Study. J Clin Oncol. 2005Aug10;23(23):5294-304
41. Alvarez-Breckenridge CA, Yu J, Price R, Wei M, Wang Y, Nowicki MO, Ha YP, Bergin S, Hwang C, Fernandez SA, Kaur B, Caligiuri MA. et al. The histone deacetylase inhibitor valproic acid lessens NK cell action against oncolytic virus-infected glioblastoma cells by inhibition of STAT5/T-BET signaling and generation of gamma interferon. J Virol. 2012Apr;86(8):4566-77
42. Shi Y, Hu G, Su J, Li W, Chen Q, Shou P, Xu C, Chen X, Huang Y, Zhu Z, Huang X, Han X. et al. Mesenchymal stem cells: a new strategy for immunosuppression and tissue repair. Cell Res. 2010May;20(5):510-8
43. Wang L, Shi J, van Ginkel FW, Lan L, Niemeyer G, Martin DR, Snyder EY, Cox NR. Neural stem/progenitor cells modulate immune responses by suppressing T lymphocytes with nitric oxide and prostaglandin E2. Exp Neurol. 2009Mar;216(1):177-83
44. Hai C, Jin YM, Jin WB, Han ZZ, Cui MN, Piao XZ, Shen XH, Zhang SN, Sun HH. Application of mesenchymal stem cells as a vehicle to deliver replication-competent adenovirus for treating malignant glioma. Chin J Cancer. 2012May;31(5):233-40
45. Yong RL, Shinojima N, Fueyo J, Gumin J, Vecil GG, Marini FC, Bogler O, Andreeff M, Lang FF. Human bone marrow-derived mesenchymal stem cells for intravascular delivery of oncolytic adenovirus Delta24-RGD to human gliomas. Cancer Res. 2009Dec1;69(23):8932-40
46. Doucette T, Rao G, Yang Y, Gumin J, Shinojima N, Bekele BN, Qiao W, Zhang W, Lang FF. Mesenchymal stem cells display tumor-specific tropism in an RCAS/Ntv-a glioma model. Neoplasia. 2011Aug;13(8):716-25
47. Dvorak HF. Tumors: wounds that do not heal. Similarities between tumor stroma generation and wound healing. N Engl J Med. 1986Dec25;315(26):1650-9
48. Hata N, Shinojima N, Gumin J, Yong R, Marini F, Andreeff M, Lang FF. Platelet-derived growth factor BB mediates the tropism of human mesenchymal stem cells for malignant gliomas. Neurosurgery. 2010Jan;66(1):144-56
49. Schichor C, Birnbaum T, Etminan N, Schnell O, Grau S, Miebach S, Aboody K, Padovan C, Straube A, Tonn JC, Goldbrunner R. Vascular endothelial growth factor A contributes to glioma-induced migration of human marrow stromal cells (hMSC). Exp Neurol. 2006Jun;199(2):301-10
50. Thaci B, Ahmed AU, Ulasov IV, Tobias AL, Han Y, Aboody KS, Lesniak MS. Pharmacokinetic study of neural stem cell-based cell carrier for oncolytic virotherapy: targeted delivery of the therapeutic payload in an orthotopic brain tumor model. Cancer Gene Ther. 2012Jun;19(6):431-42
51. Zhao D, Najbauer J, Garcia E, Metz MZ, Gutova M, Glackin CA, Kim SU, Aboody KS. Neural stem cell tropism to glioma: critical role of tumor hypoxia. Mol Cancer Res. 2008Dec;6(12):1819-29
52. Ahmed AU, Thaci B, Alexiades NG, Han Y, Qian S, Liu F, Balyasnikova IV, Ulasov IY, Aboody KS, Lesniak MS. Neural stem cell-based cell carriers enhance therapeutic efficacy of an oncolytic adenovirus in an orthotopic mouse model of human glioblastoma. Mol Ther. 2011Sep;19(9):1714-26
53. Josiah DT, Zhu D, Dreher F, Olson J, McFadden G, Caldas H. Adipose-derived stem cells as therapeutic delivery vehicles of an oncolytic virus for glioblastoma. Mol Ther. 2010Feb;18(2):377-85
54. Herrlinger U, Woiciechowski C, Sena-Esteves M, Aboody KS, Jacobs AH, Rainov NG, Snyder EY, Breakefield XO. Neural precursor cells for delivery of replication-conditional HSV-1 vectors to intracerebral gliomas. Mol Ther. 2000Apr;1(4):347-57
55. Ahmed AU, Tyler MA, Thaci B, Alexiades NG, Han Y, Ulasov IV, Lesniak MS. A comparative study of neural and mesenchymal stem cell-based carriers for oncolytic adenovirus in a model of malignant glioma. Mol Pharm. 2011Oct3;8(5):1559-72
56. Zuk P. Adipose-derived stem cells in tissue regeneration: A review. ISRN Stem Cells 2012 Oct 23. 2013
57. Wainwright DA, Dey M, Chang A, Lesniak MS. Targeting Tregs in Malignant Brain Cancer: Overcoming IDO. Front Immunol. 2013;4:116
58. Bracci L, Moschella F, Sestili P. et al. Cyclophosphamide enhances the antitumor efficacy of adoptively transferred immune cells through the induction of cytokine expression, B-cell and T-cell homeostatic proliferation, and specific tumor infiltration. Clin Cancer Res. 2007Jan15;13(2 Pt 1):644-53
59. Loeffler M, Kruger JA, Reisfeld RA. Immunostimulatory effects of low-dose cyclophosphamide are controlled by inducible nitric oxide synthase. Cancer Res. 2005Jun15;65(12):5027-30
60. Sistigu A, Viaud S, Chaput N, Bracci L, Proietti E, Zitvogel L. Immunomodulatory effects of cyclophosphamide and implementations for vaccine design. Semin Immunopathol. 2011Jul;33(4):369-83
61. Schiavoni G, Sistigu A, Valentini M, Mattei F, Sestili P, Spadaro F, Sanchez M, Lorenzi S, D'Urso MT, Belardelli F, Gabriele L, Proietti E. et al. Cyclophosphamide synergizes with type I interferons through systemic dendritic cell reactivation and induction of immunogenic tumor apoptosis. Cancer Res. 2011Feb1;71(3):768-78
62. Cerullo V, Diaconu I, Kangasniemi L, Rajecki M, Escutenaire S, Koski A, Romano V, Rouvinen N, Tuuminen T, Laasonen L, Partanen K, Kauppinen S. et al. Immunological effects of low-dose cyclophosphamide in cancer patients treated with oncolytic adenovirus. Mol Ther. 2011Sep;19(9):1737-46
63. Doloff JC, Waxman DJ. VEGF receptor inhibitors block the ability of metronomically dosed cyclophosphamide to activate innate immunity-induced tumor regression. Cancer Res. 2012Mar1;72(5):1103-15
64. Ogbomo H, Zemp FJ, Lun X, Zhang J, Stack D, Rahman MM, McFadden G, Mody CH, Forsyth PA. Myxoma virus infection promotes NK lysis of malignant gliomas in vitro and in vivo. PLoS One. 2013;8(6):e66825
65. Prestwich RJ, Errington F, Steele LP, Ilett EJ, Morgan RS, Harrington KJ, Pandha HS, Selby PJ, Vile RG, Melcher AA. Reciprocal human dendritic cell-natural killer cell interactions induce antitumor activity following tumor cell infection by oncolytic reovirus. J Immunol. 2009Oct1;183(7):4312-21
66. Donnelly OG, Errington-Mais F, Steele L, Hadac E, Jennings V, Scott K, Peach H, Phillips RM, Bond J, Pandha H, Harrington K, Vile R. et al. Measles virus causes immunogenic cell death in human melanoma. Gene Ther. 2013Jan;20(1):7-15
67. Miyamoto S, Inoue H, Nakamura T, Yamada M, Sakamoto C, Urata Y, Okazaki T, Marumoto T, Takahashi A, Takayama K, Nakanishi Y, Shimizu H. et al. Coxsackievirus B3 is an oncolytic virus with immunostimulatory properties that is active against lung adenocarcinoma. Cancer Res. 2012May15;72(10):2609-21
68. Geletneky K, Kiprianova I, Ayache A, Koch R, Herrero YC, Deleu L, Sommer C, Thomas N, Rommelaere J, Schlehofer JR. Regression of advanced rat and human gliomas by local or systemic treatment with oncolytic parvovirus H-1 in rat models. Neuro Oncol. 2010Aug;12(8):804-14
69. Kiprianova I, Thomas N, Ayache A, Fischer M, Leuchs B, Klein M, Rommelaere J, Schlehofer JR. Regression of glioma in rat models by intranasal application of parvovirus h-1. Clin Cancer Res. 2011Aug15;17(16):5333-42
70. Wang F, Jiang X, Lu W. Profiles of methotrexate in blood and CSF following intranasal and intravenous administration to rats. Int J Pharm. 2003Sep16;263(1-2):1-7
71. Thorne RG, Frey WH. Delivery of neurotrophic factors to the central nervous system: pharmacokinetic considerations. Clin Pharmacokinet. 2001;40(12):907-46
72. Thorne RG, Pronk GJ, Padmanabhan V, Frey WH. Delivery of insulin-like growth factor-I to the rat brain and spinal cord along olfactory and trigeminal pathways following intranasal administration. Neuroscience. 2004;127(2):481-96
73. Wang D, Gao Y, Yun L. Study on brain targeting of raltitrexed following intranasal administration in rats. Cancer Chemother Pharmacol. 2006Jan;57(1):97-104
74. Sakane T, Yamashita S, Yata N, Sezaki H. Transnasal delivery of 5-fluorouracil to the brain in the rat. J Drug Target. 1999;7(3):233-40
75. Lun X, Chan J, Zhou H, Sun B, Kelly JJ, Stechishin OO, Bell JC, Parato K, Hu K, Vaillant D, Wang J, Liu TC. et al. Efficacy and safety/toxicity study of recombinant vaccinia virus JX-594 in two immunocompetent animal models of glioma. Mol Ther. 2010Nov;18(11):1927-36
76. Workenhe ST, Simmons G, Pol JG, Lichty BD, Halford WP, Mossman KL. Immunogenic HSV-mediated oncolysis shapes the antitumor immune response and contributes to therapeutic efficacy. Mol Ther. 2014Jan;22(1):123-31
77. Prestwich RJ, Ilett EJ, Errington F, Diaz RM, Steele LP, Kottke T, Thompson J, Galivo F, Harrington KJ, Pandha HS, Selby PJ, Vile RG. et al. Immune-mediated antitumor activity of reovirus is required for therapy and is independent of direct viral oncolysis and replication. Clin Cancer Res. 2009Jul1;15(13):4374-81
78. Reisoli E, Gambini E, Appolloni I, Gatta V, Barilari M, Menotti L, Malatesta P. Efficacy of HER2 retargeted herpes simplex virus as therapy for high-grade glioma in immunocompetent mice. Cancer Gene Ther. 2012Nov;19(11):788-95
79. Wu A, Wei J, Kong LY, Wang Y, Priebe W, Qiao W, Sawaya R, Heimberger AB. Glioma cancer stem cells induce immunosuppressive macrophages/microglia. Neuro Oncol. 2010Nov;12(11):1113-25
80. Bartlett DL, Liu Z, Sathaiah M, Ravindranathan R, Guo Z, He Y, Guo ZS. Oncolytic viruses as therapeutic cancer vaccines. Mol Cancer. 2013;12(1):103
81. Krysko DV, Garg AD, Kaczmarek A, Krysko O, Agostinis P, Vandenabeele P. Immunogenic cell death and DAMPs in cancer therapy. Nat Rev Cancer. 2012Dec;12(12):860-75
82. Kroemer G, Galluzzi L, Kepp O, Zitvogel L. Immunogenic cell death in cancer therapy. Annu Rev Immunol. 2013;31:51-72
83. Garg AD, Martin S, Golab J, Agostinis P. Danger signalling during cancer cell death: origins, plasticity and regulation. Cell Death Differ. 2014Jan;21(1):26-38
84. Moehler M, Zeidler M, Schede J, Rommelaere J, Galle PR, Cornelis JJ, Heike M. Oncolytic parvovirus H1 induces release of heat-shock protein HSP72 in susceptible human tumor cells but may not affect primary immune cells. Cancer Gene Ther. 2003Jun;10(6):477-80
85. Koks CA, Garg AD, Ehrhardt M, Riva M, Vandenberk L, Boon L, Vleeschouwer SD, Agostinis P, Graf N, Van Gool SW. Newcastle disease virotherapy induces long-term survival and tumor-specific immune memory in orthotopic glioma through the induction of immunogenic cell death. Int J Cancer. 2014 Sep 10
86. Apetoh L, Ghiringhelli F, Tesniere A, Obeid M, Ortiz C, Criollo A, Mignot G, Maiuri MC, Ullrich E, Saulnier P, Yang H, Amigorena S. et al. Toll-like receptor 4-dependent contribution of the immune system to anticancer chemotherapy and radiotherapy. Nat Med. 2007Sep;13(9):1050-9
87. Verschuere T, Toelen J, Maes W, Poirier F, Boon L, Tousseyn T, Mathivet T, Gerhardt H, Mathieu V, Kiss R, Lefranc F, Van Gool SW. et al. Glioma-derived galectin-1 regulates innate and adaptive antitumor immunity. Int J Cancer. 2013 Aug 9
88. Grekova SP, Raykov Z, Zawatzky R, Rommelaere J, Koch U. Activation of a glioma-specific immune response by oncolytic parvovirus Minute Virus of Mice infection. Cancer Gene Ther. 2012Jul;19(7):468-75
89. Kleijn A, Kloezeman J, Treffers-Westerlaken E, Fulci G, Leenstra S, Dirven C, Debets R, Lamfers M. The in vivo therapeutic efficacy of the oncolytic adenovirus Delta24-RGD is mediated by tumor-specific immunity. PLoS One. 2014;9(5):e97495
90. Lam HY, Yeap SK, Rasoli M, Omar AR, Yusoff K, Suraini AA, Alitheen NB. Safety and clinical usage of newcastle disease virus in cancer therapy. J Biomed Biotechnol. 2011;2011:718710
91. Prestwich RJ, Errington F, Ilett EJ, Morgan RS, Scott KJ, Kottke T, Thompson J, Morrison EE, Harrington KJ, Pandha HS, Selby PJ, Vile RG. et al. Tumor infection by oncolytic reovirus primes adaptive antitumor immunity. Clin Cancer Res. 2008Nov15;14(22):7358-66
92. Errington F, Steele L, Prestwich R, Harrington KJ, Pandha HS, Vidal L, de BJ, Selby P, Coffey M, Vile R, Melcher A. Reovirus activates human dendritic cells to promote innate antitumor immunity. J Immunol. 2008May1;180(9):6018-26
93. Hall K, Scott KJ, Rose A, Desborough M, Harrington K, Pandha H, Parrish C, Vile R, Coffey M, Bowen D, Errington-Mais F, Melcher AA. Reovirus-mediated cytotoxicity and enhancement of innate immune responses against acute myeloid leukemia. Biores Open Access. 2012Jan;1(1):3-15
94. Lam HY, Yeap SK, Rasoli M, Omar AR, Yusoff K, Suraini AA, Alitheen NB. Safety and clinical usage of newcastle disease virus in cancer therapy. J Biomed Biotechnol. 2011;2011:718710
Author contact
![]() Corresponding author: Carolien Koks, Laboratory of Pediatric Immunology, Herestraat 49, 3000 Leuven, Belgium. Phone: +32 (0)16 34 61 65; Fax: +32 (0)16 34 60 35; E-mail: Carolien.kokskuleuven.be
Corresponding author: Carolien Koks, Laboratory of Pediatric Immunology, Herestraat 49, 3000 Leuven, Belgium. Phone: +32 (0)16 34 61 65; Fax: +32 (0)16 34 60 35; E-mail: Carolien.kokskuleuven.be