Impact Factor ISSN: 1837-9664

 Global reach, higher impact
Global reach, higher impactJ Cancer 2015; 6(4):382-386. doi:10.7150/jca.11187 This issue Cite
Short Research Communication
The Effect of Tumor Microenvironment on Autophagy and Sensitivity to Targeted Therapy in EGFR-Mutated Lung Adenocarcinoma
Yuan-Yuan Li, Sze-Kwan Lam, Chun-Yan Zheng, James Chung-Man Ho ![]()
Division of Respiratory Medicine, Department of Medicine, The University of Hong Kong, Queen Mary Hospital, Hong Kong SAR
Received 2014-11-27; Accepted 2015-1-1; Published 2015-2-25
Abstract
Lung cancer is the top cancer killer worldwide. Tyrosine kinase inhibitors (TKIs), for example erlotinib, are commonly used to target epidermal growth factor receptor (EGFR)-mutated lung adenocarcinoma (ADC). Autophagy is a cellular response to stress, serving as a protective mechanism during anticancer therapy. The tumor microenvironment (TME) is composed of non-tumor cells that include fibroblasts. Our study aimed to investigate the effect of TME on autophagy and TKI sensitivity. Following cell sorting after direct co-culturing, autophagy and cytokine production were observed in both HCC827 and MRC-5 cells. The synergistic combination of erlotinib and chloroquine (autophagy inhibitor) was observed under TME. Tumor growth was significantly suppressed with combined erlotinib/chloroquine compared with erlotinib in HCC827 xenografts.
Keywords: Tumor microenvironment, autophagy, tyrosine kinase inhibitors, non-small cell lung carcinoma
Introduction
Recently, we have shown that autophagy can be induced by erlotinib in EGFR-mutated lung adenocarcinoma (ADC) cells, while inhibition of autophagy with chloroquine or siRNA targeting key autophagic proteins enhances the apoptotic effect of erlotinib [1]. Nonetheless this cancer cell line model may only be a simplified representation of real-life lung cancer without the influence of the complex tumor microenvironment (TME). During tumor development, there are close interactions between tumor cells and the host leading to formation of a unique TME that is composed of different stromal cells, immune cells, vasculature and extracellular matrix. Through crosstalk between different cells, TME participates in almost every stage of tumor development and results in increased tumor progression and metastatic potential [2, 3]. Few reports have described the association between autophagy and TME. In an experimental model of breast cancer cells co-cultured with fibroblasts, autophagy was induced in fibroblasts and contributed to cancer progression [4, 5]. The effect of TME on drug sensitivity in cancers has been demonstrated in bone marrow stromal cells via mediators of CXCR4/SDF-1 axis, soluble factors and adhesion proteins [6]. Recently, hepatocyte growth factor produced by stromal fibroblasts has also been shown to render in vitro and in vivo resistance to EGFR TKI treatment in lung cancer [7].
In this study, we hypothesized that autophagy and cytokine production would occur in the presence of TME in an in vitro cell line model. The effects of an autophagic TME on TKI sensitivity in EGFR-mutated lung ADC were investigated. Finally, the in vivo combination effect of chloroquine (autophagy inhibitor) and erlotinib in EGFR-mutated lung ADC was studied in a tumor xenograft model.
Materials and Methods
Cell lines and cultures
Human lung ADC HCC827 cells (EGFR exon 19 deletion) and human lung fibroblast MRC-5 cells (American Type Culture Collection, Manassas, VA, USA) were obtained. For direct co-culture experiments, MRC-5 and HCC827 cells were co-plated in 6-well plates. MRC-5 cells were plated first, followed within 2 hours by HCC827 cells in 6-well plates. After co-culturing 7 days, cells were harvested for protein and RNA extraction [1, 8].
Apoptotic assay
Cells were incubated at 4oC in the dark for 1 hour with PE-conjugated annexin-V (BD biosciences, New Jersey, USA), PI and APC-conjugated EpCAM. Cells were then subjected to flow cytometry (Beckman Coulter, Inc., USA).
Real-time polymerase chain reaction (RT-PCR)
Total RNA was isolated using Trizol (Invitrogen, Life technologies). Reverse transcription was performed using a Qiagen RT kit. Primers for IL-6, IL-8 and GAPDH were synthesized (Tech Dragon Limited, HK): IL-6 forward and reverse primers (ATGCAATAACCACCCCTGAC and GAGGTGCCCATGCTACATTT); IL-8 forward and reverse primers (TAGCAAAATTGAGGCCAAGG and AGGCACAGTGGAACAAGGAC); GAPDH forward and reverse primers (AGCCACATCGCTCAGACACC and GTACTCAGCGCCAGCATCG). Amplification of targets was carried out using Power SYBR green PCR mix with Step One Plus Real-time PCR system (Applied Biosystems). Quantification of gene expression was calculated by the delta Ct method [9]. Data represent the mean ± SD of three independent experiments.
Cell sorting experiments
EpCAM is utilized as a surface marker to isolate lung cancer cells from cancer-associated fibroblasts [10]. After 7 days of co-culturing, cells were incubated with PE-conjugated anti-EpCAM antibody. PE-conjugated IgG Isotype was added to cells as a control for gating during cell sorting. EpCAM+ and EpCAM- cells were isolated by a BD fluorescence-activated cell sorting (FACS) Aria cell sorting system (BD Biosciences).
HCC827 tumor xenograft model
Tumor xenografts were established with HCC827 cells in nude mice (females, 4-weeks-old, 10-12 grams, BALB/cAnN-nu, Charles River Laboratories, Wilmington, USA) [11]. Cells were mixed with matrigel matrix (BD Biosciences, USA) before being injected subcutaneously into the right flank of each mouse. Tumor diameter was measured with calipers and the volume calculated (volume = width x length x height/2) [12]. When the tumor volume reached about 100 mm3, the mice were divided into 4 treatment groups: control (phosphate buffered saline) (n = 9), erlotinib (25 mg/kg) (n = 8), chloroquine (50 mg/kg) (n = 9) and combined erlotinb/chloroquine (n = 9). All treatments were administered by daily intraperitoneal injection. The growth curve was plotted for each group during 24 days of treatment. The study protocol was approved by the institutional Animal Ethics Committee (approval reference number: CULATR 2724-12) and standard humane endpoints for animal research were applied.
Statistical analysis
Data from triplicate experiments are presented in mean ± standard deviation (SD). Comparison between groups was performed using Student's two-tailed t-test by Prism (GraphPad Software, La Jolla, Southern California, USA). A p-value <0.05 was considered statistically significant.
Results
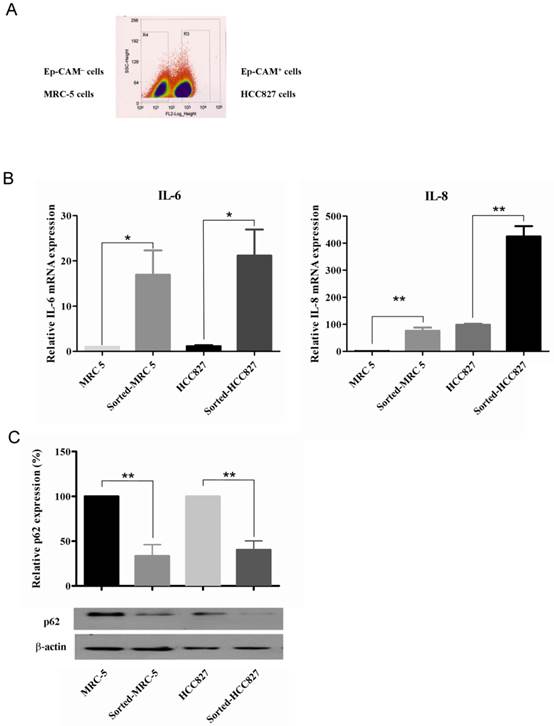
Co-cultured cells were sorted into 2 populations (Figure 1A). After 7 days of co-culturing, IL-6 and IL-8 mRNA was elevated (Figure 1B) and p62 expression was decreased (Figure 1C) in both sorted MRC-5 and HCC827 cells compared with their corresponding homotypical counterparts.
Cytokine production and autophagy induction in both MRC-5 and HCC827 cells in the tumor microenvironment. (A) After staining for EpCAM, the co-cultured cells were divided into 2 populations. (B) The IL-6 and IL-8 mRNA expression in sorted cells was significantly increased compared with corresponding homotypical cells. (C) Autophagy was induced in sorted cells compared with their homotypical counterparts, as evidenced by p62 degradation. Western blot data were quantified by fold-change compared with homotypical MRC-5 cells or homotypical HCC827 cells. Comparisons are made between sorted cells with respective homotypical cells. Statistical significance (*: p <0.05, **: p <0.01) indicates comparison with control.
In both MRC-5 and HCC827 cells, treatment with recombinant human IL-6 and IL-8 (20 ng/ml) and conditioned medium obtained after 7 days of co-culture decreased p62 expression and increased ATG5/12 conjugation. Dexamethasone (10 μM) decreased IL-6 and IL-8, followed by partial reversal of p62 downregulation in co-cultured cells (data not shown).
PI-annexin-V and APC-EpCAM staining performed on homotypical HCC827 cells and co-cultured cells indicated that 22% of homotypical HCC827 cells and 34.8% of co-cultured HCC827 cells underwent apoptosis. Combination of erlotinib and chloroquine potentiated apoptotic cell death in both homotypical and co-cultured HCC827 cells (Figure 2A).
(A) Apoptosis in HCC827 or co-cultured HCC827 cells after erlotinib ± chloroquine treatment. In homotypical HCC827 cells, combination of erlotinib with chloroquine significantly increased apoptotic cell death compared with erlotinib alone. In co-culture conditions, the sensitivity to erlotinib was preserved and synergistic combination of erlotinib/chloroquine was evident. Statistical significance (*: p <0.05, **: p <0.01) indicates comparison with control. (B) Antitumor effect of erlotinib, chloroquine (CQ) or combined erlotinib/CQ in a HCC827 xenograft model in nude mice. Longitudinal tumor size with different treatment groups. The relative tumor volume was calculated as fold-change to the baseline tumor volume prior to treatment. Progressive tumor growth was noted in both control and CQ treatment group, while significant tumor growth suppression was evident in erlotinib treatment and combination group. Statistical significance (*: p <0.05, **: p <0.01) indicates comparison between erlotinib with control and combination with erlotinib.
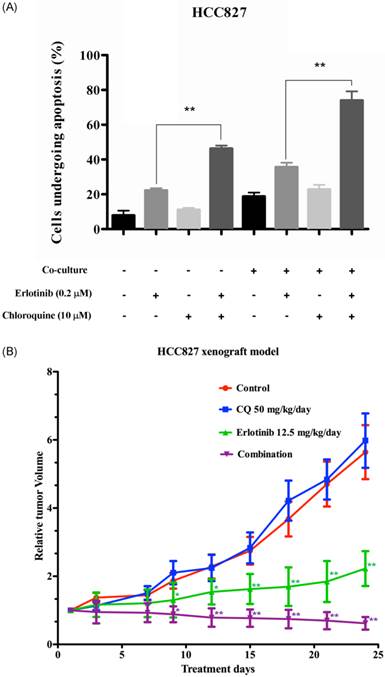
Progressive tumor growth was observed in control and treatment groups over 24 days of treatment. As expected, erlotinib alone significantly suppressed tumor growth throughout the treatment period (p < 0.05 on day 9 and 12; p < 0.001 from day 15 onwards). Combination of erlotinib and chloroquine resulted in even more significant tumor growth suppression than erlotinib alone (p < 0.01 on day 9 and 12; p < 0.001 from day 15 onwards) (Figure 2B). In addition, increased cleaved caspase-9 immunostaining was observed with erlotinib/chloroquine combination compared with either treatment alone or control (data not shown).
Discussion
In this study, we mimicked a TME by co-culturing HCC827 lung cancer cells with MRC-5 fibroblasts. Cytokine elaboration and autophagy occurred in both HCC827 and MRC-5 cells under co-culture conditions. The cytokines (IL-6 and IL-8) produced in TME served as one of the driving forces for autophagy. Nonetheless, sensitivity of HCC827 cells to erlotinib was not dampened, and combined chloroquine and erlotinib induced more apoptotic cell death compared with erlotinib alone in TME. Synergism between chloroquine and erlotinib was also demonstrated using a HCC827 xenograft model.
Co-culturing of tumor cells with stromal cells [4] allowing in vitro interactions between cancer cells and neighboring cells or soluble factors. Cancer-associated fibroblasts are often studied by co-culturing with initiated human epithelial cells, conferring faster growth kinetics and tumor progression in the epithelial component [13].
By directly co-culturing MRC-5 with HCC827 cells, an activated TME was generated. This finding differed to a prostatic cancer model wherein only stromal fibroblasts could drive tumor progression by co-culturing with prostatic epithelial cells [14], but was consistent with a previous breast cancer model [4]. Our findings suggest the presence of intermingled interactions between lung tumor cells and fibroblasts in TME.
Autophagy status in tumors is highly dependent on tumor type and stage [15]. Interestingly, autophagy occurred in both cancerous and stromal compartments of the TME, and might provide new insight into the possible relationship between autophagy and TME. Nonetheless previous studies have only demonstrated autophagy induction in cancer-associated fibroblasts via oxidative stress derived from cancer cells [5]. The possible role of elaborated cytokines in TME on autophagy was also studied. Exogenous application of IL-6 or IL-8 increased ATG5/12 conjugation and decreased p62 in both HCC827 and MRC-5 cells, indicating cytokine-induced autophagy in TME. Pretreatment with dexamethasone, an anti-inflammatory agent shown to downregulate IL-6 and IL-8 expression in fibroblasts [16], partially reversed p62 degradation in the co-culture condition, supporting the contributory role of cytokines to heightened autophagy in TME.
Stromal fibroblasts have been implicated in the development of EGFR TKI resistance in lung cancer [7, 17, 18]. Nonetheless our findings suggest that TKI sensitivity of EGFR-mutated lung ADC is not reduced in the presence of TME. Increased cytokines in our TME model may contribute to other cancer-related characteristics, such as metastasis and aggressiveness [19], rather than TKI resistance. On the other hand, sensitivity to erlotinib and synergistic combination of chloroquine/erlotinib was consistently demonstrated in our HCC827 xenograft model.
There are two limitations of our study. First, we used only a single lung ADC cell line in our in vitro and in vivo model. Second, other components of TME (e.g. vasculature, immunological cells) have not been studied.
Conflict of interest
None of the authors have conflict of interest to declare.
References
1. Li YY, Lam SK, Mak JC. et al. Erlotinib-induced autophagy in epidermal growth factor receptor mutated non-small cell lung cancer. Lung Cancer. 2013;81:354-61
2. Whiteside TL. The tumor microenvironment and its role in promoting tumor growth. Oncogene. 2008;27:5904-12
3. Mbeunkui F, Johann DJ Jr. Cancer and the tumor microenvironment: a review of an essential relationship. Cancer Chemother Pharmacol. 2009;63:571-82
4. Martinez-Outschoorn UE, Whitaker-Menezes D, Lin Z. et al. Cytokine production and inflammation drive autophagy in the tumor microenvironment: role of stromal caveolin-1 as a key regulator. Cell Cycle. 2011;10:1784-93
5. Martinez-Outschoorn UE, Trimmer C, Lin Z. et al. Autophagy in cancer associated fibroblasts promotes tumor cell survival: Role of hypoxia, HIF1 induction and NFkappaB activation in the tumor stromal microenvironment. Cell Cycle. 2010;9:3515-33
6. Meads MB, Hazlehurst LA, Dalton WS. The bone marrow microenvironment as a tumor sanctuary and contributor to drug resistance. Clin Cancer Res. 2008;14:2519-26
7. Wang W, Li Q, Yamada T. et al. Crosstalk to stromal fibroblasts induces resistance of lung cancer to epidermal growth factor receptor tyrosine kinase inhibitors. Clin Cancer Res. 2009;15:6630-8
8. Baelde HJ, Cleton-Jansen AM, van Beerendonk H. et al. High quality RNA isolation from tumours with low cellularity and high extracellular matrix component for cDNA microarrays: application to chondrosarcoma. J Clin Pathol. 2001;54:778-82
9. Yuan JS, Reed A, Chen F. et al. Statistical analysis of real-time PCR data. BMC Bioinformatics. 2006;7:85
10. Mink SR, Vashistha S, Zhang W. et al. Cancer-associated fibroblasts derived from EGFR-TKI-resistant tumors reverse EGFR pathway inhibition by EGFR-TKIs. Mol Cancer Res. 2010;8:809-20
11. Lam SK, Mak JC, Zheng CY. et al. Downregulation of thymidylate synthase with arsenic trioxide in lung adenocarcinoma. Int J Oncol. 2014;44:2093-102
12. Tomayko MM, Reynolds CP. Determination of subcutaneous tumor size in athymic (nude) mice. Cancer Chemother Pharmacol. 1989;24:148-54
13. Kalluri R, Zeisberg M. Fibroblasts in cancer. Nat Rev Cancer. 2006;6:392-401
14. Olumi AF, Grossfeld GD, Hayward SW. et al. Carcinoma-associated fibroblasts direct tumor progression of initiated human prostatic epithelium. Cancer Res. 1999;59:5002-11
15. Chen HY, White E. Role of autophagy in cancer prevention. Cancer Prev Res (Phila). 2011;4:973-83
16. Tobler A, Meier R, Seitz M. et al. Glucocorticoids downregulate gene expression of GM-CSF, NAP-1/IL-8, and IL-6, but not of M-CSF in human fibroblasts. Blood. 1992;79:45-51
17. Kutikov A, Makhov P, Golovine K. et al. Interleukin-6: a potential biomarker of resistance to multitargeted receptor tyrosine kinase inhibitors in castration-resistant prostate cancer. Urology. 2011;78:968 e7-11
18. Yao Z, Fenoglio S, Gao DC. et al. TGF-beta IL-6 axis mediates selective and adaptive mechanisms of resistance to molecular targeted therapy in lung cancer. Proc Natl Acad Sci U S A. 2010;107:15535-40
19. Abulaiti A, Shintani Y, Funaki S. et al. Interaction between non-small-cell lung cancer cells and fibroblasts via enhancement of TGF-beta signaling by IL-6. Lung Cancer. 2013;82:204-13
Author contact
![]() Corresponding author: Dr. James C. Ho M.D. FRCP, Department of Medicine, The University of Hong Kong, Queen Mary Hospital, Pokfulam, Hong Kong SAR, China. Tel: (852) 2255 4999; Fax: (852) 2872 5828; Email: jhocmhk
Corresponding author: Dr. James C. Ho M.D. FRCP, Department of Medicine, The University of Hong Kong, Queen Mary Hospital, Pokfulam, Hong Kong SAR, China. Tel: (852) 2255 4999; Fax: (852) 2872 5828; Email: jhocmhk