Impact Factor ISSN: 1837-9664

 Global reach, higher impact
Global reach, higher impactJ Cancer 2015; 6(7):643-651. doi:10.7150/jca.11913 This issue Cite
Research Paper
The Blockage of KCa3.1 Channel Inhibited Proliferation, Migration and Promoted Apoptosis of Human Hepatocellular Carcinoma Cells
Yu Liu1 *, Liang Zhao2 *, Wenya Ma2*, Xuefeng Cao3, Hongyang Chen2, Dan Feng2, Jing Liang2, Kun Yin2, Xiaofeng Jiang1 ![]()
1. Department of Laboratory Medicine, Affiliated Fourth Hospital of Harbin Medical University, Harbin 150081, PR China
2. Department of Pharmacology, Harbin Medical University, Harbin 150081, Heilongjiang Province, China
3. Department of Anesthesiology, Affiliated Hospital of Chengde Medical University, Chengde, Hebei Province, China
* contributed equally to this work.
Received 2015-2-16; Accepted 2015-4-15; Published 2015-5-26
Abstract
The intermediate conductance calcium-activated potassium channel KCa3.1 plays an important role in regulating cell proliferation and migration. However, the role of KCa3.1 channel in human hepatocellular carcinoma remained unknown. This study was therefore performed to investigate the effects of KCa3.1 potassium channel blocker on the proliferation, apoptosis and migration of human hepatocellular cancer cells HepG2. KCa3.1 mRNA and protein were detected in HepG2. Furthermore, KCa3.1 potassium channel blocker TRAM-34 was capable to inhibit the proliferation and induce the apoptosis of HepG2 cells, which can be partially attenuated by 1-EBIO, an activator of KCa3.1 channel. Moreover, the migration of HepG2 was obviously inhibited by TRAM-34. Consistently, knockdown of KCa3.1 channel using its siRNA was also able to induce apoptosis and suppress proliferation and migration of HepG2. Meanwhile, intracellular ROS level was found augmented in HepG2 treated with TRAM-34. More importantly, p53 protein was found translocation from the cytoplasm into the nuclei of HepG2. Collectively, inhibition of KCa3.1 channel suppressed the growth and migration, and promoted the apoptosis of human hepatocellular carcinoma cells by regulating intracellular ROS level and promoting p53 activation. This data suggests TRAM-34 as a promising anti-tumor drug for liver cancer.
Keywords: TRAM-34, Migration, Apoptosis, Proliferation, p53, ROS
Introduction
Hepatocellular cell carcinoma is one of the commonest cancers all over the world, especially in Asia. It accords for more than 60,000 deaths, and about 750,000 cases are diagnosed newly every year [1-2]. Nowadays, liver transplantation has been recognized as the most effective therapy of hepatocellular carcinoma, but only small number of patients is available to liver transplantation [3]. Although hepatocellular carcinoma is now increasingly diagnosed at the earlier stage due to the routine screening, effective chemotherapeutic agents for hepatocellular carcinoma are still in the lack [4]. Therefore, development of new therapeutic drug for hepatocellular cancer is urgently needed.
The intermediate conductance calcium-activated potassium channel (KCa3.1) plays an important role in regulating intracellular calcium homeostasis [5-6]. The blocker of KCa3.1 potassium channel has been implicated in therapeutic potential in a variety of diseases including cancer and autoimmune disorders [5, 7-8]. For example, TRAM-34, a specific KCa3.1 blocker was shown to prevent acute angioplasty-induced coronary smooth muscle phenotypic modulation and limits stenosis [9]. Targeted disruption of KCa3.1 potassium channel also was shown to attenuate renal fibrosis [10]. Recently, it was reported that blockage of KCa3.1 potassium channel played an inhibitory role in human endometrial cancer cell growth [11]. Additionally, TRAM-34 injection reduced the degeneration of retinal ganglion cells after optic nerve transaction [11]. Notably, the long-term treatment with TRAM-34 at therapeutic concentrations was shown not to cause discernible toxicity [5]. But, whether KCa3.1 channel blocker exerts therapeutic effects on hepatocellular cell carcinoma and its molecular mechanisms have not been elucidated yet. This study was therefore conducted to clarify this notion.
In this study, we reported that KCa3.1 channel blocker TRAM-34 and KCa3.1 channel siRNA inhibited the growth and migration, and induced apoptosis of hepatocellular cell carcinoma, and the molecular mechanisms were associated with the enhancement of intracellular ROS and activation of p53 protein. These findings provide the evidence for KCa3.1 channel as a therapeutic target for liver cancer.
Materials and methods
Reagents
TRAM-34 and 1-EBIO were purchased from Sigma-Aldrich (St Louis, MO). Antibodies to KCa3.1 and p53 were purchased from Cell Signaling (Danvers, MA) and antibody against GADPH was purchased from Santa Cruz Biotechnology (Santa Cruz, CA). All other chemicals were purchased from Sigma-Aldrich (St Louis, MO). TRAM-34 and 1-EBIO were dissolved in DMSO (less than 0.1%, v/v, without detectable effects) in all experiments of this study.
Cell culture
Human hepatocellular carcinoma cell line HepG2 and human hepatic cell lines LO2 were bought from American Type Culture Collection (ATCC, Manassas, VA, USA), and maintained as subconfluent monolayers in DMEM supplemented with 10% fetal bovine serum (Hyclone, Logan, UT), and 1% penicillin-streptomycin (Invitrogen, Carlsbad, CA). HepG2 cells were cultured in an incubator at 37°C in a humidified atmosphere of 5% CO2 and 95% air. The culture medium was changed every two days.
Cell proliferation assay
CCK-8 cell proliferation kit was used to evaluate the proliferation of HepG2 cells. The HepG2 cells were seeded in 96-well plates and incubated overnight. Afterward, the cells were rendered quiescent by serum-free media, and were then exposed to the indicated concentrations of TRAM-34 or with 1-EBIO for 48 h. The 10 μl WST-8 from the CCK-8 Kit (Boster, Wuhan, China) was then added to each well and incubated at 37 °C for 1 h. The absorbance was measured at 450 nm.
AO/EB double staining
Morphological observation by Acridine orange/ethidium bromide (AO/EB) double staining was used to determine apoptosis of HepG2 cells. The protocol to carry out AO/EB staining was according to the manufacturer's instruction. In brief, HepG2 cells were harvested and incubated with 10 µl prepared AO/EB working solution (100 µg/ml AO and 100 µg/ml EB in PBS) for 5 min. The nuclear changes and apoptotic body formation of HepG2 were visualized using an inverted fluorescence microscope (Eclipse TE300, Nikon, Japan).
TUNEL assay
Apoptosis of HepG2 cells was determined using a Terminal deoxynucleotidyl transferase-mediated dUTP nick end labeling (TUNEL) detection kit (Roche, Penzberg, Germany) according to the manufacturer's protocol. After exposed to TRAM-34 alone or with 1-EBIO, HepG2 cells fixed with 4% paraformaldehyde in PBS for 30 min at room temperature, and permeabilized with 0.1% Triton X-100 in 0.1% sodium citrate for 2 min on ice. After washing in PBS, sections were incubated with the TUNEL reaction mixture for 1 h at 37 °C. After washing with PBS, the stained cells were visualized using a fluorescence microscopy (Eclipse TE300, Nikon, Japan).
Wound healing assay
HepG2 cells were cultured in a 6-well plate for 24 h. Wound healing assay was carried out by introducing a small linear scratch with a pipette tip. Then the cells in the each well were exposed to serum-free DMEM medium containing the indicated concentrations of TRAM-34 or plus 1-EBIO for 48 h. The intervals after the scratch in cultured cells were photographed under a phase-contrast microscope (x200) to monitor the cell migration ability.
Transwell assay
Transwell assay was performed by using a Transwell chamber with pore size of 8.0 μm (Millipore). The cells were resuspended in serum-free medium, and then planted into the upper chamber in 5% CO2 at 37℃. After treatment with different concentrations of TRAM-34 or TRAM-34+1-EBIO for 48 h, the cells in the upper were removed, and the attached cells in the lower section were stained with 0.1% crystal violet. The migration rate was quantified by counting the migration cells in six random fields under a light microscope.
Intracellular reactive oxygen species (ROS) measurement
The intracellular ROS levels were measured by using the DCF-DA probe method. The cells were treated with TRAM-34 or TRAM-34+1-EBIO, and then were washed three times with fresh medium. The 100 μM DCF-DA was added to each well and incubated for 30 min at 37°C. After washing three times in PBS, the stained cells were mounted, and the ROS accumulation was determined using a laser scanning Confocal microscope (Olympus FV-300).
Immunofluorescence microscopy
After treatment with TRAM-34 alone or plus 1-EBIO, HepG2 cells were then fixed in freshly prepared 4% paraformaldehyde and then rinsed three times in PBS. HepG2 cells on the coverslips were blocked with 1% bovine serum albumin (Sigma), and then incubated with anti-p53 antibody in a humidified chamber for 1 h and then washed three times in PBS. The primary antibody was labeled with Alexa-488 conjugated secondary antibody while p53 protein was labeled with goat-anti-mouse antibody. Coverslips with staining cells were mounted, and the images were taken with a laser scanning Confocal microscope (Olympus FV-300).
KCa3.1 siRNA transfection
Transfection of Kca3.1 siRNA was carried out using Lipofectamine 2000 reagent (Invitrogen, CA) according to the manufacturer's instructions. KCa3.1 siRNA oligonucleotide was synthesized by Sangon (Shanghai, China). The sequences of KCa3.1 siRNA are as followed: 5'-GCACCUUUCAGACACACUU-3'.
Western blot
The HepG2 cells and LO2 cells were homogenized on ice, and the cell lysates were prepared through centrifugation at 14000×g for 10 min at 4°C. The protein concentration was quantified using the bicinchoninic acid assay (BCA, Pierce). Equal amounts of proteins were separated using SDS-PAGE and then transferred onto a nitrocellulose membrane. The blots were blocked with 5% non-fat milk and further incubated with the monoclonal KCa3.1 antibody and Horseradish peroxidase-conjugated secondary antibodies. The Odyssey infrared fluorescent scanning system (LICOR) and Odyssey V1.2 software were used to quantify the expression of KCa3.1 based on the intensity of the bands.
Statistical analysis
Data used for statistical analysis are expressed as the mean±S.E.M. The significance of differences among groups was determined using ANOVA. All statistical analysis was done by SPSS 13.0 software. P<0.05 was considered as statistical significance.
Results
Detection of KCa3.1 expression in HepG2 cells
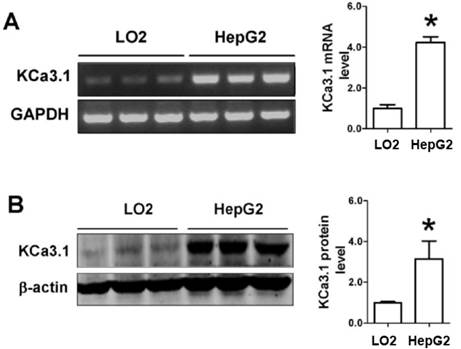
Firstly, we used RT-PCR and western blot to detect whether KCa3.1 channel was expressed in HepG2 cells. As shown in Figure 1A, HepG2 cells have a higher expression of KCa3.1 mRNA as compared to LO2 cells. Consistently, the expression of KCa3.1 protein was also detected higher in HepG2 cells than LO2 cells (Figure 1B). This data suggests that KCa3.1 channel was expressed in HepG2 cells.
The expression of KCa3.1 mRNA and protein in HepG2. (A), RT-PCR was used to detect KCa3.1 mRNA expression in two kinds of cells. Values are given normalized to band intensity of GAPDH used as internal control. (B), KCa3.1 protein expression was detected using Western blot in HepG2 cells. n=3 independent experiments. *P<0.05.
TRAM-34 inhibited the growth of HepG2 cells
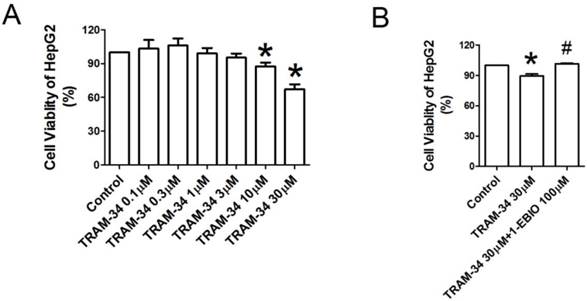
To further quantify the effects of KCa3.1 channel blocker on the proliferation of HepG2 cells, CCK-8 cell proliferation assay was performed. We observed the influences of TRAM-34 0.1, 0.3, 1, 3, 10 and 30 μM on the cellular viability of HepG2 cells. Figure 2A showed the viability of HepG2 cells was reduced after treatment with TRAM-34 10 and 30 μM for 48 h as compared to control cells (p<0.05), but TRAM-34 0.1, 0.3, 1 and 3 μM did not significantly affect the viability of HepG2 cells. The inhibition of the proliferation of HepG2 cells by TRAM-34 30 μM was suppressed in the presence of 1-EBIO 100 μM, an activator of KCa3.1 potassium channel (Figure 2B).
Anti-proliferative effect of TRAM-34 on HepG2. (A), Cell viability of HepG2 after exposed to the indicated concentrations of TRAM-34 for 48 h. TRAM-34 treatment obviously inhibited the proliferation of HepG2 cells. CCK-8 assay was used to assess cell viability. (B), Cell viability of HepG2 treated with TRAM-34 30 μM alone or TRAM-34 30 μM plus 1-EBIO for 48 h. In the presence of 1-EBIO 100 μM, the inhibition of proliferation of HepG2 by TRAM-34 30 μM was attenuated. * p < 0.05 vs control. # p < 0.05 vs TRAM-34.
TRAM-34 caused the apoptosis of HepG2 cells
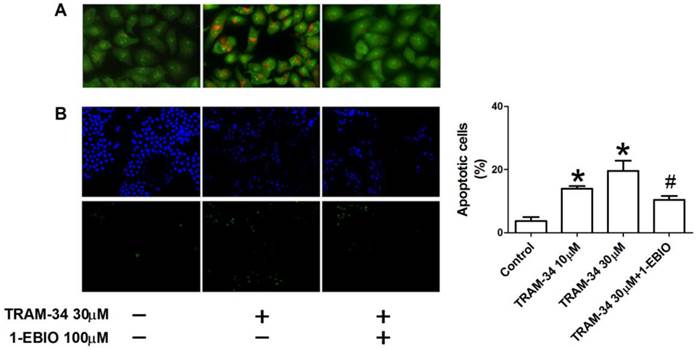
To further investigate if the apoptosis of HepG2 cells was induced by TRAM-34 treatment, apoptotic changes of HepG2 cells were observed using AO/EB staining and TUNEL apoptosis detection kits. Apoptotic appearance of HepG2 was found after TRAM-34 treatment under a fluorescence microscope by AO/EB staining (Figure 3A). Nevertheless, 1-EBIO was able to reduce the number of apoptotic cells after TRAM-34 (Figure 3A). TRAM-34-induced the apoptosis of HepG2 cells were further confirmed by TUNEL staining. The results showed that only few apoptotic HepG2 cells were observed in control group. However, TRAM-34 10 and 30 μM treatment resulted in an increase of TUNEL-positive HepG2 cells. HepG2 cells with TRAM-34 30 μM plus 1-EBIO 100 μM showed less TUNEL-positive fluorescence compared with TRAM-34 group (Figure 3B). These results suggest that TRAM-34 is able to cause the apoptosis of human hepatocellular cancer cells.
The effects of TRAM-34 on the apoptosis of HepG2. (A), Apoptotic morphological changes in the nuclear chromatin were observed in TRAM-34-treated HepG2 cells using AO/EB staining. (B), Quantification of cellular apoptosis by counting TUENL-positive cells in control, the indicated concentrations of TRAM-34, and TRAM-34+1-EBIO groups. TUNEL staining showed that apoptotic HepG2 cells were increased in the presence of TRAM-34 at 10 and 30 μM. * p < 0.05 vs control. # p < 0.05 vs TRAM-34.
TRAM-34 inhibited the migration of HepG2 cells
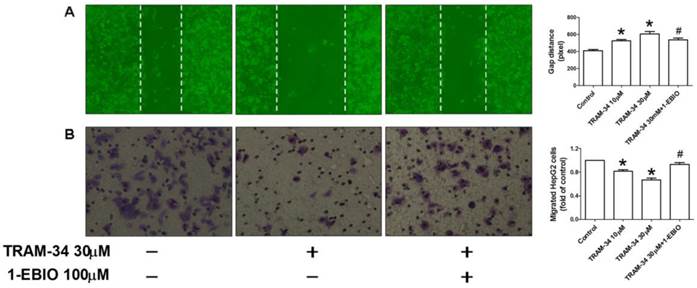
In addition to cellular proliferation, we also examined the effect of TRAM-34 on the migration activity of HepG2 cells. The wound healing assay in vitro was carried out to determine cell migration of HepG2 in the absence and presence of TRAM-34. Figure 4A showed the quantified wound closure in HepG2 cells after TRAM-34 10 and 30 μM treatment. Wound closure was slowed in TRAM-34 (30 μM)-treated HepG2 at 48 h after scratch, compared with control group. And, this change can be attenuated by 1-EBIO 100 μM treatment. Moreover, Transwell migration assay was also performed in HepG2 after TRAM-34 10 and 30 μM treatment. In agreement with wound healing assay, treatment with TRAM-34 30 μM produced a reduction of migrated cells (Figure 4B), which can be partially inhibited by 1-EBIO 100 μM. This data demonstrated that the blockage of KCa3.1 potassium channel induced the suppression of migration of HepG2 cells.
Inhibition of migration by TRAM-34 treatment. (A), Slowed migration was observed in HepG2 cells after TRAM-34 treatment under a phase-contrast microscope by wound healing assay. (B), Transwell chamber was used to determine the migration of HepG2 cells after cells were treated with difference concentrations of TRAM-34 alone or with 1-EBIO. It was demonstrated that TRAM-34 can cause a significant inhibition of migration of HepG2 cells. * p < 0.05 vs control. # p < 0.05 vs TRAM-34.
TRAM-34 increased intracellular ROS level in HepG2 cells
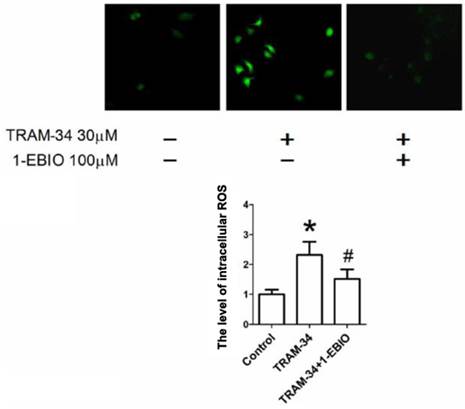
Previous studies have reported that the increase of ROS production led to cell apoptosis and inhibited cell migration in tumor cells [12-13]. To determine whether exposure to TRAM-34 enhances ROS generation in human hepatocellular carcinoma cells, the cells were exposed to TRAM-34 and the changes of DCF-DA fluorescence intensity were measured. As shown in Figure 5, treatment with TRAM-34 30 μM caused an increase of ROS generation, and this change can be partially attenuated by the treatment with 1-EBIO 100 μM.
Intracellular ROS generation by TRAM-34 treatment. The ROS level in HepG2 cells was increased after exposure to TRAM-34 30 μM. The level of intracellular ROS was measured in HepG2 cells using DCF-DA probe. * p < 0.05 vs control. # p < 0.05 vs TRAM-34.
TRAM-34 promoted the translocation of p53
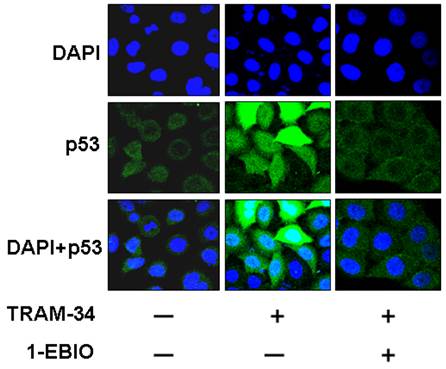
Lots of studies showed that p53 is an important tumor suppressor gene and its inactivation is involved in tumorigenesis and chemotherapy resistance [14]. The activation of p53 or translocation of p53 into nuclei plays an inhibitory role in tumor inhibition [12, 15]. So, we further investigated the effects of TRAM-34 on the localization of p53 protein in HepG2 cells. Figure 6 demonstrated that TRAM-34 30 μM significantly increased the expression of p53 protein in the nuclei of HepG2 cells. HepG2 cells treated by TRAM-34 30 μM plus 1-EBIO 100 μM showed the less accumulation of p53 protein in nuclei (p<0.05). These results suggest that p53 pathway is involved in TRAM-34-induced the inhibition of growth and migration of HepG2 cells.
Effect of TRAM-34 on the localization of p53 protein in the cytoplasm and nuclear of HepG2 cells. Immunofluorescence results demonstrated that p53 protein was mainly located in the cytoplasm in HepG2 cells without TRAM-34 30 μM treatment. Exposure to TRAM-34 30 μM led p53 protein to the nuclear of HepG2 cells.
KCa3.1 siRNA inhibited proliferation, migration and promoted apoptosis of HepG2 cells
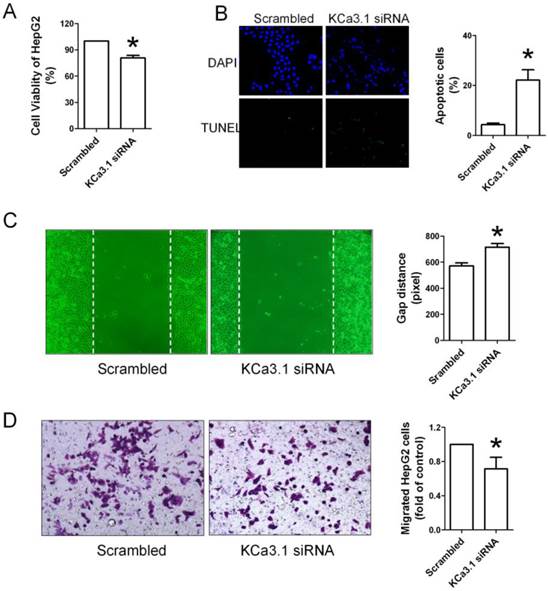
To further confirm that KCa3.1 channel is involved in regulating biological behaviors of HepG2 cells, we used KCa3.1 channel siRNA to inhibit the expression of KCa3.1 channel in HepG2 cells. As shown in Figure 7A, KCa3.1 siRNA inhibited the proliferation of HepG2 cells. TUNEL staining also showed that KCa3.1 siRNA was capable to induce the apoptosis of HepG2 cells (Figure 7B). Wound healing assay and Transwell assay also showed that the migration of HepG2 cells was obviously inhibited by KCa3.1 siRNA (Figure 7C and 7D).
Effects of KCa3.1 siRNA on the proliferation, apoptosis and migration of HepG2 cells. (A), KCa3.1 siRNA inhibited the proliferation of HepG2 cells assessed by CCK-8 assay. (B), TUNEL staining showed that KCa3.1 siRNA was capable to induce the apoptosis of HepG2 cells. (C and D), Wound healing assay and Transwell assay showed that the migration of HepG2 cells was obviously inhibited by KCa3.1 siRNA. * p < 0.05 vs scrambled.
Discussion
Hepatocellular cancer is one common type of cancer, and more than 750,000 patients are diagnosed every year all over the world [1]. In particularly, the incidence of hepatocellular carcinoma has gradually increased. But, the current chemotherapy often failed to control the recurrence of hepatocellular cancer, because hepatocellular cancer is insensitive to most chemotherapy drugs [16]. So it is necessary to develop some new drugs to treat hepatocellular cell carcinoma.
The intermediate-conductance calcium-activated potassium channel KCa3.1 is expressed in multiple cell types including excitable and nonexcitable cells, and is critically involved in regulating calcium homeostasis and membrane potential [6]. It has been showed that the blocker of KCa3.1 potassium channel has many biological activities such as anti-cancer, anti-inflammatory and anti-fibrosis effects, and thus was implicated in the treatment of cancer, renal fibrosis, asthma and hypertension [5, 8, 10-11]. Recently, it was shown that TRAM-34 induced the apoptosis of human endometrial cancer, pancreatic cancer and prostate cancer, with a promising application potential in clinics [11, 17-18]. For example, the mRNA and protein expression of KCa3.1 channel were upregulated in human prostate tissues, and the blockage of KCa3.1 potassium channel suppressed the proliferation of prostate cells via regulating calcium entry [18]. A tremendous increase of KCa3.1 mRNA we also found in primary pancreatic tumors, and the proliferation of BxPC-3 and MiaPaCa-2 cells was completely inhibited by KCa3.1 blocker clotrimazole and TRAM-34 [17]. However, the impact of KCa3.1 on the growth and apoptosis of human hepatocellular cancer cells remained unknown.
Our study showed the antiproliferative, proapoptotic and anti-migration effects of KCa3.1 channel blocker on human hepatocellular cancer cells. In consistence with previous studies [11, 18], we found that TRAM-34 significantly inhibited the growth of HepG2 cells at the concentration of 10 and 30 μM. Likewise, knockdown of KCa3.1 using its siRNA also inhibited the proliferation of HepG2. We then tested whether TRAM-34 treatment and KCa3.1 siRNA induced the apoptosis in HepG2 cells, which may explain the observed loss of viability of HepG2 cells. AO/EB staining displayed the apoptotic nuclear changes in HepG2 cells. Treatment with TRAM-34 or KCa3.1 siRNA both can increase the percentages of apoptotic cells assessed by TUNEL staining. These findings confirmed that the inhibition of KCa3.1 channel was able to cause apoptosis of human liver cancer cells. We further tested whether the blockage of KCa3.1 channel led to the inhibition of migration in HepG2 cells. The results confirmed that TRAM-34 was able to inhibit the migration of human hepatocellular cancer cells. We also used KCa3.1 siRNA to confirm the role of KCa3.1 channel in regulating the migration of HepG2 cells. Consistently, KCa3.1 siRNA was also able to inhibit the migration of HepG2 cells.
Then, several assays were carried out to understand the molecular mechanism underlying anti-tumor effect of TRAM-34 on human hepatocellular cancer. Carcinogenesis or tumorigenesis is closely correlated to the uncontrolled growth and migration of tumor cells. Intracellular ROS level were found involved in the initiation and aggression of a variety of tumors [19-20]. Enhancement of ROS level was suggested as an important approach for treating cancers [20]. In this study, we found that TRAM-34 treatment can induce the augment of intracellular ROS level in hepatocellular cancer cells.
Besides, lots of studies showed that p53 is an important tumor suppressor gene, and regulates cell cycle, apoptosis, metastasis and senescence [21]. Inactivation or mutations of p53 have been well documented in human tumors. On the contrary, overexpression or activation of p53 can induce cell apoptosis and attenuate cancer cell migration and invasion through regulating many targets such as Bcl-2 and Bax [22-24]. Also, p53 induced the apoptosis of tumor cells via activating proapoptotic gene expression [25]. Our study showed that TRAM-34 induced a translocation of p53 protein from the cytoplasm into the nuclei. It implies that TRAM-34 inhibits the growth and cell migration of hepatocellular cancer in vitro through regulating p53 pathway.
In this study, low concentration (0.1, 0.3 and 1µM) of TRAM-34 did not significant affect the proliferation of HepG2 cells. Consistently, previous studies [11, 17] also reported that TRAM-34 at 10 and 30 µM displayed antitumor effects. For example, TRAM-34 10 µM was reported to inhibit cell growth of human pancreatic cancer cell lines by blocking KCa3.1 channel [17]. The moderate concentration of TRAM-34 (3-30µM) used in these studies were explained by: (1), the increase of intracellular calcium in cancer cells was recognized to antagonize TRAM-34; (2) the KCa3.1 channel expressed in cancer cells is less sensitive or mature than that in cardiovascular system.
In conclusion, the intermediate conductance Ca2+-activated potassium channel inhibitor TRAM-34 inhibited the proliferation and migration, and promoted the apoptosis of human hepatocellular carcinoma cells, which was associated with the increased intracellular ROS level and activated p53 protein. This study suggests TRAM-34 as a new therapeutic agent for hepatocellular carcinoma.
Acknowledgements
This work was supported by the National Natural Science Foundation of China (81100483), and The Young Investigator Foundation of Affiliated Fourth Hospital of Harbin Medical University.
Conflict of Interests
The authors have declared that no conflict of interest exists.
References
1. Pinyol R, Llovet JM. Hepatocellular carcinoma: Genome-scale metabolic models for hepatocellular carcinoma. Nat Rev Gastroenterol Hepatol. 2014. doi:nrgastro. 2014 doi:10.1038/nrgastro.2014.70
2. Parkin DM, Bray F, Ferlay J, Pisani P. Global cancer statistics, 2002. CA Cancer J Clin. 2005;55:74-108
3. Mazzaferro V, Regalia E, Doci R, Andreola S, Pulvirenti A, Bozzetti F. et al. Liver transplantation for the treatment of small hepatocellular carcinomas in patients with cirrhosis. N Engl J Med. 1996;334:693-9 doi:10.1056/NEJM199603143341104
4. Chalasani N, Said A, Ness R, Hoen H, Lumeng L. Screening for hepatocellular carcinoma in patients with cirrhosis in the United States: results of a national survey. Am J Gastroenterol. 1999;94:2224-9 doi:10.1111/j.1572-0241.1999.01297.x
5. Toyama K, Wulff H, Chandy KG, Azam P, Raman G, Saito T. et al. The intermediate-conductance calcium-activated potassium channel KCa3.1 contributes to atherogenesis in mice and humans. J Clin Invest. 2008;118:3025-37 doi:10.1172/JCI30836
6. Neylon CB, Lang RJ, Fu Y, Bobik A, Reinhart PH. Molecular cloning and characterization of the intermediate-conductance Ca(2+)-activated K(+) channel in vascular smooth muscle: relationship between K(Ca) channel diversity and smooth muscle cell function. Circ Res. 1999;85:e33-43
7. Klein H, Garneau L, Trinh NT, Prive A, Dionne F, Goupil E. et al. Inhibition of the KCa3.1 channels by AMP-activated protein kinase in human airway epithelial cells. Am J Physiol Cell Physiol. 2009;296:C285-95 doi:10.1152/ajpcell.00418.2008
8. Chou CC, Lunn CA, Murgolo NJ. KCa3.1: target and marker for cancer, autoimmune disorder and vascular inflammation? Expert Rev Mol Diagn. 2008;8:179-87 doi:10.1586/14737159.8.2.179
9. Tharp DL, Wamhoff BR, Wulff H, Raman G, Cheong A, Bowles DK. Local delivery of the KCa3.1 blocker, TRAM-34, prevents acute angioplasty-induced coronary smooth muscle phenotypic modulation and limits stenosis. Arterioscler Thromb Vasc Biol. 2008;28:1084-9 doi:10.1161/ATVBAHA.107.155796
10. Grgic I, Kiss E, Kaistha BP, Busch C, Kloss M, Sautter J. et al. Renal fibrosis is attenuated by targeted disruption of KCa3.1 potassium channels. Proc Natl Acad Sci U S A. 2009;106:14518-23 doi:10.1073/pnas.0903458106
11. Wang ZH, Shen B, Yao HL, Jia YC, Ren J, Feng YJ. et al. Blockage of intermediate-conductance-Ca(2+) -activated K(+) channels inhibits progression of human endometrial cancer. Oncogene. 2007;26:5107-14 doi:10.1038/sj.onc.1210308
12. Bellomo G, Perotti M, Taddei F, Mirabelli F, Finardi G, Nicotera P. et al. Tumor necrosis factor alpha induces apoptosis in mammary adenocarcinoma cells by an increase in intranuclear free Ca2+ concentration and DNA fragmentation. Cancer Res. 1992;52:1342-6
13. Chandra-Kuntal K, Lee J, Singh SV. Critical role for reactive oxygen species in apoptosis induction and cell migration inhibition by diallyl trisulfide, a cancer chemopreventive component of garlic. Breast Cancer Res Treat. 2013;138:69-79 doi:10.1007/s10549-013-2440-2
14. Miyashita T, Krajewski S, Krajewska M, Wang HG, Lin HK, Liebermann DA. et al. Tumor suppressor p53 is a regulator of bcl-2 and bax gene expression in vitro and in vivo. Oncogene. 1994;9:1799-805
15. Bishayee K, Ghosh S, Mukherjee A, Sadhukhan R, Mondal J, Khuda-Bukhsh AR. Quercetin induces cytochrome-c release and ROS accumulation to promote apoptosis and arrest the cell cycle in G2/M, in cervical carcinoma: signal cascade and drug-DNA interaction. Cell Prolif. 2013;46:153-63 doi:10.1111/cpr.12017
16. Nies AT, Konig J, Pfannschmidt M, Klar E, Hofmann WJ, Keppler D. Expression of the multidrug resistance proteins MRP2 and MRP3 in human hepatocellular carcinoma. Int J Cancer. 2001;94:492-9 doi:10.1002/ijc.1498 [pii]
17. Jager H, Dreker T, Buck A, Giehl K, Gress T, Grissmer S. Blockage of intermediate-conductance Ca2+-activated K+ channels inhibit human pancreatic cancer cell growth in vitro. Mol Pharmacol. 2004;65:630-8 doi:10.1124/mol.65.3.630
18. Lallet-Daher H, Roudbaraki M, Bavencoffe A, Mariot P, Gackiere F, Bidaux G. et al. Intermediate-conductance Ca2+-activated K+ channels (IKCa1) regulate human prostate cancer cell proliferation through a close control of calcium entry. Oncogene. 2009;28:1792-806 doi:10.1038/onc.2009.25
19. Hsu JL, Pan SL, Ho YF, Hwang TL, Kung FL, Guh JH. Costunolide induces apoptosis through nuclear calcium2+ overload and DNA damage response in human prostate cancer. J Urol. 2011;185:1967-74 doi:10.1016/j.juro.2010.12.091
20. Kim KW, Choi CH, Kim TH, Kwon CH, Woo JS, Kim YK. Silibinin inhibits glioma cell proliferation via Ca2+/ROS/MAPK-dependent mechanism in vitro and glioma tumor growth in vivo. Neurochem Res. 2009;34:1479-90 doi:10.1007/s11064-009-9935-6
21. Drygin D, O'Brien SE, Hannan RD, McArthur GA, Von Hoff DD. Targeting the nucleolus for cancer-specific activation of p53. Drug Discov Today. 2013 doi:10.1016/j.drudis.2013.08.012
22. Yaguchi T, Nakano T, Gotoh A, Nishizaki T. Adenosine promotes GATA-2-regulated p53 gene transcription to induce HepG2 cell apoptosis. Cell Physiol Biochem. 2011;28:761-70 doi:10.1159/000335770
23. Sipeki S, Bander E, Ways DK, Farago A. Activation of Erk1/Erk2 and transiently increased p53 levels together may account for p21 expression associated with phorbol ester-induced transient growth inhibition in HepG2 cells. Cell Signal. 2002;14:115-21
24. Lee YS, Jin DQ, Kwon EJ, Park SH, Lee ES, Jeong TC. et al. Asiatic acid, a triterpene, induces apoptosis through intracellular Ca2+ release and enhanced expression of p53 in HepG2 human hepatoma cells. Cancer Lett. 2002;186:83-91
25. Barlev NA, Liu L, Chehab NH, Mansfield K, Harris KG, Halazonetis TD. et al. Acetylation of p53 activates transcription through recruitment of coactivators/histone acetyltransferases. Mol Cell. 2001;8:1243-54
Author contact
![]() Corresponding author: Prof. Xiaofeng Jiang, MD, PhD, Department of Laboratory Medicine, Affiliated Fourth Hospital of Harbin Medical University, PR. China. Tel: +86-451-82576987; Fax: +86-451-82576987; Email: liuy.doctorcom
Corresponding author: Prof. Xiaofeng Jiang, MD, PhD, Department of Laboratory Medicine, Affiliated Fourth Hospital of Harbin Medical University, PR. China. Tel: +86-451-82576987; Fax: +86-451-82576987; Email: liuy.doctorcom