Impact Factor ISSN: 1837-9664

 Global reach, higher impact
Global reach, higher impactJ Cancer 2015; 6(8):701-708. doi:10.7150/jca.11785 This issue Cite
Research Paper
Anticancer Effect of a Novel Proteasome Inhibitor, YSY01A, via G2/M Arrest in PC-3M Cells in vitro and in vivo
Mei Zhang1,3, Xia Yuan1, Bo Xu1, Wei Guo1, Fu-Xiang Ran1, Run-Tao Li2, Jing-Rong Cui1 ![]()
1. The State Key Laboratory of Natural and Biomimetic Drugs
2. Department of Chemical Biology, School of Pharmaceutical Sciences, Peking University, Beijing 100191, China
3. Department of pharmacology, School of Pharmaceutical Sciences, Shihezi University, Xinjiang 832000, China
Received 2015-2-4; Accepted 2015-5-5; Published 2015-6-11
Abstract
YSY01A is a new tripeptideboronic acid and an analog of PS341. However, YSY01A's antitumor effects and mechanism have not yet been elucidated. This study demonstrates that YSY01A inhibited proteasome activity by combining with the chymotrypsin-like (CT-L) site (β5i/β5), the post-glutamyl peptide hydrolase (PGPH) site (β1i/β1) and the trypsin-like (T-L) site (β2i/β2) in special fluorgonic substrates and proteasome probe tests. We explored the anticancer effect using methyl thiazolyltetrazolium (MTT) or sulforhodamine B (SRB), and PC-3M cells were sensitive to YSY01A among the four cancer cell types tested. The YSY01A antiproliferative effect was stronger than that of PS341. In vivo, YSY01A (1.25, 2.25, and 3.25 mg/kg) inhibited PC-3M cell xenograft tumor growth, and the tumor volume inhibition rate was approximately 40% to 60%. YSY01A arrested PC-3M cells in the G2/M phase of the cell cycle by flow cytometry (FCM). Many proteins related to the cell cycle were analyzed using western blot, and YSY01A was shown to increase p21, p27, cyclinB1, P-cdc2 (tyr15) and wee1 protein expression in both cells and tumor tissue in a concentration-dependent manner. YSY01A, a proteasome inhibitor, exerts anticancer effects on PC-3M cells in vitro and in vivo. The mechanism of the YSY01A-mediated antitumor effect is that the cell cycle is arrested at the G2/M stage. This study suggests that YSY01A may be a novel therapeutic agent for prostate cancer.
Keywords: Anticancer, proteasome inhibitor, tripeptideboronic acid, PC-3M cell, cell cycle
Introduction
The ubiquitin proteasome system exerts a crucial action on protein elimination in eukaryotic cells. The system degrades various proteins, usually including damaged and abnormal proteins and proteins with errors, along with many short- and long-life proteins involved in the cell cycle, immune responses and signal transduction [1-3]. This pathway plays a critical role in physiological and pathophysiological processes.
Ubiquitin-proteasome system dysfunction is correlated with various tumors, including prostate cancer [4-7]. Prostate cancer is the most common cancer in male patients. It is estimated that prostate cancer is the most frequent cancer in males, and it has the second highest mortality rate in the United States [8]. Currently, therapeutic approaches for prostate cancer are limited and target drugs are rare because of the complex pathophysiology of this disease and a high death rate after development of resistance. Thus, there is a need for better drugs to treat patients with prostate cancer.
In the early 2000's, proteasomes became new targets for anticancer therapeutics. PS341 (Bortezomib, MLN-341, Velcade) was the first anticancer proteasome inhibitor and it was approved for refractory multiple myeloma [9]. Anticancer effects and mechanisms of many proteasome inhibitors have been identified in vivo and in vitro [10]. Some of these compounds have been considered to be potential anticancer drugs, and a few, such as marizomib (NPI-0052) and delanzomib (CEP-18770), have entered clinic trials [11, 12]. In addition, carfilzomib (PR-171), a novel epoxyketone-based proteasome inhibitor, has been used for relapsed and refractory multiple myeloma [13].
The present study shows that PS341 exerts widespread anticancer activity via cell cycle arrest, apoptosis, the NF-κB pathway and autophagy [14-16]. However, antitumor effects of bortezomib alone or in combination with chemotherapy agents are not significant in patients with prostate cancer [17-19]. To some extent, the widespread clinical use of bortezomib continues to be hampered by dose-limiting toxicities and drug resistance [20], and this drug usually causes serious side effects [21].
YSY01A(N-(2-Pyrazinecarbonyl)-L-leucine-L-(2-naphthyl)-alanine-L-leucine boronic acid) is an agent with powerful anticancer activity in vitro [22]. However, YSY01A anticancer activity and its mechanism of action remain unclear. To determine in vitro activity, HL-60 cells, Bcap-37 cells, PC-3M cells and Bel-7704 cells were used. We then used the xenograft tumour model with PC-3M cells to investigate antitumor effects. In addition, the influence of YSY01A on the cell cycle was determined in PC-3M cells and tumour tissue. This study provides information for drug development against prostate cancer.
Materials and methods
Cell lines and cell culture
HL-60, Bcap37, Bel-7704 (Cell Resource Center of Shanghai Institutes for Biological Sciences, CAS, China) and PC-3M cells (Cell Resource Center, IBMS, CAMS/PUMC, China) were used in the study. These cells were cultured in RPMI 1640 medium, supplemented with 10% FBS and 1% penicillin/streptomycin at 37°C and 5% CO2.
Drugs and reagents
YSY01A and PS341 were synthetized by Professor Li Run Tao's laboratory. Me4BodipyFL-Ahx3Leu3VS, Suc-LLVY-AMC, Boc-LRR-AMC, Z-LLE-AMC, and Z-RR-AMC were purchased from Boston Biochem, USA. Polyclonal antibodies (ubiquitin, wee 1, cyclinB1) and monoclonal antibodies (p27Kip1, p21Waf1/Cip1, GAPDH) were obtained from Cell Signaling Technology Inc, USA. Fetal bovine serum (GIBCO, USA), RPMI 1640, penicillin, and streptomycin were purchased from M&C Gene Technology Inc, China.
Animals
Balb/c male nude mice (4-5 weeks) were housed under specific pathogen-free conditions. All animal protocols were approved and monitored by the Institutional Animal Care and Use Committee. The animals were obtained from the Department of Laboratory Animal Science at Peking University Health Science Center (China; certificate number SYXK (Jing) 2008-0021; SCXK (Jing) 2011-0012).
26S proteasome activity assays
The 26S proteasomes were extracted and examined as previously described [23, 24]. The cells were harvested and washed with cold PBS and centrifuged, and the pellets were lysed in homogenization buffer (50 mM Tris-HCl, pH 7.5, 1 mM DTT, 5 mM MgCl2, 250 mM sucrose and 2 mM ATP) on ice for 30 min. Cell debris were removed by centrifugation at 10000rpm for 15 min at 4°C, and samples underwent ultracentrifugation at 15000rpm and 4°C for 1 h. Protein content was measured using the Bradford assay and bovine serum albumin as a standard.
Proteasome activity was measured by monitoring the release of 7-amido-4-methylcoumarin (AMC) from peptide substrates specific for each activity. Proteasome inhibition was determined as previously described [25-27]. 26S proteasomes (12.5 μg) and various concentrations of YSY01A were incubated with a 50 μM peptide substrate solution (Suc-LLVY-AMC for CT-L, Boc-LRR-AMC for T-L and Z-LLE-AMC for PGPH) in 100 μL of assay buffer (50 mM Tris-HCl, pH 7.5, 5 mM MgCl2, 0.5 mM ATP, 1 mM DTT and 0.5 mg/mL BSA) at 37°C for 1h.
Proteasome activity profiling in intact cells
Me4BodipyFL-Ahx3Leu3VS is a cell-permeable fluorescent proteasome probe that allows accurate profiling of proteasomal activity in cells with high selectivity. Proteasome activity profiling in intact cells using fluorescent probes was performed as described previously [28, 29]. The cells were exposed to various concentrations of YSY01A for 3h, washed with PBS and harvested. The 26S proteasomes were extracted as described above. For competitive binding experiments, the lysates were incubated with Me4BodipyFL-Ahx3Leu3VS (5 μM) at 37°C for 1h, and proteins were boiled in sample buffer. Then, 20 μg of protein/lane was resolved using 12.5% SDS-PAGE. Labeled proteasome subunits were visualized by in-gel fluorescence scanning on a variable-mode imager.
Cancer cells proliferation
PC-3M, Bel-7704, HL-60 or Bcap37 cells were placed in 96-well plates. The cells were treated with various concentrations of YSY01A or PS341 for 48h. Cell survival was tested using sulforhodamine B (SRB) or methyl thiazolyltetrazolium (MTT) for HL-60.
The xenograft tumor model
The PC-3M cells was harvested, washed and suspended with PBS. PC-3M cells (3×106) were injected subcutaneously into the right flank of Balb/c male nude mice. When a palpable tumor appeared (approximate 0.2 cm3), tumor-bearing mice were randomly distributed into 5 groups (7 mice/group), as follows: model group (normal saline), positive group (PS341, 1.25 mg/kg) and 3 YSY01A dose groups (1.25, 2.25 or 3.25 mg/kg). These nude mice received the treatments twice weekly by intraperitoneal injection. The mice were weighed and the tumor size was measured before administration, and the tumor volume was calculated using the following formula: Volume =a×b 2/2 (where a=length and b=width).
Mice were treated 11 times over 36 days. On day 37, tumor size and body weight were measured before the mice were sacrificed. Tumors and organs (heart, liver, lung, brain, kidney, and spleen) were removed and weighed.
Cell cycle assay
PC-3M cells were exposed to various YSY01A concentrations for 48h, washed with twice cold PBS and harvested. The cell aggregate then was suspended in 70% ice-cold ethanol at -20°C overnight. The fixed cells were incubated with RNAase A for 30 min at 37°C, and stained with PI (propidiumidodide). The DNA content was detected using flow cytometry.
Western blot analysis
PC-3M cells were either seeded with various YSY01A concentrations for 48h or obtained after grinding the tumor tissue. The cells were harvested and lysed with buffer. Whole proteins were extracted and tested. Proteins then were resolved using 10% SDS-PAGE following by blotting on nitrocellulose membranes. The membranes were blocked with 5% milk and incubated with a primary antibody at 4°C overnight. Anti-mouse and anti-rabbit antibodies were used as secondary antibodies. Enhanced chemiluminescence western blot analysis was performed using imaging equipment.
Statistical methods
The experimental data are presented as the mean ± standard deviation (SD). Significant differences were determined using the Student's t-test. Statistical analysis was performed using SAS (P<0.05 was considered significant).
Results
YSY01A inhibited proteasome activity
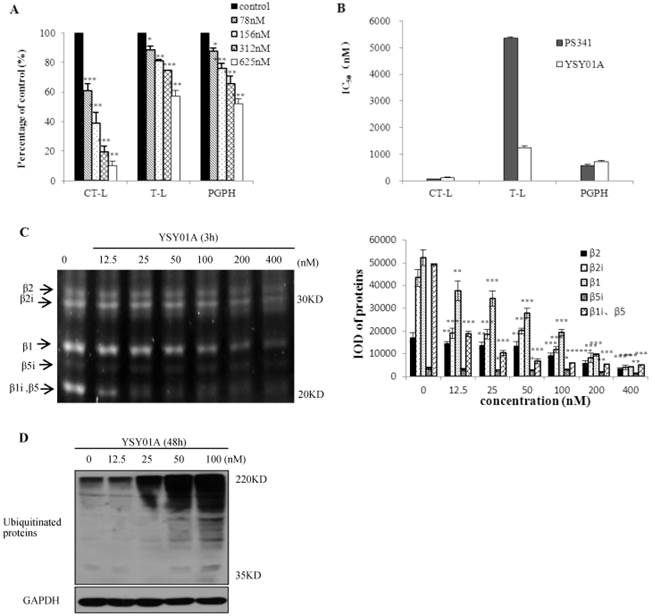
To observe the effect of YSY01A on proteasomes, we first measured the proteasome activity in vitro and in whole cells. YSY01A inhibited CT-L, T-L and PGPH activity in 26S proteasomes in vitro (Fig. 1A). After 26S proteasomes were treated with various YSY01A concentrations for 1h, CT-L activity was inhibited by 34-86%, T-L activity by 12-43% and PGPH activity by 12-48%. The IC50 values of YSY01A on the three catalytic sites are shown in Fig. 1B. YSY01A IC50 values for CT-L, T-L and PGPH were 123±18 nM, 1243±77 nM and 714±44 nM, respectively, and the corresponding PS341 IC50 values were 71±1 nM, 5349±57 nM and 564±62 nM, respectively. The inhibition effect of YSY01A on T-L activity was higher than 3-fold that of PS341. The proteasome activity profile shows β5i, β2, β2i, β1, and β1i fluorgenic bands that were visualized in the PC-3M cells (Fig. 1C). Compared with control group, these expressions of different doses of YSY01A were significant (p<0.05). Ubiquitin is the target signal for protein degradation by the 26S proteasomes. If the proteasomes are blocked, ubiquitin proteins will accumulate. Ubiquitinated proteins were enhanced in PC-3M cells (Fig.1D). 25nM YSY01A caused protein accumulation, and the expression increased with increasing concentration. These data indicate that YSY01A inhibits 26S proteasome activity in a concentration-dependent manner in vitro and in whole cells.
Effects of YSY01A on 26S proteasomes. A. YSY01A inhibited 26S proteasomes from PC-3M cells. B. IC50 values of YSY01A and PS341 on three catalytic sites. C. PC-3M cells were exposed to YSY01A (12.5-400 nM) for 3 h, expression of β catalytic sites and IOD of these bands were shown. D. Ubiquitinated proteins were displayed after YSY01A (12.5-100 nM) treated PC-3M cells for 48h. Every panel is the representative image of three independent experiments. Columns, mean of three independent experiments; bars, SD; compared with control, * p<0.05, ** p<0.01, *** p<0.001
YSY01A inhibited proliferation of cancer cells
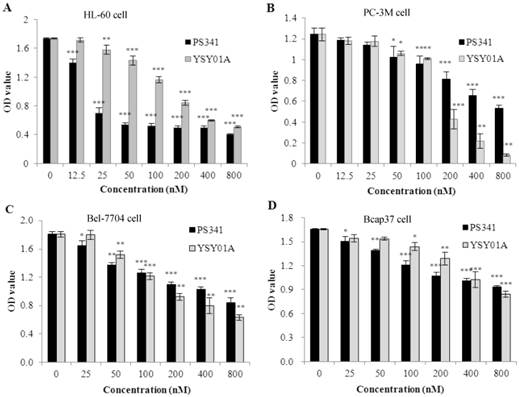
To evaluate YSY01A anticancer activity in vitro, we tested cell survival in four types of cancer cells which were treated for 48 h. These data indicated that YSY01A inhibited proliferation in the cancer cells (Fig. 2 A, B, C, D). The IC50 values were 149.8 ± 9.0 nM in PC-3M cells, 170.1 ±9.9 nM in HL-60 cells, 285.9 ± 13.2 nM in Bel-7740 cells and 878.6±18.2 nM in Bcap37 cells. In these cells, PC-3M was most sensitive and the YSY01A inhibition efficacy was three times highter that of PS341. However, PS341 inhibited proliferation more strongly than YSY01A in other types of cancer cells.
Effects of YSY01A on proliferation of several types cancer cells. A. YSY01A and PS341 inhibited HL-60 cell proliferation. B. YSY01A and PS341 inhibition in PC-3M cells is shown. C. YSY01A and PS341 inhibited Bel-7704 cell proliferation. D. YSY01A and PS341 inhibited proliferation in Bcap37 cells. Columns, mean of three independent experiments; bars, SD; compared with control, * p<0.05, ** p<0.01, *** p<0.001
Antitumor effect in vivo
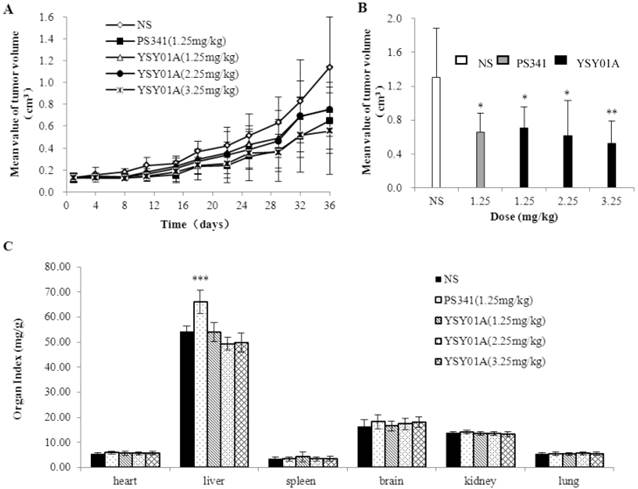
We used PC-3M cell xenograft nude mice to investigate YSY01A anticancer activity in vivo. Tumor size was measured before each administration, and the tumor volume curve was created (Fig. 3A). Tumors in the control group grew faster, and PS341 (1.25 mg/kg) and YSY01A (1.25, 2.25, 3.25 mg/kg) slowed the tumor growth. These curves indicated that YSY01A and PS341 inhibited xenograft tumor growth. Final tumor volume is shown in Fig. 3B. YSY01A (1.25, 2.25 and 3.25 mg/kg) inhibited tumor growth by 45.7%, 52.8% and 60.1%, respectively, and that of PS341 was 49.6%. The toxic effect is reflected in the viscera index (Fig. 3C). The viscera index was calculated and we found that all three YSY01A doses slightly impacted on the viscera (body, heart, liver, brain, kidney, spleen and lung) in tumor-bearing mice (P>0.05). However, PS341 (1.25 mg/kg) increased the liver index by 20% (P<0.001). The present findings suggest that YSY01A induces antitumor effects in PC-3M cell xenografts, and that its toxicity is lower than PS341.
YSY01A inhibited PC-3M cell xenograft tumor growth. A. The tumor volume curve before each administration. B. The end tumor volume of each group was compared on 37th day after YSY01A treatment. C. The organ index was shown that all three YSY01A doses slightly effects on the viscera. Columns, mean of each group; bars, SD; compared with control, * p<0.05, ** p<0.01,*** p<0.001
YSY01A caused cell cycle arrest in the G2/M stage
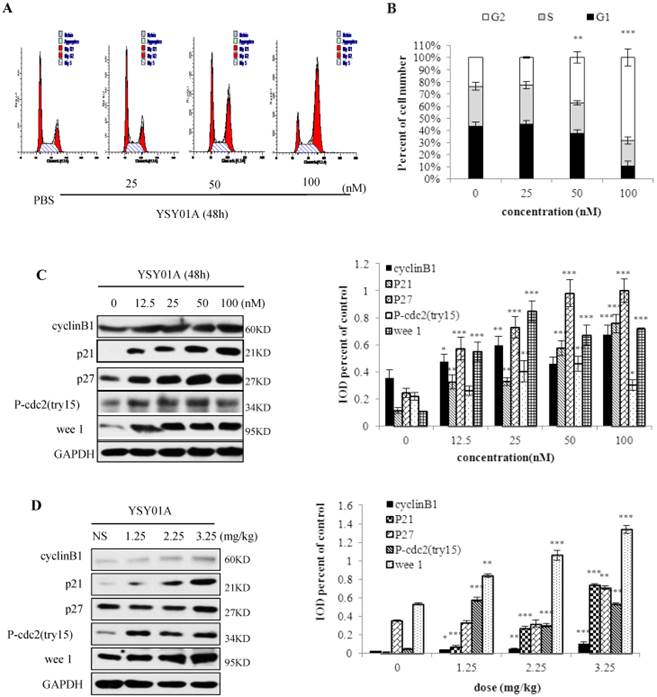
Because many cell cycle proteins are degraded by proteasomes, proteasome inhibition may cause cell cycle arrest. Thus, we determined the effect of YSY01A on PC-3M cell cycle (Fig. 4 A, B). When 50 nM YSY01A was applied to PC-3M cells for 48 h, cells in the G2/M phase increased by 14%, and when 100 nM YSY01A was used, G2/M increased by 44%. In PC-3M cells treated with YSY01A (12.5-100 nM) for 48 h (Fig.4C), compared with control group, protein (cyclinB1, p21, p27, P-cdc2 (tyr15), wee 1) expression increased and have significant difference (p<0.05). Moreover, in fig. 4D, expression of these proteins from tumor tissue was enhanced and have significant difference (p<0.05). These show a concentration-dependent arrest of the cell cycle.
YSY01A arrests the PC-3M cells in G2/M phase. A. After PC-3M cells were treated with 25, 50 or 100 nM for 48h, the cell cycle was arrested in the G2/M phase. B. The percentage of cells in the G2/M phase gradually increased with increasing YSY01A concentration, and it decreased in the S phase. C. YSY01A increased the cyclinB1, p21, p27, P-cdc2 (tyr15) and wee 1 protein expression in a concentration-dependent manner. D. YSY01A increased levels of cyclinB1, p21, p27, P-cdc2 (tyr15) and wee 1 in PC-3M cell xenograft tumors. Every panel is the representative image of three independent experiments. Columns, mean of three independent experiments; bars, SD; compared with control, * p<0.05, ** p<0.01, *** p<0.001
Discussion
The ubiquitin-proteasome pathway has been an important target for anticancer therapeutics. Currently, PS341 and carfilzomib (both proteasome inhibitors) have been successfully used to treat patients with multiple myeloma. YSY01A caused the most cytotoxicity in the serial administration of tripeptideboronic acid. Based on previous studies, we expect that YSY01A results in an antineoplastic effect for prostate cancer.
The β1, β2 and β5 sites have different proteolytic activities in 26S proteasomes, whereas β1i, β2i and β5i have the same effect as their homologues in 26S immunoproteasomes [30, 31]. In our results, the β5i, β2, β2i, β1 and β1i/β5 fluorgenic bands decreased because YSY01A was bound to these sites on the PC-3M cells. YSY01A inhibited three catalytic activity sites of the 26S proteasomes, and the YSY01A-induced inhibition of CT-L was the strongest, followed by that of PGPH and T-L. The effect of YSY01A is four times greater than that of PS341 on the T-L site. Moreover, YSY01A caused the accumulation ubiquitin proteins. Overall, we demonstrated that YSY01A inhibits proteasomes and has different selectivity for the β active subunit compared with PS341.
In this study, YSY01A inhibited proliferation in four types of cancer cells, but PC-3M was the most sensitive. We also found that YSY01A inhibited PC-3M xenograft tumor growth. These results indicate that YSY01A has anticancer activity in vitro and in vivo, and that it has the same potency as PS341. However, the toxicity of YSY01A was lower than that of PS341. These results show YSY01A may be a novel potent anticancer agent.
Proteasome inhibitors exert antitumor effects through cell cycle arrest, induction of cell apoptosis, enhanced autophagy and inhibition of angiogenesis [32-34]. It is known that p27 (Kip1) is a member of the Cip/Kip family of cyclin-dependent kinase inhibitors. The tumor suppressor protein p21 Waf1/Cip1 acts as an inhibitor of cell cycle progression [35]. Proteasomes degrade some proteins in the cell cycle, such as p27, p21, cyclinB1 and wee1 [36-39]. Phosphorylation at Tyr 15 and Thr14 results in cdc2 inhibition can be carried out by wee1 and Myt1 protein kinases [38]. We showed the anticancer mechanism of YSY01A by analyzing cell cycle arrest. YSY01A caused PC-3M cell cycle arrest in the G2/M phase and p21, p27, cyclinB1, P-cdc2 (tyr15) and wee1 protein expression significantly increased in cells or tumor tissue; PS341 produced the same effects [40]. YSY01A blocks proteasomal activity and elimination of the substrates, resulted in accumulation of these proteins and cyclinB1/cdc2 complex decrease. PC-3M cell are then inhibited in the G2/M phase of the cell cycle and cell death follows. Further study is required to determine other mechanisms of YSY01A action.
In summary, we demonstrated that YSY01A acted on proteasome, and inhibited PC-3M cell growth in vitro and in vivo. Antitumor effect of YSY01A was approximately equal to that of PS341, but its toxicity was lower. We also revealed that the anticancer mechanism of YSY01A was closely related to cell cycle arrest at the G2/M stage. Therefore, this study has provided information for the development of YSY01A as a potential therapeutic drug for prostate cancer and a theoretical basis for antineoplastic therapy.
Acknowledgements
This work was supported by grants from Eleventh Five Year Plan for National Science and Technology Major Project (No. 2009ZX0930010), the National Natural Science Foundation of China (No. 81172915) and a grant from Major New Drugs Research and Development Platform of Peking University (No. 2009ZX09301010).
Conflicts of interest
There are no conflicts of interest.
References
1. Goldberg AL, Stein R, Adams J. New insights into proteasome function: from archaebacteria to drug development. Chemistry & biology. 1995;2:503-8
2. Samy TS, Schwacha MG, Chung CS, Cioffi WG, Bland KI, Chaudry IH. Proteasome participates in the alteration of signal transduction in T and B lymphocytes following trauma-hemorrhage. Biochimica et biophysica acta. 1999;1453:92-104
3. Tohe A. Structure and function of the yeast 26S proteasome. Seikagaku The Journal of Japanese Biochemical Society. 1999;71:173-81
4. Sun Y. E3 ubiquitin ligases as cancer targets and biomarkers. Neoplasia. 2006;8:645-54
5. Morelva Tde M, Antonio LB. Immunohistochemical expression of ubiquitin and telomerase in cervical cancer. Virchows Archiv: an international journal of pathology. 2009;455:235-43
6. Xiang T, Li L, Yin X, Yuan C, Tan C, Su X. et al. The ubiquitin peptidase UCHL1 induces G0/G1 cell cycle arrest and apoptosis through stabilizing p53 and is frequently silenced in breast cancer. PLoS One. 2012;7:e29783
7. Voutsadakis IA, Papandreou CN. The ubiquitin-proteasome system in prostate cancer and its transition to castration resistance. Urologic oncology. 2012;30:752-61
8. Siegel R, Naishadham D, Jemal A. Cancer statistics, 2013. CA: a cancer journal for clinicians. 2013;63:11-30
9. Adams J. The development of proteasome inhibitors as anticancer drugs. Cancer cell. 2004;5:417-21
10. de Bettignies G, Coux O. Proteasome inhibitors: Dozens of molecules and still counting. Biochimie. 2010;92:1530-45
11. Obaidat A, Weiss J, Wahlgren B, Manam RR, Macherla VR, McArthur K. et al. Proteasome regulator marizomib (NPI-0052) exhibits prolonged inhibition, attenuated efflux, and greater cytotoxicity than its reversible analogs. The Journal of pharmacology and experimental therapeutics. 2011;337:479-86
12. Gallerani E, Zucchetti M, Brunelli D, Marangon E, Noberasco C, Hess D. et al. A first in human phase I study of the proteasome inhibitor CEP-18770 in patients with advanced solid tumours and multiple myeloma. Eur J Cancer. 2013;49:290-6
13. Thompson JL. Carfilzomib: a second-generation proteasome inhibitor for the treatment of relapsed and refractory multiple myeloma. The Annals of pharmacotherapy. 2013;47:56-62
14. Hutter G, Rieken M, Pastore A, Weigert O, Zimmermann Y, Weinkauf M. et al. The proteasome inhibitor bortezomib targets cell cycle and apoptosis and acts synergistically in a sequence-dependent way with chemotherapeutic agents in mantle cell lymphoma. Annals of hematology. 2012;91:847-56
15. Yao F, Wang G, Wei W, Tu Y, Tong H, Sun S. An autophagy inhibitor enhances the inhibition of cell proliferation induced by a proteasome inhibitor in MCF-7 cells. Molecular medicine reports. 2012;5:84-8
16. An J, Sun Y, Fisher M, Rettig MB. Maximal apoptosis of renal cell carcinoma by the proteasome inhibitor bortezomib is nuclear factor-kappaB dependent. Molecular cancer therapeutics. 2004;3:727-36
17. Morris MJ, Kelly WK, Slovin S, Ryan C, Eicher C, Heller G. et al. A phase II trial of bortezomib and prednisone for castration resistant metastatic prostate cancer. The Journal of urology. 2007;178:2378-83 discussion 83-4
18. Papandreou CN, Daliani DD, Nix D, Yang H, Madden T, Wang X. et al. Phase I trial of the proteasome inhibitor bortezomib in patients with advanced solid tumors with observations in androgen-independent prostate cancer. Journal of clinical oncology: official journal of the American Society of Clinical Oncology. 2004;22:2108-21
19. Dreicer R, Petrylak D, Agus D, Webb I, Roth B. Phase I/II study of bortezomib plus docetaxel in patients with advanced androgen-independent prostate cancer. Clinical cancer research: an official journal of the American Association for Cancer Research. 2007;13:1208-15
20. Chen D, Frezza M, Schmitt S, Kanwar J, Dou QP. Bortezomib as the first proteasome inhibitor anticancer drug: current status and future perspectives. Current cancer drug targets. 2011;11:239-53
21. Davis NB, Taber DA, Ansari RH, Ryan CW, George C, Vokes EE. et al. Phase II trial of PS-341 in patients with renal cell cancer: a University of Chicago phase II consortium study. Journal of clinical oncology: official journal of the American Society of Clinical Oncology. 2004;22:115-9
22. Zhu Y, Yao S, Xu B, Ge Z, Cui J, Cheng T. et al. Design, synthesis and biological evaluation of tripeptide boronic acid proteasome inhibitors. Bioorganic & medicinal chemistry. 2009;17:6851-61
23. Altun M, Galardy PJ, Shringarpure R, Hideshima T, LeBlanc R, Anderson KC. et al. Effects of PS-341 on the activity and composition of proteasomes in multiple myeloma cells. Cancer research. 2005;65:7896-901
24. Kisselev AF, Callard A, Goldberg AL. Importance of the different proteolytic sites of the proteasome and the efficacy of inhibitors varies with the protein substrate. The Journal of biological chemistry. 2006;281:8582-90
25. Braun HA, Umbreen S, Groll M, Kuckelkorn U, Mlynarczuk I, Wigand ME. et al. Tripeptide mimetics inhibit the 20 S proteasome by covalent bonding to the active threonines. The Journal of biological chemistry. 2005;280:28394-401
26. Manam RR, McArthur KA, Chao TH, Weiss J, Ali JA, Palombella VJ. et al. Leaving groups prolong the duration of 20S proteasome inhibition and enhance the potency of salinosporamides. Journal of medicinal chemistry. 2008;51:6711-24
27. Wan SB, Chen D, Dou QP, Chan TH. Study of the green tea polyphenols catechin-3-gallate (CG) and epicatechin-3-gallate (ECG) as proteasome inhibitors. Bioorganic & medicinal chemistry. 2004;12:3521-7
28. Clerc J, Li N, Krahn D, Groll M, Bachmann AS, Florea BI. et al. The natural product hybrid of Syringolin A and Glidobactin A synergizes proteasome inhibition potency with subsite selectivity. Chem Commun (Camb). 2011;47:385-7
29. Berkers CR, Verdoes M, Lichtman E, Fiebiger E, Kessler BM, Anderson KC. et al. Activity probe for in vivo profiling of the specificity of proteasome inhibitor bortezomib. Nature methods. 2005;2:357-62
30. Fenteany G, Standaert RF, Lane WS, Choi S, Corey EJ, Schreiber SL. Inhibition of proteasome activities and subunit-specific amino-terminal threonine modification by lactacystin. Science. 1995;268:726-31
31. Kingsbury DJ, Griffin TA, Colbert RA. Novel propeptide function in 20 S proteasome assembly influences beta subunit composition. The Journal of biological chemistry. 2000;275:24156-62
32. Meinel FG, Mandl-Weber S, Baumann P, Leban J, Schmidmaier R. The novel, proteasome-independent NF-kappaB inhibitor V1810 induces apoptosis and cell cycle arrest in multiple myeloma and overcomes NF-kappaB-mediated drug resistance. Molecular cancer therapeutics. 2010;9:300-10
33. Selimovic D, Porzig BB, El-Khattouti A, Badura HE, Ahmad M, Ghanjati F. et al. Bortezomib/proteasome inhibitor triggers both apoptosis and autophagy-dependent pathways in melanoma cells. Cellular signalling. 2013;25:308-18
34. Yue CX, Ma J, Zhou HJ, Tang QL, Li LL, Bi F. et al. [The effect of RhoA and proteasome inhibitor MG132 on angiogenesis in tumors]. Sichuan da xue xue bao Yi xue ban = Journal of Sichuan University Medical science edition. 2011;42:445-50
35. Wang Y, Prives C. Increased and altered DNA binding of human p53 by S and G2/M but not G1 cyclin-dependent kinases. Nature. 1995;376:88-91
36. Lloyd RV, Erickson LA, Jin L, Kulig E, Qian X, Cheville JC. et al. p27kip1: a multifunctional cyclin-dependent kinase inhibitor with prognostic significance in human cancers. The American journal of pathology. 1999;154:313-23
37. Chehab NH, Malikzay A, Stavridi ES, Halazonetis TD. Phosphorylation of Ser-20 mediates stabilization of human p53 in response to DNA damage. Proceedings of the National Academy of Sciences of the United States of America. 1999;96:13777-82
38. Sheaff RJ, Singer JD, Swanger J, Smitherman M, Roberts JM, Clurman BE. Proteasomal turnover of p21Cip1 does not require p21Cip1 ubiquitination. Molecular cell. 2000;5:403-10
39. Watanabe N, Arai H, Nishihara Y, Taniguchi M, Hunter T, Osada H. M-phase kinases induce phospho-dependent ubiquitination of somatic Wee1 by SCFbeta-TrCP. Proceedings of the National Academy of Sciences of the United States of America. 2004;101:4419-24
40. Tamura D, Arao T, Tanaka K, Kaneda H, Matsumoto K, Kudo K. et al. Bortezomib potentially inhibits cellular growth of vascular endothelial cells through suppression of G2/M transition. Cancer science. 2010;101:1403-8
Author contact
![]() Corresponding author: Jing-Rong Cui, Professor, the State Key Laboratory of Natural and Biomimetic Drugs, School of Pharmaceutical Sciences, Peking University Health Science Center, Xueyuan Rd, Haidian District, Beijing, china, 100191. Telephone: 861082802467; Email: jrcuiedu.cn
Corresponding author: Jing-Rong Cui, Professor, the State Key Laboratory of Natural and Biomimetic Drugs, School of Pharmaceutical Sciences, Peking University Health Science Center, Xueyuan Rd, Haidian District, Beijing, china, 100191. Telephone: 861082802467; Email: jrcuiedu.cn