Impact Factor ISSN: 1837-9664

 Global reach, higher impact
Global reach, higher impactJ Cancer 2015; 6(9):832-842. doi:10.7150/jca.12010 This issue Cite
Research Paper
Genetic Characterization of Ten-Eleven-Translocation Methylcytosine Dioxygenase Alterations in Human Glioma
Theo F. J. Kraus ![]() , Andrea Greiner, Martina Steinmaurer, Vanessa Dietinger, Virginie Guibourt, Hans A. Kretzschmar
, Andrea Greiner, Martina Steinmaurer, Vanessa Dietinger, Virginie Guibourt, Hans A. Kretzschmar
Center for Neuropathology and Prion Research (ZNP), Ludwig-Maximilians-University, Feodor-Lynen-Str. 23, D-81377 Munich, Bavaria, Germany.
Received 2015-2-27; Accepted 2015-6-23; Published 2015-7-15
Abstract
The molecular mechanisms leading to brain tumors still remain unclear. Nevertheless, there is increasing evidence that epigenetic effects play crucial roles in tumor development and progression. Thereby, 5-hydroxymethylcytosine (5hmC) represents a further base modification of cytosine besides 5-methylcytosine (5mC). In addition to the role of 5hmC as an intermediate in demethylation, 5hmC is of reasonable importance for cellular control. Previous studies showed that loss of 5hmC is a hallmark of human malignancies, e.g. in glioma, melanoma, and myeloid tumors. In myeloid malignancies studies showed that loss of 5hmC was due to mutations within ten-eleven-translocation (TET) genes, enzymes being responsible for conversion of 5mC to 5hmC. Nevertheless, till date there are no genetic characterization data of TET enzymes available for glioma.
In this study, we genetically characterized TET2 and TET3 alterations in 50 human gliomas (WHO-Grade II-IV) and in 19 healthy brain samples. We identified 7 genetic alterations within TET2 (p.V218M, p.G355N, p.P363L, p.L1721W, p.P1723S, p.I1762V, p.H1778R). Additionally, we performed quantification of 5hmC amount and added functional prediction analysis of identified TET alterations to evaluate the biological impact of these alterations on the hydroxymethylome. An analysis of TET3 showed no non-synonymous alterations.
In summary, we did not find correlations of TET alterations with 5hmC amount. Thus, our data emphasize that, in contrast to leukemia, loss of 5hmC in glioma is not caused by TET gene alterations. Moreover, other disturbances, such as disrupted gene expressions or functional inhibitions of TET proteins may be responsible for the aberrant epigenome of human glioma.
Keywords: Epigenetics, Glioma, Methylation, Demethylation, 5hmC, Ten-Eleven-Translocation.
Introduction
The mechanisms leading to human brain tumors still remain unclear. Nevertheless, there is strong evidence that epigenetic changes significantly contribute to tumor development and progression [1-6]. One prominent example of an epigenomic hallmark that is highly relevant in human glioma is the silencing of O6-methylguanine DNA-methyltransferase (MGMT) [1] by promoter methylation. This phenomenon has been described as a strong predictive factor of treatment response to chemotherapy in anaplastic astrocytoma (AA) and glioblastoma (GBM) [5].
In the recent years, a new base modification besides 5-methylcytosine (5mC) has been described: 5-hydroxymethylcytosine (5hmC) [7]. Till date, the distinct function of 5hmC still remains unclear but there is evidence, that 5hmC is involved in gene regulation, developmental control and active demethylation processes [8-10]. Whereas 5mC is generated out of cytosine (C) by DNMT (DNA methyltransferase) enzymes, 5hmC is converted of 5mC by the TET (ten-eleven-translocation) enzymes [8]. After the generation, 5hmC can be further converted to 5-formylcytosine (5fC) and 5-carboxycytosine (5caC) by the TET enzymes and finally decarboxylated by TDG (thymine-DNA glycosylase) to unmodified C [8]. This sequence of modifications describes an active demethylation pathway of methylated cytosine [8].
Besides the role as an intermediate in active demethylation pathways, other studies showed that 5hmC also states a functional importance in gene regulation [10, 11]: Spruije et al. showed that there are dynamic readers for 5mC and 5hmC that recognize either 5mC or 5hmC [10]. Interestingly, these readers change during cellular differentiation from embryonic stem cell (ESC) state to neuronal progenitor cells (NPC), and adult brain tissue [10]. There are also differences of 5hmC amount detectable depending on the investigated tissue type and on age [9, 12, 13], emphasizing the complexity and plasticity of the epigenome.
Whereas human brain tissue exhibits high amounts of 5hmC, there is a loss of 5hmC in brain tumors leading to a disturbance of the hydroxymethylome [12]: In contrast to normal human brain tissue, that exhibits up to 1.1 % 5hmC of all cytosines, brain tumors and especially highly malignant WHO-Grade IV glioblastoma (GBM) exhibit only vanishingly low amounts of 5hmC (mean amounts of 0.078 % 5hmC of all cytosines) [12].
Besides brain tumors, a similar phenomenon has also been described for other malignancies, e.g. melanoma, oral squamous cell carcinoma and myeloid malignancies [14-19]. Thereby, studies revealed that TET2 mutations are accompanied by decreased 5hmC amounts [20]. In a wide range of myeloid neoplasms, such as chronic myelomonocytic leukemia, acute myeloic leukemia and myelodysplastic syndrome, heterozygous deletions and mutations of TET2 were found in high frequencies [16, 21-24]. Furthermore, functional studies revealed that the loss of 5hmC in these tumor entities is directly caused by TET2 mutations within the functional domains of the TET2 protein [15-17, 20].
Till date, there are no comparable studies available investigating the significance of TET mutations in glioma by validating the consequences of TET mutations on glioma 5hmC amount. This problem is now addressed by us. Therefore, this is the first comprehensive study that genetically characterizes TET alterations in the context of human glioma by direct sequencing of the TET2 and TET3 genes. Thus, the data generated in this approach are of fundamental importance to better understand the role of TET proteins in the context of brain tumor formation and they will help to better understand the molecular mechanisms leading to disruption of the glioma epigenome.
In this study, we investigated 50 human glioma of WHO-Grades II to IV and genetically characterized TET2 and TET3 sequence alterations. Additionally, we sequenced the TET2 and TET3 genes in 19 human control brain samples. Furthermore, we quantified 5hmC amount in all 69 investigated tissue specimen and correlated 5hmC amount with TET mutations. A prediction analysis of the functional effects of identified TET alterations was performed using the PolyPhen-2 algorithm in order to evaluate the biological significance of identified TET alterations.
Materials and Methods
Sample collection
In the current study, we investigated 69 different tissue specimen including 50 glioma samples (WHO-Grade II-IV) and 19 brain tissue samples as unaffected control brain tissue.
All tumor samples were provided by the Brain Tumor Bank of the Center for Neuropathology. Written informed consent was obtained according to the guidelines of the local ethics committee. The surgical tumor samples used in this study were snap-frozen and stored at -80°C. Routine histological procedures included H&E (Haematoxylin and Eosin), EvG (Elastica van Gieson), PAS (Periodic acid-Schiff), Gomori silver stain as well as immunohistochemistry using antibodies against GFAP (glial fibrillary acidic protein, monoclonal mouse antibody, clone 6F2, Dako), MAP2 (microtubule-associated protein 2, clone HM.2, Sigma) and Ki67 proliferation associated antigen (monoclonal mouse antibody, clone MIB1, Dako). Routine diagnostics was performed on tumor tissue that has been formalin-fixed (4 % buffered formalin) and paraffin embedded. All tumor samples were classified according to the current WHO (world health organization) classification of tumors of the central nervous system, 4th edition, 2007 [25-27]. Mean age of patients was 53 years. The female to male ratio was 13 to 37. Detailed information of patients can be found in Table 1.
Detailed information on glioma samples used in this study. We included 50 glioma patients in our study, including three patients with WHO-Grade II glioma, ten patients with WHO-Grade III glioma and 37 patients with WHO-Grade IV glioblastoma. The mean age of patients was 53 years. The female to male ratio was 13 to 37. MGMT promoter methylation: 0: unmethylated; 1: methylated; 2: partially methylated; 3: not analysable; 9: not analysed; IDH1 and IDH2 mutation status: 0: IDH1 no mutation, IDH2 no mutation; 1: IHD1 mutation, IDH2 no mutation; 2: IDH1 no mutation, IDH2 mutation; 3: IDH1 mutation, IDH2 mutation; 9: not analysed; LOH 1p/19q status: 0: no LOH 1p/19q; 1; partial LOH 1p; 2: partial LOH 19q; 3: LOH 1p/19q; 4: isolated partial LOH 1p; 5: isolated partial LOH 19q; 6: other aberration; 9: not analysed; TET2 and TET3 analysis: 1: exploratory set; 2: validation set; 3: verification set; 9: not analysed.
| Case | Sex | Age at surgery [y] | Diagnosis | WHO-Grade | MGMT promoter methylation | IDH1/2 | LOH 1p/19q | 5hmC positive cells [%] | TET2 analysis | TET3 analysis | TET2 sequence variation | TET2 variation in amino acid sequence | TET3 sequence variation | TET3 variation in amino acid sequence |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | m | 33 | Diffuse Astrocytoma | II | 9 | 1 | 9 | 13.07 | 1 | 9 | c.1064G>A, c.5162T>G | p.G355N, p.L1721W | - | - |
| 2 | m | 59 | Diffuse Astrocytoma | II | 9 | 1 | 9 | 11.80 | 2 | 9 | c.5284A>G | p.I1762V | - | - |
| 3 | m | 32 | Protoplasmatic Astrocytoma | II | 9 | 1 | 9 | 3.89 | 2 | 9 | c.5284A>G | p.I1762V | - | - |
| 4 | m | 36 | Anaplastic Oligodendroglioma | III | 9 | 0 | 2 | 8.59 | 2 | 9 | c.5284A>G | p.I1762V | - | - |
| 5 | f | 58 | Anaplastic Astrocytoma | III | 9 | 0 | 9 | 5.27 | 1 | 1 | c.1088C>T, c.5162T>G | p.P363L, p.L1721W | - | - |
| 6 | m | 53 | Anaplastic Astrocytoma | III | 0 | 0 | 0 | 3.63 | 1 | 9 | c.5284A>G | p.I1762V | - | - |
| 7 | m | 45 | Anaplastic Astrocytoma | III | 9 | 1 | 0 | 1.59 | 2 | 9 | - | - | - | - |
| 8 | m | 32 | Anaplastic Astrocytoma | III | 9 | 1 | 0 | 3.40 | 2 | 9 | c.5162T>G, c.5284A>G | p.L1721W, p.I1762V | - | - |
| 9 | m | 33 | Anaplastic Astrocytoma | III | 9 | 1 | 9 | 11.80 | 2 | 9 | c.5284A>G | p.I1762V | - | - |
| 10 | f | 27 | Anaplastic Astrocytoma | III | 9 | 1 | 9 | 1.81 | 2 | 9 | c.1064G>A, c.5162T>G | p.G355N, p.L1721W | - | - |
| 11 | m | 24 | Anaplastic Astrocytoma | III | 2 | 1 | 0 | 5.09 | 2 | 9 | c.5162T>G | p.L1721W | - | - |
| 12 | m | 26 | Anaplastic Astrocytoma | III | 1 | 1 | 0 | 3.58 | 2 | 9 | c.5284A>G | p.I1762V | - | - |
| 13 | m | 62 | Anaplastic Astrocytoma | III | 1 | 1 | 0 | 16.93 | 2 | 9 | c.5284A>G | p.I1762V | - | - |
| 14 | f | 67 | Glioblastoma with oligo component | IV | 0 | 0 | 9 | 0.56 | 1 | 9 | c.5162T>G | p.L1721W | - | - |
| 15 | m | 65 | Glioblastoma | IV | 0 | 0 | 5 | 3.48 | 1 | 1 | c.5284A>G | p.I1762V | c.2131T>C | p.L711L |
| 16 | f | 69 | Glioblastoma | IV | 0 | 0 | 6 | 0.25 | 1 | 1 | - | - | - | - |
| 17 | m | 39 | Glioblastoma | IV | 0 | 0 | 0 | 1.33 | 1 | 1 | c.5284A>G | p.I1762V | - | - |
| 18 | m | 54 | Glioblastoma | IV | 1 | 0 | 0 | 0.08 | 2 | 1 | c.5284A>G | p.I1762V | c.2131T>C | p.L711L |
| 19 | m | 64 | Glioblastoma | IV | 1 | 0 | 4 | 0.97 | 2 | 1 | c.5284A>G | p.I1762V | c.2131T>C | p.L711L |
| 20 | f | 44 | Glioblastoma | IV | 0 | 0 | 0 | 0.28 | 2 | 1 | c.1064G>A, c.5162T>G, c.5284A>G | p.G355N, p.L1721W, p.I1762V | - | - |
| 21 | f | 64 | Glioblastoma | IV | 2 | 0 | 0 | 1.61 | 2 | 9 | c.5284A>G | p.I1762V | - | - |
| 22 | m | 68 | Glioblastoma | IV | 1 | 0 | 0 | 17.26 | 2 | 9 | - | - | - | - |
| 23 | m | 54 | Glioblastoma | IV | 0 | 0 | 9 | 0.37 | 3 | 9 | - | - | - | - |
| 24 | m | 73 | Glioblastoma | IV | 0 | 0 | 0 | 0.37 | 3 | 9 | - | - | - | - |
| 25 | m | 62 | Glioblastoma | IV | 0 | 9 | 0 | 3.23 | 3 | 9 | c.1064G>A | p.G355N | - | - |
| 26 | m | 63 | Glioblastoma | IV | 1 | 0 | 0 | 0.00 | 3 | 9 | - | - | - | - |
| 27 | f | 65 | Glioblastoma | IV | 3 | 1 | 0 | 0.74 | 3 | 9 | - | - | - | - |
| 28 | m | 70 | Glioblastoma | IV | 1 | 0 | 0 | 1.82 | 3 | 9 | - | - | - | - |
| 29 | m | 39 | Glioblastoma | IV | 0 | 0 | 0 | 0.29 | 3 | 9 | - | - | - | - |
| 30 | m | 52 | Glioblastoma | IV | 2 | 9 | 2 | 1.82 | 3 | 9 | - | - | - | - |
| 31 | m | 49 | Glioblastoma | IV | 0 | 9 | 9 | 0.10 | 3 | 9 | - | - | - | - |
| 32 | m | 78 | Glioblastoma | IV | 1 | 0 | 0 | 0.00 | 3 | 9 | - | - | - | - |
| 33 | m | 75 | Glioblastoma | IV | 1 | 9 | 0 | 1.22 | 3 | 9 | - | - | - | - |
| 34 | m | 65 | Glioblastoma | IV | 0 | 0 | 0 | 3.33 | 3 | 9 | - | - | - | - |
| 35 | f | 45 | Glioblastoma | IV | 0 | 0 | 6 | 4.32 | 3 | 9 | - | - | - | - |
| 36 | m | 64 | Glioblastoma | IV | 1 | 0 | 1 | 2.35 | 3 | 9 | - | - | - | - |
| 37 | m | 72 | Glioblastoma | IV | 1 | 0 | 0 | 0.59 | 3 | 9 | - | - | - | - |
| 38 | f | 60 | Glioblastoma | IV | 2 | 0 | 0 | 0.16 | 3 | 9 | - | - | - | - |
| 39 | m | 54 | Glioblastoma | IV | 0 | 0 | 4 | 1.10 | 3 | 9 | - | - | - | - |
| 40 | f | 48 | Glioblastoma | IV | 1 | 0 | 0 | 3.56 | 3 | 9 | - | - | - | - |
| 41 | m | 37 | Glioblastoma | IV | 1 | 0 | 1 | 0.58 | 3 | 9 | - | - | - | - |
| 42 | m | 50 | Glioblastoma | IV | 1 | 0 | 3 | 0.16 | 3 | 9 | - | - | - | - |
| 43 | f | 50 | Glioblastoma | IV | 0 | 0 | 4 | 0.49 | 3 | 9 | - | - | - | - |
| 44 | m | 56 | Glioblastoma | IV | 1 | 0 | 4 | 0.78 | 3 | 9 | - | - | - | - |
| 45 | f | 9 | Glioblastoma | IV | 1 | 0 | 4 | 0.04 | 3 | 9 | - | - | - | - |
| 46 | f | 33 | Glioblastoma | IV | 0 | 0 | 0 | 0.32 | 3 | 9 | - | - | - | - |
| 47 | m | 56 | Glioblastoma | IV | 0 | 0 | 0 | 0.02 | 3 | 9 | - | - | - | - |
| 48 | m | 68 | Glioblastoma | IV | 0 | 0 | 2 | 0.01 | 3 | 9 | - | - | - | - |
| 49 | m | 73 | Glioblastoma | IV | 1 | 0 | 0 | 0.02 | 3 | 9 | c.1064G>A, c. 652G>A | p.G355N, p.V218M | - | - |
| 50 | m | 73 | Glioblastoma | IV | 2 | 0 | 3 | 0.00 | 3 | 9 | - | - | - | - |
Brain tissue specimen that served as control samples were provided by the Neurobiobank Munich (NBM) and were collected and clinically as well as neuropathologically characterized according to the standard protocols of the NBM established by BrainNet Europe and BrainNet Germany. Written informed consent was obtained according to the guidelines of the local ethics committee. Mean age of patients was 64 years. The female to male ratio was 8 to 11. Detailed information on patients can be found in Table 2.
Detailed information on control samples used in this study. We included 19 control brain samples with mean age of 64 years. The female to male ratio was 8 to 11. TET2 and TET3 analysis: 1: exploratory set; 2: validation set; 3: verification set; 9: not analysed.
| Case | Sex | Age [y] | 5hmC positive cells [%] | TET2 analysis | TET3 analysis | TET2 sequence variation | TET2 variation in amino acid sequence | TET3 sequence variation | TET3 variation in amino acid sequence |
|---|---|---|---|---|---|---|---|---|---|
| 1 | f | 46 | 84.38 | 1 | 1 | c.5284A>G | p.I1762V | c.2131T>C | p.L711L |
| 2 | f | 58 | 41.11 | 1 | 1 | c.5284A>G | p.I1762V | - | - |
| 3 | f | 40 | 79.66 | 2 | 9 | c.5162T>G, c.5284A>G | p.L1721W, p.I1762V | - | - |
| 4 | m | 60 | 81.21 | 2 | 9 | c.5284A>G | p.I1762V | - | - |
| 5 | m | 87 | 54.32 | 2 | 9 | c.5167C>T, c.5333A>G | p.P1723S, p.H1778R | - | - |
| 6 | m | 61 | 55.46 | 2 | 9 | - | - | - | - |
| 7 | f | 85 | 55.05 | 2 | 9 | c.5284A>G | p.I1762V | - | - |
| 8 | m | 46 | 64.29 | 2 | 9 | - | - | - | - |
| 9 | f | 77 | 82.21 | 2 | 9 | c.5162T>G, c.5284A>G | p.L1721W, p.I1762V | - | - |
| 10 | m | 74 | 83.41 | 2 | 9 | c.5284A>G | p.I1762V | - | - |
| 11 | f | 83 | 80.33 | 2 | 9 | c.5284A>G | p.I1762V | - | - |
| 12 | f | 77 | 81.32 | 3 | 9 | - | - | - | - |
| 13 | m | 75 | 77.77 | 3 | 9 | - | - | - | - |
| 14 | m | 63 | 82.14 | 3 | 9 | - | - | - | - |
| 15 | m | 53 | 83.11 | 3 | 9 | - | - | - | - |
| 16 | m | 71 | 86.66 | 3 | 9 | - | - | - | - |
| 17 | f | 55 | 78.89 | 3 | 9 | - | - | - | - |
| 18 | m | 60 | 84.19 | 3 | 9 | - | - | - | - |
| 19 | m | 54 | 54.17 | 3 | 9 | c.1064G>A | p.G355N | - | - |
Molecular genetic characterization of gliomas
Determinations of IDH1 (isocitrate dehydrogenase 1) and IDH2 (isocitrate dehydrogenase 2) mutations were performed using the pyrosequencing technique as described previously [12, 28]. To detect IDH1 mutations, we amplified a 88 bp long DNA fragment that includes the mutation hot spot at codon 132 using the primer pair 5'-biotin-AAAAATATCCCCCGGCTTG-3' and 5'-TGCCAACATGACTTACTTGATCC-3' and the sequencing primer 5'-TGATCCCCATAAGCA-3'. To detect IDH2 mutations, we amplified a 83 bp long DNA fragment including the mutation hot spot at codon 172 using the primer pair 5'-TCCCACGCCTAGTCCCTG-3' and 5'-biotin-CTCTCCACCCTGGCCTACC-3' and the sequencing primer 5′-AGCCCATCACCATTG-3′. MGMT promoter methylation was determined as described previously [29]. DNA was bisulfite treated using the EpiTect Bisulfite kit (Qiagen, Hilden Germany) according to the manufacturer's protocol. Afterwards, we performed methylation specific PCR (MSP) and Sanger sequencing. MSP was performed using the primer pair 5'-TTTGTGTTTTGATGTTTGTAGGTTTTTGT-3' and 5'-AACTCCACACTCTTCCAAAAACAAAACA-3' for amplification of the unmethylated promoter region and the primer pair 5'-TTTCGACGTTCGTAGGTTTTCGC-3' and 5'-GCACTCTTCCGAAAACGAAACG-3' for amplification of the methylated promoter region. Sanger sequencing of the MGMT promoter region was performed by amplifying a 316 bp long PCR product that covers 25 CpGs using the primer pair 5'-ATGTTGGGATAGTTCGCGTTTTTAGA-3' and 5'-CCAATCCACAATCACTACTTC-3'. As described previously we considered the MGMT promoter as methylated if more than half of the CpGs were methylated (i.e. 13 of 25), as partially methylated if 9 to 12 CpGs were methylated and as unmethylated if less than 9 CpGs were methylated [29]. LOH1p/19q (loss of heterozygousity of chromosome 1p and 19q) was detected as described previously [30]. In brief, we used the 5 microsatellite markers D1S1608, D1S548, D1S1592, D1S1184, and D1S1161 for the short arm of chromosome 1 (1p) and the 5 microsatellite markers D19S433, D19S431, D19S718, D19S559, and D19S601 for the long arm of chromosome 19 (19q). PCR amplicons of microsatellite markers were subsequently loaded onto high resolution SYBR Gold gels for interpretation [30].
DNA extraction
Representative H&E stained cryo-slides were prepared and microscopically assessed. For further analysis, only samples with solid, viable tumor consisting of at least 90% tumor cells were included. DNA extraction was performed on 50 mg of micro-dissected tumor tissue. In case of control brain tissue, 50 mg of frozen brain tissue was used in this study. DNA extractions were performed using the QIAamp DNA Micro Kit (Qiagen, Hilden, Germany) according to the manufacturer's protocol.
Polymerase chain reaction (PCR)
PCR was performed using the HotStarTaq DNA Polymerase Master Mix Kit (Qiagen, Hilden, Germany) according to the manufacturer's protocol and standard PCR setups. In order to identify TET2 mutations, we used the primers published by Delhommeau et al. [16], in order to identify TET3 mutations, we used the primers published by Abdel-Wahab et al. [15], respectively.
Sequencing analysis
Sequencing reactions of PCR amplified DNA fragments were performed using standard protocols. In brief we purified PCR fragments using the QIAquick PCR Purification Kit (Qiagen, Hilden, Germany) and performed a subsequent amplification using the BigDye Terminator V3.1 Cycle Sequencing kit (life technologies, Carlsbad, USA). The product was purified using the DyeEX 2.0 Spin Kit (Qiagen, Hilden, Germany) and sequenced on a 3130 Genetic Analyzer (life technologies, Carlsbad, USA). Data analysis was performed using SeqScape Software (life technologies, Carlsbad, USA), ApE (http://biologylabs.utah.edu/jorgensen/wayned/ape/) and Chromas Lite (http://technelysium.com.au/). Functional effects of identified alterations were analysed using PolyPhen-2 algorithm [31].
Quantification of 5hmC amount
Quantification of 5hmC amount was performed as described previously [9, 12]. In brief, 4 µm FFPE (formaline-fixed, paraffin-embedded) tissue slides were deparaffinized and treated with 2N HCl. After pre-incubation with 3% I-Block Protein-Based Blocking Reagent (Applied Biosystems, Darmstadt, Germany) sections were incubated with polyclonal rabbit anti-5-hydroxymethylcytosine antibody (1:1,000, Active Motif, Rixensart, Belgium) for 18 hours at 4°C. Afterwards, sections were treated with Super Sensitive Polymer-HRP Detection System (BioGenix, San Ramon, USA), followed by incubation in DAB (3,3'-diaminobenzidine) and counter stained using hemalum. Quantification of positive nuclei was performed using an Olympus BX50 microscope (Olympus, Tokyo, Japan) and a Color View III camera system (Soft Imaging System, Münster, Germany) using 10x and 20x objectives (Olympus, Tokyo, Japan). Representative areas without technical artefacts were selected for quantifications.
Results
Genetic Analysis of Ten-Eleven-Translocation 2 and 3 in Glioma
In this study, we analysed 50 human glioma of WHO-Grades II-IV, including 3 WHO-Grade II gliomas, 10 WHO-Grade III gliomas and 37 WHO-Grade IV glioblastomas (GBM) on genetic alterations of the TET2 and TET3 genes. Sequencing was performed by a step-wise approach, including (1) an exploratory set, (2) a validation set and (3) a verification set (Table 1). Glioma samples were randomly matched to the three test sets. Samples of the exploratory set were subjected to complete sequencing of TET2 or TET3, respectively, samples of the validation set were subjected to sequencing of only genomic fragments that showed alterations in the exploratory set, and samples of the verification set were subjected to targeted re-sequencing of most abundant genetic variations of the exploratory and validation set.
Using this approach, we identified 5 different TET2 (p.V218M, p.G355N, p.P363L, p.L1721W, p.I1762V) variants in 21 glioma samples (Table 1, Figure 1). 29 gliomas did not show TET2 alterations. Interestingly, no variant was located within one of the two conserved domains of TET2 (Figure 2). The most frequent alteration was the p.I1762V exchange that occurred in 50 % of all alterations. Further details on the distribution of alterations can be found in Figure 3a.
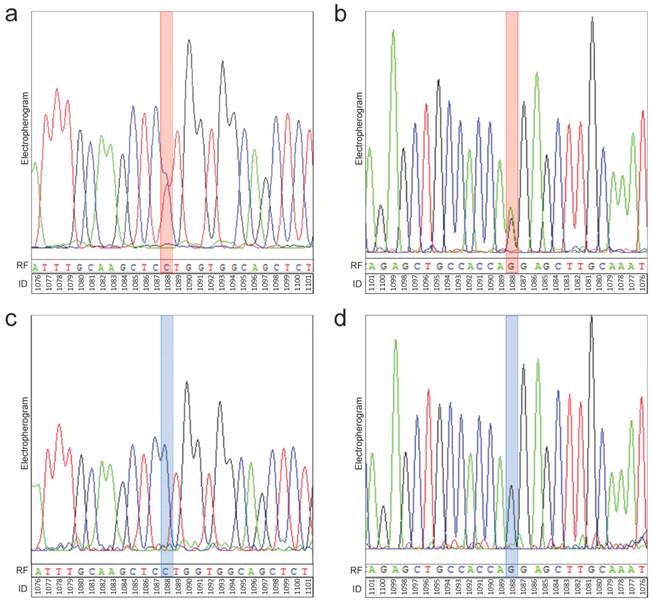
Representative electropherograms of TET2 sequencing. Identification of alterations was performed by classical dideoxy sequencing. Thereby, sequencing of both the forward and reverse strand was performed. As representative electropherograms, the sequencing analysis of TET2 gene of glioma sample # 5 (anaplastic astrocytoma, WHO-Grade III) (a, b) is shown. There is a heterozygous position within the TET2 sequence (red box) both in the forward (a) and reverse (b) sequencing analysis. This heterozygous position corresponds with position 1088 of the DNA sequence and results in a CCT to CTT exchange (c.1088C>T) (a). After translation there is a proline (P) to leucine (L) exchange in the amino acid sequence resulting in a p.P336L mutation of TET2. The analysis of the reverse strand confirms the identified heterozygosity at position 1088 (AAG to AGG exchange in the reverse complement sequence corresponding to a CTT to CCT exchange in the forward sequencing reaction). Representative electropherograms of the sequencing analysis of a control sample (control case # 5) shows that there is no sequence variation at position 1088 (blue box) in the forward (c) and reverse (d) sequencing analysis of the TET2 gene. Electropherograms: black peaks represent guanine (G), blue peaks represent cytosine (C), green peaks represent adenine (A) and red peaks represent thymine (T). RF: reference sequence. ID: identifier of base position in gene.
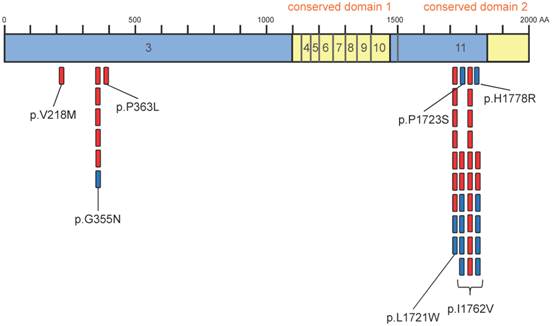
Overview of identified variants within the coding region of TET2. Sequencing of 50 glioma and 19 controls revealed 28 TET2 variants in glioma and 13 variants in controls. Interestingly, most variants accumulate within two short fragments of TET2 (355-363 and 1723-1778). There are no alterations within the 2 conserved domains. Indicated is the coding region of TET2 (exons 3 to 11). Conserved domains 1 and 2 are shown in yellow. Variants occurring in glioma are indicated by red marks, variants occurring in controls are indicated by blue marks.
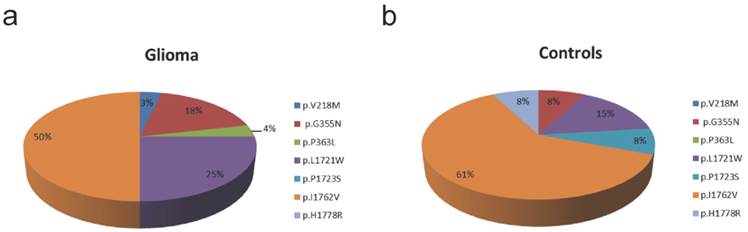
Frequency of TET2 variants in glioma and controls. a) In glioma we identified 5 different TET2 variants (p.V218M, p.G355N, p.P363L, p.L1721W, p.I1762V) with p.I1762V being most frequent one. b) In control tissue, we identified 5 different TET2 variants (p.G355N, p.L1721W, p.P1723S, p.I1762V, p.H1778R) with p.I1762V being the most frequent variant.
An analysis of TET3 showed three glioma samples with synonymous p.L711L alterations. We did not identify any non-synonymous alterations within TET3.
Genetic Analysis of Ten-Eleven-Translocation 2 and 3 in Brain Tissue
To evaluate the frequency of identified TET alterations in healthy human brain tissue, we performed an equivalent analysis as performed in case of glioma using 19 human control brain samples.
In case of TET2 we found 5 different TET2 variants (p.G355N, p.L1721W, p.P1723S, p.I1762V, p.H1778R) within 13 control brain samples (Table 2). Six controls did not show any TET2 alterations. No variant was located within one of the two conserved domains of TET2 (Figure 2). In control tissue, the p.I1762V exchange was the most frequent variation that was found in 61 % of all alterations. Further details on the distribution of alterations can be found in Figure 3b.
An analysis of TET3 showed one control sample with a synonymous p.L711L. We did not identify any non-synonymous alterations within TET3.
Quantification of 5hmC Amount and Evaluation of Functional Effects of Identified Alterations
In order to evaluate the biological relevance of TET alterations identified in glioma we quantified the amount of 5hmC in all 50 investigated glioma and in all 19 healthy control samples.
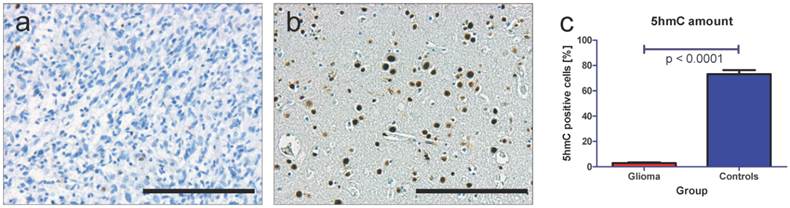
Using immunohistochemistry (IHC) and specific antibodies against 5hmC we found that glioma almost stain negative for 5hmC (Figure 4a). These data are in good correlation with our previous findings [12, 32]. In contrast to this finding, healthy brain tissue shows numerous 5hmC positive cells (Figure 4b). These data also correlate well with our previous findings [9, 12]. The quantification of 5hmC amount in all 69 samples revealed mean amounts of 2.88 % 5hmC positive cells in glioma and 73.14 % 5hmC positive cells in healthy brain tissue (p < 0.0001, unpaired t-test, Figure 4c).
Quantification of 5hmC amount in glioma and control tissue. Using specific antibodies against 5hmC we see only very few positively stained cells in human glioma samples (a) but numerous positively stained cells in healthy control brain tissue (b). Quantification of 5hmC positive cells in all 50 glioma and 19 control samples reveals a significant loss of 5hmC in glioma (mean of 2.88 % 5hmC positive cells) compared to healthy brain tissue (mean of 73.14 % 5hmC positive cells, p < 0.0001, unpaired t-test). These results are well in line with our previous findings [12, 32]. Indicated are mean and standard error of the mean (SEM). Scale bar: 200µm.
A sample specific comparison of the presence of TET2 alterations with the amount of 5hmC did not show correlations of 5hmC amounts and TET2 altered gliomas on individual level (Figure 5a). In healthy brain samples, there was no sample specific correlation of low 5hmC amounts and TET2 alterations (Figure 5b).
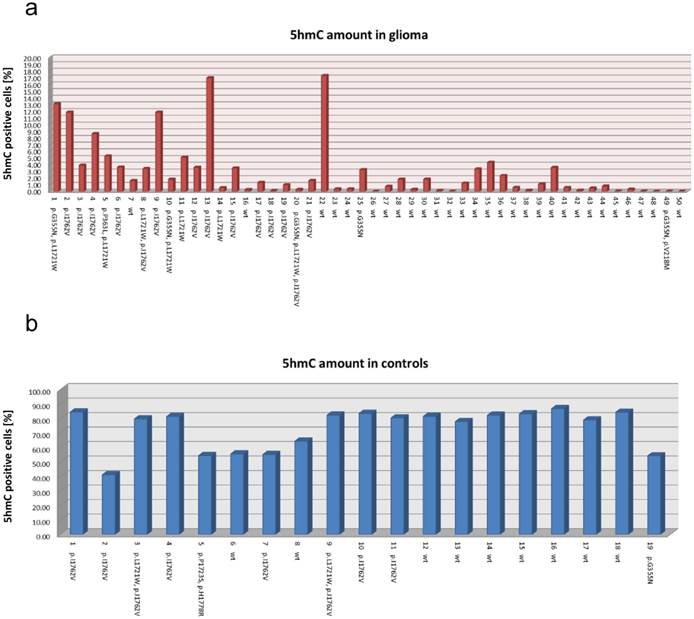
5hmC amount in all 69 investigated specimens. Bar graph representing the individual amounts of 5hmC positive cells in all 50 glioma samples (a) and in all 19 control samples (b). Additionally, the case numbers and all identified TET2 alterations are indicated at the bottom of the bar graphs. Interestingly, the amount of 5hmC is independent of TET2 alterations. wt: wild type.
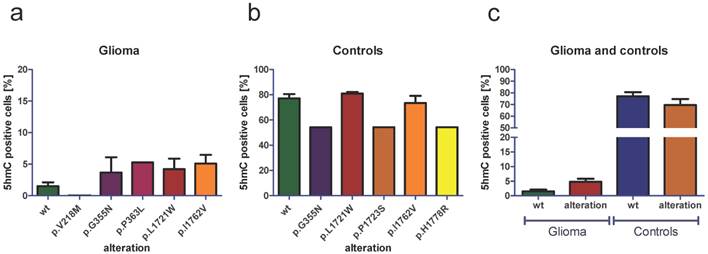
In order to determine the general effect of TET2 alterations on 5hmC amounts, we next performed alteration-specific analysis of 5hmC amounts. We found that there were no general correlations of TET2 alterations and low 5hmC amounts: TET2 wild type (wt) glioma showed mean amounts of 1.50 % 5hmC positive cells (Figure 6a). Glioma with the TET2 p.V218M alteration (one sample) showed 0.02 % 5hmC positive cells, glioma with the p.G355N alteration (five samples) showed mean amounts of 3.68 % 5hmC positive cells, glioma with the p.P363L alteration (one sample) showed 5.27 % 5hmC positive cells, glioma with the p.L1721W alteration (seven samples) showed mean amounts of 4.21 % 5hmC positive cells and glioma with the p.I1762V alteration (14 samples) showed mean amounts of 5.10 % 5hmC positive cells (Figure 6a). In control tissue, TET2 wild type samples showed mean amounts of 77.10 % 5hmC positive cells (Figure 6b). Samples with the TET2 p.G355N alteration (one sample) showed 54.17 % 5hmC positive cells, samples with the p.L1721W alteration (two cases) showed mean amounts of 80.94 % 5hmC positive cells, cases with the p.P1723S alteration (one sample) showed 54.32 % 5hmC positive cells, cases with the p.I1762V alteration (eight cases) showed mean mounts of 73.42 % 5hmC positive cells and cases with the p.H1778R alteration (one case) showed 54.32 % 5hmC positive cells (Figure 6b). Comparing the amount of 5hmC in all TET2 wild type glioma with the amount of 5hmC in all TET2 altered glioma showed slightly increased amounts of 5hmC in glioma with TET2 alteration (Figure 6c). A comparison of the 5hmC amounts in all healthy brain tissue without TET2 alteration with the samples with TET2 alteration showed slightly decreased amounts of 5hmC in samples with TET2 alteration (Figure 6c).
Analysis of 5hmC amount depending on distinct TET2 alterations. An analysis of the 5hmC amount in glioma without TET2 alteration (wt) and with the TET2 alterations p.V218M, p.G355N, p.P363L, p.L1721W and p.I1762V demonstrate that TET2 alterations are not associated with decreased amounts of 5hmC (a). Analysing the amount of 5hmC in control brain tissue without TET2 alterations (wt) and with the TET2 alterations p.G355N, p.L1721W, p.P1723S, p.I1762V and p.H1778R, we do not find that TET2 alterations are associated with a statistically significant loss of 5hmC (b). A comparison of 5hmC amount in TET2 wild type glioma with the amount of 5hmC in all TET2 altered glioma shows slightly increased amount of 5hmC in TET2 altered glioma (c). In healthy brain tissue, we see slightly decreased amounts of 5hmC in specimens with TET2 alterations compared with TET2 wild type specimens (c). Indicated are mean and standard error of the mean (SEM). wt: wild type.
Additionally, we performed a prediction analysis of functional effects using the PolyPhen-2 (Polymorphism Phenotyping v2) algorithm via the online web server [31]. The PolyPhen-2 software predicts the possible impact of amino acid substitutions on the function and stability of human proteins by the use of structural and comparative evolutionary considerations [31]. Thereby, the algorithm estimates the probability of missense mutations to be damaging and integrates the UCSC Genome Browser's human genome annotations and MultiZ multiple alignments of vertebrate genomes with the human genome [31]. After processing a query, PolyPhen-2 reports a score ranging from 0 to 1 indicating the likelihood of a functional effect of the analysed mutation. Thereby, low scores (near 0) indicate that the mutations is likely to be not of functional importance (“benign”) while high scores (near 1) indicate that the mutation is likely to be of functional importance (“possibly damaging” or “probably damaging”) [31].
The analysis showed that the p.V218M variant found in 2 % of glioma has a predicted “benign” effect (PolyPhen-2 score 0.000) (Table 3). The p.G355N variant that we found in 10 % of glioma has a predicted “possibly damaging” effect (PolyPhen-2 score 0.795). The p.P363L variant that we found in 2 % of glioma has a predicted “benign” effect (PolyPhen-2 score 0.0421). The p.L1721W variant that we found in 14 % of glioma has a predicted “possibly damaging” effect (PolyPhen-2 score 0.794). The p.I1762V variant that we found in 28 % of glioma has a predicted “benign” effect (PolyPhen-2 score 0.012).
Analysis of functional effects of identified glioma-associated alterations. Using the PolyPhen-2 algorithm we analysed the functional effect of all identified glioma-associated TET2 alterations. Indicated are the frequencies of all identified variants in glioma and control tissue as well as the evaluation of the biological effect of the variants using the PolyPhen-2 algorithm.
| Variation | Frequency in glioma | Frequency in controls | PolyPhen-2 analysis of variation |
|---|---|---|---|
| p.V218M | 2 % (1/50) | 0 % (0/19) | benign (0.000) |
| p.G355N | 10 % (5/50) | 5.26 % (1/19) | possibly damaging (0.795) |
| p.P363L | 2 % (1/50) | 0 % (0/19) | benign (0.0421) |
| p.L1721W | 14 % (7/50) | 10.53 % (2/19) | possibly damaging (0.794) |
| p.I1762V | 28 % (14/50) | 42.11 % (8/19) | benign (0.012) |
Discussion
Human gliomas are characterized by a disturbed epigenomic landscape [12, 32]. Especially in the context of 5hmC, a further base modification of cytosine besides 5mC, high grade glioma (WHO-Grade III and IV) show a tremendous loss of 5hmC compared to normal brain tissue [12, 32]. Till date, the causal mechanisms leading to this disturbance remain unclear [12, 32].
5hmC is generated out of 5mC and can be further converted to 5fC and 5caC representing an active demethylation pathway [8]. These steps are catalyzed by the ten-eleven-translocation methylcytosine dioxygenases TET [8]. Thus, a disturbance of the conversion of 5mC to 5hmC may explain the loss of 5hmC in glioma.
In 2011, Xu et al. described that the alpha-ketoglutarate (aKG) dependent dioxigenases TET can be competitively inhibited by 2-hydroxyglutatrate (2-HG) [33]. As mutations of the isocitrate-dehydrogenases (IDH) lead to the production of 2-HG it is likely that tumors with IDH mutations show a loss of 5hmC due to inhibition of TET enzyme activity. In WHO-Grade II and III gliomas, IDH-mutations are frequent events, thus these IDH-mutations may explain the loss of 5hmC in WHO-Grade II and III glioma. Investigations examining the effect of IDH-mutations and the amount of 5hmC revealed that, indeed, IDH mutations result in decreased levels of 5hmC in WHO-Grade II diffuse astrocytoma and WHO-Grade III anaplastic astrocytoma [12]. But in WHO-Grade IV glioblastoma (GBM) there was no correlation of IDH-mutation status and amount of 5hmC detectable [12, 32]. Furthermore, GBMs show even lower amounts of 5hmC compared with WHO-Grade II and III glioma [12]. This emphasizes, that there are other disturbances occurring in IDH unmutated glioma that result in a loss of 5hmC.
One possible explanation might be an inappropriate function of TET enzymes due to mutations. Recent research in myeloid malignancies showed that there is indeed an associated of TET mutations with the loss of 5hmC [15-17]: Heterozygous deletions and mutations of TET2 were found in a wide range of myeloid cancers, such as chronic myelomonocytic leukemia, acute myeloic leukemia and myelodysplastic syndrome [16, 21, 22]. Additionally, studies revealed that TET2 mutations are accompanied with decreased 5hmC amounts [20]. As glioma also show a reduction of 5hmC amounts, the question rises if this tremendous loss of 5hmC in glioma is caused by TET mutations similar to the phenomenon observed in myeloid tumors. But till date, there are no sequencing data available for TET enzymes in human glioma. Thus, this study is of fundamental importance representing the first comprehensive analysis addressing this question in human glioma.
Our genetic characterization of TET2 and TET3 alterations in 50 glioma samples and 19 control brain samples revealed that, indeed TET2 alterations occur in glioma but the comparison with control tissue showed, that there are also genetic alteration in healthy brain tissue (Figure 3). Additionally, no identified variant was located within the conserved domains 1 or 2 of TET2 (Figure 2). There were no non-synonymous alterations within TET3. Additionally, we quantified the amount of 5hmC in glioma and control specimens and found that glioma exhibited significantly reduced amounts of 5hmC compared with healthy brain tissue (Figure 4). These results are well in line with our previous findings [12, 32]. A correlation of 5hmC amount with identified TET2 alterations demonstrated that TET2 alterations are not associated with low 5hmC amounts (Figures 5 and 6). These findings emphasize that in glioma - contrasting the findings in myeloid malignancies [15, 16, 34, 35] - TET mutations are not causal for the loss of 5hmC. Thus, other molecular events may occur in glioma leading to the decrease of 5hmC amounts in brain tumors. One possible mechanism may be a nuclear exclusion of TET proteins in glioma. Müller et al. described that nuclear exclusion of TET1 is associated with low amounts of 5hmC in some tumors [36]. However, till date there are no localization studies for TET2 and TET3 available that investigate, if there is a nuclear exclusion of TET2 and TET3 in glioma that might explain low amounts of 5hmC. Additionally, the TET2 alterations found in the current study may also influence the localization of the TET2 protein. However, additional studies analysing the functional consequences of TET2 alterations on the cellular localization of the protein will be necessary.
Another possible mechanism affecting the 5hmC amount in glioma might be a reduced TET gene expression resulting in lower amounts of functional protein in glioma. As TET proteins are responsible for the conversion of 5mC to 5hmC [8], reduced transcription levels of TET may result in decreased 5hmC amounts. Lian et al. addressed this question in malignant melanoma and found that loss of 5hmC can be explained by decreased expressions of TET genes [14] in skin tumors: Whereas TET1, TET2 and TET3 are highly transcribed in nevi, all three genes are significantly down-regulated in malignant melanoma. These data demonstrate that additional studies will be necessary that investigate TET gene expression levels in glioma.
Further possible mechanism that may be responsible for the loss of 5hmC might be a decreased TET enzyme activity (independent of IDH mutations), an inhibition of DNA accessibility for TET enzymes due to aberrant DNA occupancy or an increased demethylation rate disturbing the homeostasis between methylation and demethylation processes.
In summary, our findings demonstrate for the first time that, in contrast to myeloid malignancies, loss of 5hmC in human glioma is not caused by functionally relevant TET alterations. Thus, other molecular mechanisms, such as aberrant cellular TET localizations, inhibition of DNA accessibility or aberrant TET gene expressions may be responsible for the loss of 5hmC and the disruption of the epigenome in glioma. Thus, further research addressing these new challenges will be necessary to understand the complex mechanisms leading to the disturbed epigenomic landscape in human brain tumors.
Acknowledgements
We thank the Brain Tumor Bank of the Center for Neuropathology and the BrainBank Munich for providing tissue specimens.
Competing Interests
None declared.
References
1. Hegi ME, Diserens AC, Gorlia T, Hamou MF, de Tribolet N, Weller M. et al. MGMT gene silencing and benefit from temozolomide in glioblastoma. The New England journal of medicine. 2005;352:997-1003 doi:10.1056/NEJMoa043331
2. Tabatabai G, Stupp R, van den Bent MJ, Hegi ME, Tonn JC, Wick W. et al. Molecular diagnostics of gliomas: the clinical perspective. Acta neuropathologica. 2010;120:585-92 doi:10.1007/s00401-010-0750-6
3. Weller M, Stupp R, Hegi ME, van den Bent M, Tonn JC, Sanson M. et al. Personalized care in neuro-oncology coming of age: why we need MGMT and 1p/19q testing for malignant glioma patients in clinical practice. Neuro-oncology. 2012;14(Suppl 4):iv100-iv8 doi:10.1093/neuonc/nos206
4. Weller M, Stupp R, Reifenberger G, Brandes AA, van den Bent MJ, Wick W. et al. MGMT promoter methylation in malignant gliomas: ready for personalized medicine? Nature reviews Neurology. 2010;6:39-51 doi:10.1038/nrneurol.2009.197
5. Wick W, Hartmann C, Engel C, Stoffels M, Felsberg J, Stockhammer F. et al. NOA-04 randomized phase III trial of sequential radiochemotherapy of anaplastic glioma with procarbazine, lomustine, and vincristine or temozolomide. J Clin Oncol. 2009;27:5874-80 doi:10.1200/JCO.2009.23.6497
6. Sturm D, Witt H, Hovestadt V, Khuong-Quang DA, Jones DT, Konermann C. et al. Hotspot mutations in H3F3A and IDH1 define distinct epigenetic and biological subgroups of glioblastoma. Cancer cell. 2012;22:425-37 doi:10.1016/j.ccr.2012.08.024
7. Kriaucionis S, Heintz N. The nuclear DNA base 5-hydroxymethylcytosine is present in Purkinje neurons and the brain. Science. 2009;324:929-30 doi:10.1126/science.1169786
8. Branco MR, Ficz G, Reik W. Uncovering the role of 5-hydroxymethylcytosine in the epigenome. Nature reviews Genetics. 2012;13:7-13 doi:10.1038/nrg3080
9. Kraus TF, Guibourt V, Kretzschmar HA. 5-Hydroxymethylcytosine, the "Sixth Base", during brain development and ageing. Journal of neural transmission. 2014 doi:10.1007/s00702-014-1346-4
10. Spruijt CG, Gnerlich F, Smits AH, Pfaffeneder T, Jansen PW, Bauer C. et al. Dynamic readers for 5-(hydroxy)methylcytosine and its oxidized derivatives. Cell. 2013;152:1146-59 doi:10.1016/j.cell.2013.02.004
11. Mellen M, Ayata P, Dewell S, Kriaucionis S, Heintz N. MeCP2 Binds to 5hmC Enriched within Active Genes and Accessible Chromatin in the Nervous System. Cell. 2012;151:1417-30 doi:10.1016/j.cell.2012.11.022
12. Kraus TF, Globisch D, Wagner M, Eigenbrod S, Widmann D, Munzel M. et al. Low values of 5-hydroxymethylcytosine (5hmC), the "sixth base," are associated with anaplasia in human brain tumors. International journal of cancer Journal international du cancer. 2012;131:1577-90 doi:10.1002/ijc.27429
13. Munzel M, Globisch D, Carell T. 5-Hydroxymethylcytosine, the sixth base of the genome. Angewandte Chemie. 2011;50:6460-8 doi:10.1002/anie.201101547
14. Lian CG, Xu Y, Ceol C, Wu F, Larson A, Dresser K. et al. Loss of 5-hydroxymethylcytosine is an epigenetic hallmark of melanoma. Cell. 2012;150:1135-46 doi:10.1016/j.cell.2012.07.033
15. Abdel-Wahab O, Mullally A, Hedvat C, Garcia-Manero G, Patel J, Wadleigh M. et al. Genetic characterization of TET1, TET2, and TET3 alterations in myeloid malignancies. Blood. 2009;114:144-7 doi:10.1182/blood-2009-03-210039
16. Delhommeau F, Dupont S, Della Valle V, James C, Trannoy S, Masse A. et al. Mutation in TET2 in myeloid cancers. The New England journal of medicine. 2009;360:2289-301 doi:10.1056/NEJMoa0810069
17. Konstandin N, Bultmann S, Szwagierczak A, Dufour A, Ksienzyk B, Schneider F. et al. Genomic 5-hydroxymethylcytosine levels correlate with TET2 mutations and a distinct global gene expression pattern in secondary acute myeloid leukemia. Leukemia. 2011;25:1649-52 doi:10.1038/leu.2011.134
18. Gambichler T, Sand M, Skrygan M. Loss of 5-hydroxymethylcytosine and ten-eleven translocation 2 protein expression in malignant melanoma. Melanoma research. 2013;23:218-20 doi:10.1097/CMR.0b013e32835f9bd4
19. Jawert F, Hasseus B, Kjeller G, Magnusson B, Sand L, Larsson L. Loss of 5-hydroxymethylcytosine and TET2 in oral squamous cell carcinoma. Anticancer research. 2013;33:4325-8
20. Ko M, Huang Y, Jankowska AM, Pape UJ, Tahiliani M, Bandukwala HS. et al. Impaired hydroxylation of 5-methylcytosine in myeloid cancers with mutant TET2. Nature. 2010;468:839-43 doi:10.1038/nature09586
21. Mohamedali AM, Smith AE, Gaken J, Lea NC, Mian SA, Westwood NB. et al. Novel TET2 mutations associated with UPD4q24 in myelodysplastic syndrome. Journal of clinical oncology: official journal of the American Society of Clinical Oncology. 2009;27:4002-6 doi:10.1200/JCO.2009.22.6985
22. Langemeijer SM, Kuiper RP, Berends M, Knops R, Aslanyan MG, Massop M. et al. Acquired mutations in TET2 are common in myelodysplastic syndromes. Nature genetics. 2009;41:838-42 doi:10.1038/ng.391
23. Tefferi A, Lim KH, Abdel-Wahab O, Lasho TL, Patel J, Patnaik MM. et al. Detection of mutant TET2 in myeloid malignancies other than myeloproliferative neoplasms: CMML, MDS, MDS/MPN and AML. Leukemia. 2009;23:1343-5 doi:10.1038/leu.2009.59
24. Tefferi A, Lim KH, Levine R. Mutation in TET2 in myeloid cancers. The New England journal of medicine. 2009;361:1117. doi:10.1056/NEJMc091348
25. Louis DN, Ohgaki H, Wiestler OD, Cavenee WK, Burger PC, Jouvet A. et al. The 2007 WHO classification of tumours of the central nervous system. Acta neuropathologica. 2007;114:97-109 doi:10.1007/s00401-007-0243-4
26. Crocetti E, Trama A, Stiller C, Caldarella A, Soffietti R, Jaal J. et al. Epidemiology of glial and non-glial brain tumours in Europe. European journal of cancer. 2012;48:1532-42 doi:10.1016/j.ejca.2011.12.013
27. Dolecek TA, Propp JM, Stroup NE, Kruchko C. CBTRUS statistical report: primary brain and central nervous system tumors diagnosed in the United States in 2005-2009. Neuro-oncology. 2012;14(Suppl 5):v1-49 doi:10.1093/neuonc/nos218
28. Kraus TF, Greiner A, Guibourt V, Lisec K, Kretzschmar HA. Identification of Stably Expressed lncRNAs as Valid Endogenous Controls for Profiling of Human Glioma. Journal of Cancer. 2015;6:111-9 doi:10.7150/jca.10867
29. Thon N, Eigenbrod S, Grasbon-Frodl EM, Lutz J, Kreth S, Popperl G. et al. Predominant influence of MGMT methylation in non-resectable glioblastoma after radiotherapy plus temozolomide. Journal of neurology, neurosurgery, and psychiatry. 2011;82:441-6 doi:10.1136/jnnp.2010.214593
30. Thon N, Eigenbrod S, Grasbon-Frodl EM, Ruiter M, Mehrkens JH, Kreth S. et al. Novel molecular stereotactic biopsy procedures reveal intratumoral homogeneity of loss of heterozygosity of 1p/19q and TP53 mutations in World Health Organization grade II gliomas. Journal of neuropathology and experimental neurology. 2009;68:1219-28 doi:10.1097/NEN.0b013e3181bee1f1
31. Adzhubei I, Jordan DM, Sunyaev SR. Predicting functional effect of human missense mutations using PolyPhen-2. Current protocols in human genetics. 2013 Chapter7: Unit7 20. doi:10.1002/0471142905.hg0720s76
32. Kraus TF, Kolck G, Greiner A, Schierl K, Guibourt V, Kretzschmar HA. Loss of 5-hydroxymethylcytosine and intratumoral heterogeneity as an epigenomic hallmark of glioblastoma. Tumour biology: the journal of the International Society for Oncodevelopmental Biology and Medicine. 2015 doi:10.1007/s13277-015-3606-9
33. Xu W, Yang H, Liu Y, Yang Y, Wang P, Kim SH. et al. Oncometabolite 2-hydroxyglutarate is a competitive inhibitor of alpha-ketoglutarate-dependent dioxygenases. Cancer cell. 2011;19:17-30 doi:10.1016/j.ccr.2010.12.014
34. Figueroa ME, Abdel-Wahab O, Lu C, Ward PS, Patel J, Shih A. et al. Leukemic IDH1 and IDH2 mutations result in a hypermethylation phenotype, disrupt TET2 function, and impair hematopoietic differentiation. Cancer cell. 2010;18:553-67 doi:10.1016/j.ccr.2010.11.015
35. Weissmann S, Alpermann T, Grossmann V, Kowarsch A, Nadarajah N, Eder C. et al. Landscape of TET2 mutations in acute myeloid leukemia. Leukemia. 2012;26:934-42 doi:10.1038/leu.2011.326
36. Muller T, Gessi M, Waha A, Isselstein LJ, Luxen D, Freihoff D. et al. Nuclear exclusion of TET1 is associated with loss of 5-hydroxymethylcytosine in IDH1 wild-type gliomas. The American journal of pathology. 2012;181:675-83 doi:10.1016/j.ajpath.2012.04.017
Author contact
![]() Corresponding author: Theo F. J. Kraus, Center for Neuropathology and Prion Research (ZNP), Ludwig-Maximilians-University, Feodor-Lynen-Str. 23, D-81377, Munich, Bavaria, Germany. Tel: +49/(0)89/2180-78021; Fax: +49/(0)89/2180-78037; Email: theo.krausuni-muenchen.de
Corresponding author: Theo F. J. Kraus, Center for Neuropathology and Prion Research (ZNP), Ludwig-Maximilians-University, Feodor-Lynen-Str. 23, D-81377, Munich, Bavaria, Germany. Tel: +49/(0)89/2180-78021; Fax: +49/(0)89/2180-78037; Email: theo.krausuni-muenchen.de