Impact Factor ISSN: 1837-9664

 Global reach, higher impact
Global reach, higher impactJ Cancer 2015; 6(10):996-1004. doi:10.7150/jca.12135 This issue Cite
Research Paper
The Histone Deacetylase Inhibitor Vaproic Acid Induces Cell Growth Arrest in Hepatocellular Carcinoma Cells via Suppressing Notch Signaling
Guangchun Sun1, Lily V. Mackey3, David H. Coy3, Cui-Yun Yu2,3, ![]() , Lichun Sun1,2,3,
, Lichun Sun1,2,3, ![]()
1. Department of Pharmacy, The Fifth People's Hospital of Shanghai, Fudan University; 801 He-Qing Rd., Shanghai 200240, China
2. Institute of Pharmacy & Pharmacology, Department of Pharmacy, University of South China, Hengyang 421001, China
3. Department of Medicine, School of Medicine, Tulane Health Sciences Center, New Orleans, LA 70112-2699, USA
Received 2015-3-14; Accepted 2015-7-19; Published 2015-8-22
Abstract
Hepatocellular carcinoma (HCC) is a type of malignant cancer. Notch signaling is aberrantly expressed in HCC tissues with more evidence showing that this signaling plays a critical role in HCCs. In the present study, we investigate the effects of the anti-convulsant drug valproic acid (VPA) in HCC cells and its involvement in modulating Notch signaling. We found that VPA, acting as a histone deacetylase (HDAC) inhibitor, induced a decrease in HDAC4 and an increase in acetylated histone 4 (AcH4) and suppressed HCC cell growth. VPA also induced down-regulation of Notch signaling via suppressing the expression of Notch1 and its target gene HES1, with an increase of tumor suppressor p21 and p63. Furthermore, Notch1 activation via overexpressing Notch1 active form ICN1 induced HCC cell proliferation and anti-apoptosis, indicating Notch signaling played an oncogenic role in HCC cells. Meanwhile, VPA could reverse Notch1-induced increase of cell proliferation. Interestingly, VPA was also observed to stimulate the expression of G protein-coupled somatostatin receptor type 2 (SSTR2) that has been used in receptor-targeting therapies. This discovery supports a combination therapy of VPA with the SSTR2-targeting agents. Our in vitro assay did show that the combination of VPA and the peptide-drug conjugate camptothecin-somatostatin (CPT-SST) displayed more potent anti-proliferative effects on HCC cells than did each alone. VPA may be a potential drug candidate in the development of anti-HCC drugs via targeting Notch signaling, especially in combination with receptor-targeting cytotoxic agents.
Keywords: valproic acid, HDAC inhibitor, Notch signaling, hepatocellular carcinoma, receptor-targeting.
Introduction
Hepatocellular carcinoma (HCC) is a type of malignant primary liver cancer with one of the lowest five-year survival rates (1). Conventional therapeutics, including chemotherapy and radiotherapy, have very limited efficacy with severe toxic side effects. Currently, sorafenib, a dual multiple kinase inhibitor of Raf kinase and VEGFR tyrosine kinase, is the only drug approved for treating advanced HCCs. Thus, there is an urgent need to develop novel and specific HCC-targeting drugs.
Reportedly, HCC is strongly associated with hepatitis (2-4). However, various other signaling pathways like Hedgehog signaling, are also involved in HCC progression (5-7). Currently, the precise molecular mechanisms are not clearly known. Compared to other cancers, certain gene mutations in HCC cells such as PIK3CA/PTEN were less or undetectable (7). Conversely, some others such as Notch1/Notch3 are mutated more frequently in HCC cells (5, 7). These mutations result in an aberrance of the associated molecular signaling. Many of these signaling pathways are involved in the pathogenesis of malignant HCC, including Wnt signaling, p53 signaling, chromatin remodelling, histone acetylation/deacetylation, Notch signaling, JAK signaling, Hedgehog signaling and multiple growth factors (5, 7-9). Notch signaling is frequently reported aberrantly activated in HCCs and various types of human cancers (10-13) and is involved in carcinogenesis and cancer progression (12, 14, 15).
Notch signaling, besides being involved in liver development, also plays a critical role in HCC carcinogenesis (16-20). Most HCC tissues have high Notch signatures, especially the overexpression of Notch1, Notch3 and Notch4 (12, 21-23). This accumulating evidence supports that Notch signaling may play an oncogenic role in HCC cells (5, 16, 21, 23, 24). Thus, it may be a potential therapeutic strategy for developing anti-HCC drugs to target Notch signaling pathways. However, there are other studies showing that Notch signaling is controversial (25-27). In the present study, we evaluate the effects of Notch signaling activation in HCC cells.
Meanwhile, histone deacetylase (HDAC) inhibitors have been used to treat cancers and other diseases (28, 29). Several of these HDAC inhibitors display effective anti-tumor efficacy by modulating multiple signaling pathways (15, 30). The HDAC inhibitor VPA is currently being investigated for treating various cancers (30-33). Our previous studies have demonstrated VPA's anti-cancer efficacy in cervical cancer cells, with the involvement of Notch signaling (13, 34). VPA was also found to induce cell growth arrest in some other HCC cell lines (35-37). In the present study, we evaluated the role of Notch signaling and the effects of VPA in HCC HTB-52 cells having a high Notch signature. We attempt to understand whether Notch signaling is involved in VPA-mediated cell growth suppression. VPA also was evaluated for its effect on the expression of certain G protein-coupled receptors (GPCRs) and for its use in a combination treatment with the somatostatin receptor type 2 (SSTR2)-targeting peptide-drug conjugate camptothecin-somatostatin cytotoxin (CPT-SST).
Materials and Methods
Materials
Valproic acid sodium salt (VPA) was purchased from Sigma (St. Louis, MO). Antibodies to SSTR2 (Cat. No.: sc-11609), p21 (sc-756), p63 (sc-8343), histone 4 (H4, sc-10810), HDAC4 (sc-11418), acetylation of histone 4 (AcH4, sc-8660-R), HES1 (sc-25392) and β-actin (sc-1616-HRP) were purchased from Santa Cruz Biotechnology (Santa Cruz, CA), with Notch1 (Cat. No.: 4380) from Cell Signaling Technology (Danvers, MA). The plasmids are the kind gifts from Dr. Wu (University of Florida). The cytotoxic CPT-SST was synthesized in our laboratories (38).
Cell culture and cell transfection
Human HCC HTB-52 (SK-HEP-1) cells were purchased from ATCC (American Type Culture Collection, Manassas, VA) and maintained in MEM medium supplemented with 10% fetal bovine serum (FBS), 1% penicilin/streptomycin and 0.5% kanamycin. For cell transfection, 500 μl of cells (2x105cells/ml) were plated in each well of 24-well plates. Two μl of LipofectamineTM 2000 (Lipo-2000) and 0.8 μg of DNA were added separately into a vial with 50 μl Opti-MEM transfection medium, and combined together after a 5-10 minute incubation. The DNA/Lipo2000 complexes were continuously incubated for 20-30 minutes and then added to each well. The transfected cells were incubated for different time based on different assays and then tested for their cell cycle and apoptosis, or for a cell proliferation as described below. The experiments were done separately three times.
RT-PCR and real-time PCR
Total RNA was isolated from tumor cells and RT-PCR was performed as described in the protocols (Invitrogen, Carlsbad, CA). The primers and conditions for RT-PCR analyses are as previously described (13, 34). The PCR was regularly amplified with 35 cycles, with more or less cycles due to the difference in RNA abundance of these investigated genes. Primers for real-time PCR analyses were the same as described above. Real-time PCR assays were performed as described (13). β-actin was used as the internal control and results were calculated by applying 2-ΔΔCT methods. The experiments were done separately three times.
Western blot analysis
This assay was employed as described in the protocol (Santa Cruz). Briefly, cells were harvested, re-suspended in RIPA buffer with cocktail inhibitors, homogenized with a 21-gauge needle, mixed with the loading buffer and heated for 7 minutes in boiling water. Supernatants were loaded to run on a 8-16% Tris-glycine gel after centrifugation at 12,000xg for 10 minutes. Protein was transferred to a nitrocellulose membrane and blocked with a 5% dried milk/TBST solution, then washed and incubated with primary antibody. The membrane was washed again and incubated with second antibody (Santa Cruz). Membranes were developed according to the ECL system protocol (Amersham Biosciences, England). The experiments were done separately three times.
Cell apoptosis and cell cycle analysis
Cells were plated on 6-well plates and incubated overnight. The test compounds were added. Cells were incubated overnight or as required and then were harvested for the assay. For the apoptosis assay, the Annexin V-FITC apoptosis detection kit (Cat. No.: APOAF) was purchased from Sigma (Saint Louis, MO). Into five hundred μl of cell suspension was added 5 μl of Annexin V-FITC conjugate and 10 μl of propidium iodide solution. The mixture was incubated for 10 minutes and assayed to determine fluorescence using a Beckman-Coulter Gallios analyzer. Data were analyzed with Gallios software. For the cell cycle assay, the Coulter DNA Prep reagents kit (Cat. No.: PN 6607055) from Beckman Coulter (Fullerton, CA) was used. Analysis was done on a Beckman-Coulter Epics FC500 analyzer using CXP software for acquisition and the ModFit LT v3.1 (Verity Software) for Cell Cycle Modeling. Both experiments were repeated three times.
Cell proliferation assay
The cell proliferation assay (Promega, Madison, WI) was performed as described previously (38). Briefly, 50 μl aliquots of medium with different concentrations of compounds were added to 96-well plates. All compound concentrations were tested in triplicate. Fifty μl of the BON cell stock (1x105cells/ml of media) was dispensed into each test well and the plates were incubated at 37oC in a CO2 incubator for 3 days. Following the incubation period, 15 μl of the dye solution was added to each well and the plates were then incubated at 37oC for 4 hours, followed by the addition of 100 µl per well of the solubilization solution. The plates were incubated at 37oC until the contents in each well became a uniform-colored solution. The absorbance was measured and recorded at 570 nm by a Victor Plate Reader (PerkinElmer, Boston, MA).
Cell morphological change and cell attachment
One ml of cells (1x105 cells/ml) were plated in 24-well plates and treated with or without the test compound (VPA). VPA-induced cell morphological changes were inspected and photographed under an inverted light microscope at 10x magnification. The cell attachment assay (ECM205) was used to study the effects of VPA on cell attachment components including fibronectin, vitronectin, laminin, collagen I and collagen IV (ECM205, Chemicon). The assay was performed after a 72-hour incubation according to the kit instructions. The treated cells were harvested with enzyme-free cell dissociation buffer and added to each well of strips that were pre-rehydrated with PBS buffer. The strips were incubated for 45-60 minutes at 37oC in the incubator. The strips were washed 3-5 times, stained with 0.2% crystal violet for 5 minutes and washed 3-5 times. One hundred μl of solubilization buffer was added to each well and the absorbance at 570 nm was determined after the cell-bound stain was completely solubilized. The assays were normalized by cell proliferation assays.
Results
VPA has demonstrated its cancer cell growth suppression in various types of cancer cells (13, 31, 32). In its anti-cancer effects, VPA serves as a HDAC inhibitor and is also believed to act as a Notch signaling regulator (13, 32, 33). Notch signaling is aberrant in HCC cells and is strongly associated with HCC progression. Thus, in the present study, we investigated for VPA's effects on HCC cell growth and the involvement of Notch signaling in VPA-meditated HCC cell growth arrest.
1.VPA acting as a HDAC inhibitor induces cell proliferation suppression
VPA is believed to act as a HDAC inhibitor in its tumor suppression in various cancers (15, 33). Thus, we first investigated the effects of VPA on histone acetylation/deacetylation in HCC HTB-52 cells. Western blot analysis showed that VPA reduced the expression of HDAC4 and induce the acetylation of histone H4 (AcH4), with no obvious change of histone H4 (Fig.1B). We further did cell proliferation assay and found that VPA displayed anti-proliferative abilities in HCC HTB-52 cells. As shown in Fig.1, VPA significantly induced cell proliferation suppression in a dose-dependent manner, with the suppressive rates being nearly 50% (VPA: 2.5 mM) and 70% (5 mM), respectively (Fig.2A). VPA-induced decrease of the proliferation marker PCNA (0.35-fold) assayed by real-time PCR was observed as well (Fig.1C). Another HDAC inhibitor trichostatin A (TSA), and the Notch signaling inhibitor dibenzazepine (DBZ) also displayed their suppressive effect on HTB-52 cell growth, with TSA at 1µM having more potent anti-proliferation and DBZ at 20 µM less (data not shown).
The effects of VPA on gene expression in HCC HTB-52 cells. A, RT-PCR assay at mRNA level showed that VPA at a dose-dependent manner induced a decrease of Notch1 and Notch2, and an increase of p63, p21, SSTR2, GRPR and BRS3; B, western blot assay at protein level showed that VPA at a time-dependent manner induced a decrease of HDAC4, full Notch1, Notch1 fragment and Notch target gene HES1, and also an increase of AcH4, p21, p63 and SSTR2, with no obvious change of H4; C, real-time PCR assay showed that VPA regulated the expression of certain genes, with an increase, a decrease or no change while the value > 1, <1 or =1, respectively. The average change has been acquired from three independent assays, with VPA-induced increase of p63 (3.4-fold), Ecadherin (8-fold), MMP2 (9.8-fold), Fibronectin (15-fold) and SSTR2 (20.6-fold), and decrease of Notch1 (0.56-fold), Notch2 (0.7-fold), HES1 (0.26-fold), Twist(0.58-fold), c-Myc (0.6-fold), PCNA (0.35-fold) and BCL-2 (0.6-fold).
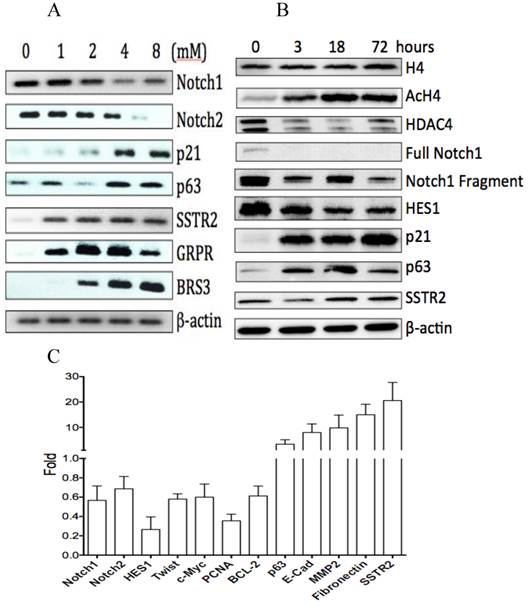
The effects of VPA on cell proliferation and cell apoptosis in HCC HTB-52 cells. A, cell proliferation assay showed that VPA induced a cell growth suppression in a dose-dependent manner, with inhibitory rates being 48.4% (2.5 mM) and 68.9% (5 mM). Asterisk (*) shows a significant difference compared to control, with P value < 0.01 (One-way ANOVA analysis); B, Apoptosis assay showed that VPA obviously induced cell apoptosis. Assays from three experiments showed an increasing apoptotic rates at early apoptosis (VPA/Control: 23.6 ± 0.5% / 17.8 ± 2.9%) and necro-apoptosis (7.5 ± 0.6% / 2.0 ± 0.3%), with P value < 0.01 (t-test analysis).
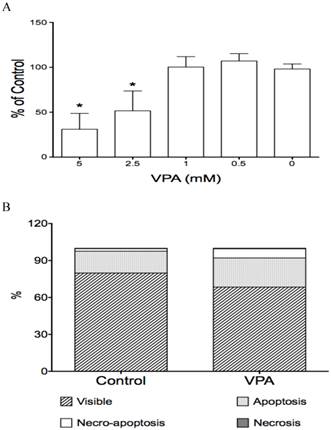
2. VPA induces cell apoptosis
We also investigated the effects of VPA on cell apoptosis in HCC HTB-52 cells and found that VPA significantly induced cell apoptosis. VPA at 4 mM and a 72-hour incubation induced apoptosis, with an apoptotic rate (early apoptosis and necro-apoptosis together) of 31% compared to 20% in the control (Fig.2B). There was also a decrease of the anti-apoptotic marker BCL-2 (0.6-fold), detected at the mRNA level by real-time PCR assay (Fig.1C). Meanwhile, the HDAC inhibitor TSA at 1 µM markedly induced an apoptotic increase with the apoptotic rate of 68%, but Notch signaling inhibitor DBZ at 20 µM obviously have no effect, being similar to the DMSO control (data not shown).
3. VPA induces cell cycle arrest
Besides its suppression of cell proliferation and induction of cell apoptosis as described above, VPA also was evaluated for its effects on the cell cycle. VPA at 4 mM and with an 18-hour incubation was found to induce cell cycle arrest at phase G2, with TSA at 1μM at G2, but DBZ did not cause arrest when compared to the effect of the DMSO control (data not shown).The time course analysis further showed the VPA-induced cell cycle arrest at phase G2 can be observed at an incubation of 18 (an increase of rate at phase G2: 10%) and 72 hours (13%) compared to control (5%), with less effect at a 3-hour incubation (7%) (Table 1). VPA also induced an increase of the tumor repressors p21 and p63 at the mRNA level as seen by RT-PCR and real-time PCR and at the protein level by Western blot analysis (Fig.1A, 1B, 1C). These assays showed that the HDAC inhibitor VPA might suppress HCC cell growth via inducing cell cycle arrest.
VPA-induced cell growth arrest at phase G2 in HCC HTB-52 cells via cell cycle analysis.
| Time (hrs) | Phase | Arrest | ||
|---|---|---|---|---|
| G1 | G2 | S | ||
| 0 | 80.80±2.25 | 4.72±1.40 | 14.99± 2.10 | |
| 3 | 75.51 ±2.86 | 6.60 ±2.12 | 17.69± 1.31 | → |
| 18 | 79.28 ± 3.65 | 10.32± 1.63 | 10.39 ± 2.40 | G2 |
| 72 | 81.52± 2.68 | 12.76 ± 0.79 | 5.50 ± 1.55 | G2 |
4. VPA induces cell morphological change and cell attachment suppression
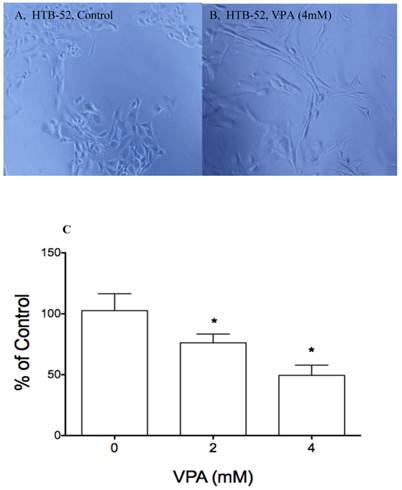
Another major function of VPA is to induce cell morphological changes that are associated with differentiation and cell migration. Previously, we observed VPA-induced cell morphological change in many different cancer cells. Thus, VPA also was evaluated for its effects on cell morphological changes and on cell attachment in HTB-52 cells with epithelial morphology. VPA at 4mM and a 72-hour incubation was observed to induce cell morphological change (Fig.3B). We further did attachment assay to evaluate the effects of VPA on cell attachment. In our previous study, we found that HCC HTB-52 cells more strongly attached to ECM component laminin, not to other components including fibronectin, vitronectin, collagen I and collagen IV. We further observed that VPA at 2 mM and 4 mM with a 72-hour incubation resulted in a significant suppression of cell-laminin attachment, with the inhibitory rates being 24 % (2 mM) and 51% (4 mM) (Fig.3C). The observation was also acquired with the effects of VPA on MMP2 (cell invasive and metastasis marker) and E-cadherin (an epithelial cell marker) that are related to cell differentiation and migration (Fig.1C).VPA was found to induce an increase of MMP2 (8-fold) and E-cadherin (7-fold) in HTB-52 cells. These findings support that VPA may inhibit HCC metastasis via blocking cell migration.
The effects of VPA on cell morphological change and cell attachment. A&B, VPA at 4 mM and 2-day incubation induced a epithelial type of morphological change (B) compared to control (A); Cell attachment assay showed that VPA at 2 and 4 mM inhibited HCC HTB-52 cells to attach to ECM component laminin, with the inhibitory rates being 24.8% (2 mM) and 50.5% (4 mM) and P value < 0.01 (One-way ANOVA analysis).
5. Notch signaling plays an oncogenic role
Notch signaling is aberrantly expressed in HCC cells and plays a critical role in HCC progression (25, 27, 39). Thus, we evaluated the effects of Notch signaling in HCC HTB-52 cells. By investigating the expression profiles of four Notch receptor genes. We first found that Notch1 and 2 receptors were expressed at a higher level in HCC HTB-52 cells compared to undetected Notch3 and Notch4 (data not shown). We further investigated the effects of Notch1 activation on HCC cell growth. HCC HTB-52 cells were transiently transfected with the Notch1 active form ICN1 and assayed for the effects of ICN1 on cell proliferation. We found that ICN1 overexpression stimulated HCC HTB-52 cell proliferation in a dose-dependent manner, with an increase of 9% (ICN1: 200 ng), 18% (400 ng) and 67% (800 ng), respectively (Fig.4A). We also observed that all the other three Notch active form ICN2, ICN3 and ICN4 stimulated HCC cell proliferation (data not shown).
The effects of Notch1 active form ICN1 on cell proliferation and cell cycle arrest. Cells (0.5 ml medium/per well) were plated in 24-well plates and cultured overnight, then transfected with ICN1 or pcDNA (control) and incubated for 5-6 hours. 1.5 ml fresh medium were added and cell were cultured for different time (3 days for cell proliferation, overnight for cell cycle). A, the proliferation assay from three experiments showed that ICN1 induced cell proliferation, with an increase of 9% (ICN1: 200 ng), 18%(400 ng) and 67% (800 ng), respectively. One-way ANOVA showed an significant increase by the dose trend and the highest (Asterisk *) compared to control, with P < 0.01. B, the cell cycle analysis from three experiments showed that ICN1 overexpression induced an increase at phase G1 of cell cycle, with a rate 82.9 ± 0.8% compared to 67.3 ± 1.3% of control, a decrease at phase G2 with a rate 1.7 ± 0.1% (control: 3.9 ± 1.7% and phase S with a rate 15.4 ± 0.9 % (control: 28.8 ± 1.6%).
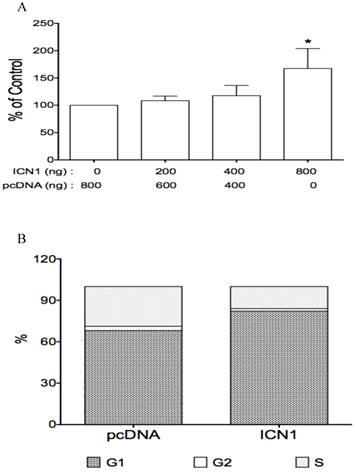
Also, ICN1 was investigated for its effects on HCC cell cycle and cell apoptosis. The cell cycle assay showed that ICN1 resulted in an significant accumulation at phase G1 with the rate over 80% compared to 68% of the control vector (transfected with pcDNA vector only) (Fig.4B), different from VPA-induced cycle arrest at phase G2 as mentioned above (Table 1). The apoptosis assay also showed that ICN1 overexpression induced anti-apoptosis, with the total apoptotic rate being 24 % compared to 48% in the control (data not shown). These findings indicate that Notch signaling stimulates cell growth by playing an oncogenic role in HCC HTB-52 cells.
6. VPA suppresses Notch signaling and reverses Notch1-stimulated cell proliferation
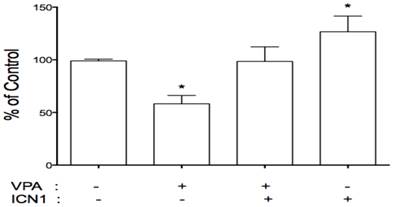
As described above, VPA acted as a tumor suppressor with Notch signaling activation playing an oncogenic role. This implies a connection between VPA and Notch signaling. We further evaluated the effects of VPA on the expression of Notch receptors in these cells. HCC cells were treated with VPA at a serial dose of 0, 1, 2, 4, and 8 mM. Further assays showed that VPA down-regulated the expression of Notch1 and Notch2 at the mRNA level as seen by RT-PCR (Fig.1A) and real-time PCR (Fig.1C). Western blot assays also confirmed VPA-induced suppression of Notch1 and the Notch downstream target gene HES1 at the protein level (Fig.1B). We further co-treated HCC HTB-52 cells with ICN1 and VPA by first transfecting ICN1 for 4-5 hours and then adding VPA at 4 mM. After a 72-hour incubation, we found that Notch1-induced cell proliferation was reversed by VPA (Fig.5), indicating that VPA functions as a tumor suppressor, possibly via blocking the oncogenic Notch signaling.
VPA reversed the stimulatory effects of Notch1 activation (ICN1) on cell proliferation. HCC cells were pre-plated in 24-well plates, transfected next day and incubated for 5-6 hours. 1.5 ml fresh medium were added with/without VPA and cells were continuously cultured for 3 days. As shown, ICN1 significantly induced cell proliferation with an increase rate 26.6% and VPA significantly reduced cell proliferation with a decrease rate 41.8%. The combination of VPA and ICN1 showed that VPA reversed the stimulatory effects of Notch1 activation, with no significant difference from control. Asterisk (*) shows a significant difference compared to control, with P value < 0.01 (t-test analysis).
7. The application of VPA in combination with SSTR2-targeting cytotoxin
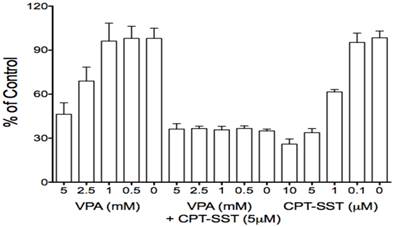
Due to its effects on certain GPCR members, particularly SSTR2 which is aberrantly expressed in many cancer cells and has been used for receptor-targeting therapeutics, VPA was used in combination therapy with receptor-targeting peptide-drug conjugates (13, 34). Thus, presently, VPA also was evaluated for its effects on some GPCRs in HCC HTB-52 cells. We found that VPA significantly increased the expression of SSTR2, bombesin receptor subtypes (GRPR and BRS3) at the mRNA level [Fig.1A], the increase of SSTR2 being confirmed by real-time PCR and Western blot assays [Fig.1B, 1C]. Thus, VPA in combination with receptor-targeting agents could be a potential strategy for HCC treatment. Our in vitro proliferation assay further identified that the combination of VPA and SSTR2-targeting CPT-SST could greatly enhance cell growth suppression compared to each alone [Fig.6]. The in vivo assay is under preparation.
The combination of VPA and SSTR2-targeted peptide-drug conjugate CPT-SST enhanced cell growth suppression compared to each alone.
Discussion
Traditional radio- and chemo-therapies, being severely toxic to the liver, are of limited use in HCC treatments except sorafenib, the only FDA-approved anti-HCC drug, being well-tolerated by HCC patients (40) (NCI website: http://www.cancer.gov). Thus, it is necessary to search and develop more effective anti-HCC drugs. VPA as an anti-convulsant drug and a HDAC inhibitor displayed wide anti-cancer activities in many different types of cancers (15, 31, 32). Our previous studies also showed that VPA suppressed cervical cancer cell growth and tumor growth (13, 34). In this study, VPA was identified as a HDAC inhibitor to suppress HDAC4 and induce acetylated histone H4 in HCC HTB-52 cells. We also found that VPA induced cell proliferation suppression, cell cycle arrest and cell apoptosis, identical to other reports showing the suppressive effects of VPA on other HCC cells (35-37). VPA was also observed to induce cell attachment in HCC HTB-52 cells. These findings support that VPA may be a potential agent for HCC treatment.
Accumulating evidence shows that Notch signaling may most likely be a HCC diagnostic marker. The activation of this signaling is frequently seen in human HCCs. There is a high Notch1 signature in most HCC tissues compared to the adjacent normal tissues (12, 14, 21). Dr. Bolondi reported high expression of Notch3 and Notch4 in most HCC tissues (22). In other reports, the high frequency of Notch1 and Notch3 were observed in HCC tissues (12, 14, 41). Moreover, HBV X protein (HBx), that is a critical factor in HCC carcinogenesis, is also found to up-regulate Notch signaling (5, 12, 42). These findings support that Notch signaling is critical for HCC development. However, different reports showed that Notch signaling may display controversial functions in HCC cells. The observations from several other studies show that Notch signaling may play a tumor-suppressive role (21, 25-27). Thus, the function of Notch signaling is not determined fully, with the precise Notch-mediated mechanisms remaining unclear.
We evaluated the effects of Notch signaling activation in HCC cells. Our assays showed that Notch1 activation stimulated cell proliferation and reduced cell apoptosis, and conversely, its knockdown could suppress cell growth. We also found the positive effects of all other three Notch receptors (ICN2, ICN3, ICN4) on multiple HCC cells and identified the role of Notch signaling in serving as an oncogenic player in HCC cells, identical to the role of Notch1.Thus, the activation of Notch signaling in HCC cells plays an oncogenic role (5, 8, 23, 24). it may be a promising anti-HCC approach to target oncogenic Notch signaling. In our present study, the HDAC inhibitor VPA was found to act as a Notch signaling inhibitor to suppress the expression of Notch1 and the Notch target gene HES1. Furthermore, VPA could reverse cell growth stimulated by Notch1 activation. Thus, the HDAC inhibitor VPA may possibly suppress HCC cell growth via acting as a Notch signaling inhibitor. Tumor suppressors like p53 and p27 also were reportedly associated with regulation of Notch signaling in HCC cells. In Dr. Giovannini's report, Notch3 knockdown increases the expression of the tumor suppressors p53 and p27, and enhances doxorubicin-induced apoptosis and DNA damage in HCC HepG2 cells (14). We also found that VPA induced an increase of tumor repressor p21 and p63, supporting VPA's suppressive effects in HCC cells.
More importantly, VPA was found to significantly increase the expression of SSTR2 and GRPR in HCC HTB-52 cells. This provides a potentially novel opportunity for a receptor-targeted chemotherapy with VPA and a SSTR2- or GRPR-targeting peptide/antibody conjugate by taking advantage of VPA's dual functions of tumor suppression and receptor upregulation. Normally, due to its limited effects, VPA more frequently serves as an adjuvant for the combination treatments. VPA in combination with other anti-cancer agents such as sorafenib and acyclic retinoid was also reported to inhibit HCC cell growth (15, 35, 39, 43). However, these combination treatments are not specific to HCC cells. Our new strategy showed that VPA in combination with receptor-targeting agents displayed a more effective synergic effect. In the present study, the combination of VPA and the SSTR2-targeting CPT-SST enhanced the suppression of HCC cell growth compared to each alone. Similar results were observed from our previous study in that the combination displayed synergic in vitro and in vivo anti-tumor effects on cervical cancer tumor growth (13, 34). Due to receptor-targeting therapeutics could more specifically induce tumor suppression via binding with the specific receptor and increasing the internalization of the cytotoxic agents (44), VPA-induced increase of the target receptors can provide an enhanced synergic effect via a combination of VPA and receptor-targeting cytotoxins. This combination strategy is under preparation for in vivo evaluation.
Conclusively, Notch signaling is highly expressed in HCC tissues, with more evidence showing it serving as a HCC oncogenic marker. The HDAC inhibitor VPA displayed its efficacy against HCC cell growth via targeting Notch signaling and tumor repressor. Particularly, the characteristics of VPA-mediated increase of receptors may provide us a novel and promising anti-HCC therapeutic approach with the combination of VPA and receptor-targeting cytotoxic agents.
Abbreviations
VPA: valproic acid; HDAC: histone deacetylase; AcH4: acetylated histone 4; HCC: hepatocellular carcinoma; SSTR: somatostatin receptor; CPT-SST: camptothecin-somatostatin conjugate; TSA: trichostatin A; DBZ: dibenzazepine; GPCRs: G protein-coupled receptors.
Acknowledgements
The authors gratefully acknowledge the financial supports from Shanghai Science and Technology Committee of China (Grant No.14411972700), Financial supports from National Natural Science Foundation of China (Grant No. 81471777 and 81102409) and Tulane Peptide Research Fund.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Siegel R, Ma J, Zou Z, Jemal A. Cancer statistics, 2014. CA Cancer J Clin. 2014;64:9-29
2. Mittal S, El-Serag HB. Epidemiology of hepatocellular carcinoma: consider the population. J Clin Gastroenterol. 2013;47(Suppl):S2-6
3. El-Serag HB. Hepatocellular carcinoma. N Engl J Med. 2011;365(12):1118-27
4. El-Serag HB. Epidemiology of viral hepatitis and hepatocellular carcinoma. Gastroenterology. 2012;142:1264-73
5. Moeini A, Cornella H, Villanueva A. Emerging signaling pathways in hepatocellular carcinoma. Liver Cancer. 2012;1:83-93
6. Morell CM, Fiorotto R, Fabris L, Strazzabosco M. Notch signalling beyond liver development: emerging concepts in liver repair and oncogenesis. Clin Res Hepatol Gastroenterol. 2013;37:447-54
7. Villanueva A, Llovet JM. Liver cancer in 2013: Mutational landscape of HCC--the end of the beginning. Nat Rev Clin Oncol. 2014;11:73-4
8. Noh JH, Bae HJ, Eun JW, Shen Q, Park SJ, Kim HS. et al. HDAC2 provides a critical support to malignant progression of hepatocellular carcinoma through feedback control of mTORC1 and AKT. Cancer Res. 2014;74:1728-38
9. Venturelli S, Berger A, Bocker A, Busch C, Weiland T, Noor S. et al. Resveratrol as a pan-HDAC inhibitor alters the acetylation status of histone [corrected] proteins in human-derived hepatoblastoma cells. PLoS One. 2013;8:e73097
10. Kunnimalaiyaan M, Chen H. Tumor suppressor role of Notch-1 signaling in neuroendocrine tumors. Oncologist. 2007;12:535-42
11. Yin L, Velazquez OC, Liu ZJ. Notch signaling: emerging molecular targets for cancer therapy. Biochem Pharmacol. 2010;80:690-701
12. Giovannini C, Gramantieri L, Chieco P, Minguzzi M, Lago F, Pianetti S. et al. Selective ablation of Notch3 in HCC enhances doxorubicin's death promoting effect by a p53 dependent mechanism. J Hepatol. 2009;50:969-79
13. Franko-Tobin LG, Mackey LV, Huang W, Song X, Jin B, Luo J. et al. Notch1-mediated tumor suppression in cervical cancer with the involvement of SST signaling and its application in enhanced SSTR-targeted therapeutics. Oncologist. 2012;17:220-32
14. Giovannini C, Baglioni M, Baron Toaldo M, Ventrucci C, D'Adamo S, Cipone M. et al. Notch3 inhibition enhances sorafenib cytotoxic efficacy by promoting GSK3b phosphorylation and p21 down-regulation in hepatocellular carcinoma. Oncotarget. 2013;4:1618-31
15. Duenas-Gonzalez A, Candelaria M, Perez-Plascencia C, Perez-Cardenas E, de la Cruz-Hernandez E, Herrera LA. Valproic acid as epigenetic cancer drug: preclinical, clinical and transcriptional effects on solid tumors. Cancer Treat Rev. 2008;34:206-22
16. Morell CM, Strazzabosco M. Notch signaling and new therapeutic options in liver disease. J Hepatol. 2014;60:885-90
17. Bogaerts E, Heindryckx F, Vandewynckel YP, Van Grunsven LA, Van Vlierberghe H. The roles of transforming growth factor-beta, Wnt, Notch and hypoxia on liver progenitor cells in primary liver tumours (Review). Int J Oncol. 2014;44:1015-22
18. Geisler F, Strazzabosco M. Emerging roles of Notch signaling in liver disease. Hepatology. 2015;61:382-92
19. Dill MT, Tornillo L, Fritzius T, Terracciano L, Semela D, Bettler B. et al. Constitutive Notch2 signaling induces hepatic tumors in mice. Hepatology. 2013;57:1607-19
20. Ahn S, Hyeon J, Park CK. Notch1 and Notch4 are markers for poor prognosis of hepatocellular carcinoma. Hepatobiliary Pancreat Dis Int. 2013;12:286-94
21. Ning L, Wentworth L, Chen H, Weber SM. Down-regulation of Notch1 signaling inhibits tumor growth in human hepatocellular carcinoma. Am J Transl Res. 2009;1:358-66
22. Gramantieri L, Giovannini C, Lanzi A, Chieco P, Ravaioli M, Venturi A. et al. Aberrant Notch3 and Notch4 expression in human hepatocellular carcinoma. Liver Int. 2007;27:997-1007
23. Villanueva A, Alsinet C, Yanger K, Hoshida Y, Zong Y, Toffanin S. et al. Notch signaling is activated in human hepatocellular carcinoma and induces tumor formation in mice. Gastroenterology. 2012;143:1660-9
24. Gao J, Dong Y, Zhang B, Xiong Y, Xu W, Cheng Y. et al. Notch1 activation contributes to tumor cell growth and proliferation in human hepatocellular carcinoma HepG2 and SMMC7721 cells. Int J Oncol. 2012;41:1773-81
25. Viatour P, Ehmer U, Saddic LA, Dorrell C, Andersen JB, Lin C. et al. Notch signaling inhibits hepatocellular carcinoma following inactivation of the RB pathway. J Exp Med. 2011;208:1963-76
26. Wang C, Qi R, Li N, Wang Z, An H, Zhang Q. et al. Notch1 signaling sensitizes tumor necrosis factor-related apoptosis-inducing ligand-induced apoptosis in human hepatocellular carcinoma cells by inhibiting Akt/Hdm2-mediated p53 degradation and up-regulating p53-dependent DR5 expression. J Biol Chem. 2009;284:16183-90
27. Qi R, An H, Yu Y, Zhang M, Liu S, Xu H. et al. Notch1 signaling inhibits growth of human hepatocellular carcinoma through induction of cell cycle arrest and apoptosis. Cancer Res. 2003;63:8323-9
28. Vigushin DM, Coombes RC. Histone deacetylase inhibitors in cancer treatment. Anticancer Drugs. 2002;13:1-13
29. Cavasin MA, Demos-Davies K, Horn TR, Walker LA, Lemon DD, Birdsey N. et al. Selective class I histone deacetylase inhibition suppresses hypoxia-induced cardiopulmonary remodeling through an antiproliferative mechanism. Circ Res. 2012;110:739-48
30. Mohammed TA, Holen KD, Jaskula-Sztul R, Mulkerin D, Lubner SJ, Schelman WR. et al. A pilot phase II study of valproic acid for treatment of low-grade neuroendocrine carcinoma. Oncologist. 2011;16:835-43
31. Greenblatt DY, Vaccaro AM, Jaskula-Sztul R, Ning L, Haymart M, Kunnimalaiyaan M. et al. Valproic acid activates notch-1 signaling and regulates the neuroendocrine phenotype in carcinoid cancer cells. Oncologist. 2007;12:942-51
32. Platta CS, Greenblatt DY, Kunnimalaiyaan M, Chen H. Valproic acid induces Notch1 signaling in small cell lung cancer cells. J Surg Res. 2008;148:31-7
33. Wedel S, Hudak L, Seibel JM, Juengel E, Tsaur I, Wiesner C. et al. Inhibitory effects of the HDAC inhibitor valproic acid on prostate cancer growth are enhanced by simultaneous application of the mTOR inhibitor RAD001. Life Sci. 2011;88:418-24
34. Tsai C, Leslie JS, Franko-Tobin LG, Prasnal MC, Yang T, Vienna Mackey L. et al. Valproic acid suppresses cervical cancer tumor progression possibly via activating Notch1 signaling and enhances receptor-targeted cancer chemotherapeutic via activating somatostatin receptor type II. Arch Gynecol Obstet. 2013;288:393-400
35. Li X, Zhu Y, He H, Lou L, Ye W, Chen Y. et al. Synergistically killing activity of aspirin and histone deacetylase inhibitor valproic acid (VPA) on hepatocellular cancer cells. Biochem Biophys Res Commun. 2013;436:259-64
36. Machado MC, Bellodi-Privato M, Kubrusly MS, Molan NA, Tharcisio T Jr, de Oliveira ER. et al. Valproic acid inhibits human hepatocellular cancer cells growth in vitro and in vivo. J Exp Ther Oncol. 2011;9:85-92
37. Tatebe H, Shimizu M, Shirakami Y, Sakai H, Yasuda Y, Tsurumi H. et al. Acyclic retinoid synergises with valproic acid to inhibit growth in human hepatocellular carcinoma cells. Cancer Lett. 2009;285:210-7
38. Sun LC, Mackey LV, Luo J, Fuselier JA, Coy DH. Targeted chemotherapy using a cytotoxic somatostatin conjugate to inhibit tumor growth and metastasis in nude mice. Clin Med Oncol. 2008;2:491-9
39. Sun L, Sun G, Yu Y, Coy DH. Is Notch Signaling a Specific Target in Hepatocellular Carcinoma?. Anticancer Agents Med Chem. 2015 [Epob ahead of print]
40. Bruix J, Raoul JL, Sherman M, Mazzaferro V, Bolondi L, Craxi A. et al. Efficacy and safety of sorafenib in patients with advanced hepatocellular carcinoma: subanalyses of a phase III trial. J Hepatol. 2012;57:821-9
41. Hu L, Xue F, Shao M, Deng A, Wei G. Aberrant expression of Notch3 predicts poor survival for hepatocellular carcinomas. Biosci Trends. 2013;7:152-6
42. Wang F, Zhou H, Yang Y, Xia X, Sun Q, Luo J. et al. Hepatitis B virus X protein promotes the growth of hepatocellular carcinoma by modulation of the Notch signaling pathway. Oncol Rep. 2012;27:1170-6
43. Matsuda Y, Wakai T, Kubota M, Osawa M, Hirose Y, Sakata J. et al. Valproic acid overcomes transforming growth factor-beta-mediated sorafenib resistance in hepatocellular carcinoma. Int J Clin Exp Pathol. 2014;7:1299-313
44. Sun LC, Coy DH. Somatostatin receptor-targeted anti-cancer therapy. Curr Drug Deliv. 2011;8:2-10
Author contact
![]() Corresponding authors: Lichun Sun: Tel: 504-988-1179; Email: lsunedu and Cui-Yun Yu: Tel: +86 734-828-2614; E-mail:yucuiyunusccom
Corresponding authors: Lichun Sun: Tel: 504-988-1179; Email: lsunedu and Cui-Yun Yu: Tel: +86 734-828-2614; E-mail:yucuiyunusccom