Impact Factor ISSN: 1837-9664

 Global reach, higher impact
Global reach, higher impactJ Cancer 2015; 6(11):1066-1078. doi:10.7150/jca.12134 This issue Cite
Research Paper
Nucleocytoplasmic Translocation of UBXN2A Is Required for Apoptosis during DNA Damage Stresses in Colon Cancer Cells
Ammara Abdullah1, Sanam Sane1, Jessica L Freeling1, Hongmin Wang1, Dong Zhang2, Khosrow Rezvani1, ![]()
1. Division of Basic Biomedical Sciences, Sanford School of Medicine, The University of South Dakota, 414 E. Clark Street, Lee Medical Building, Vermillion, SD 57069, USA
2. Department of Biomedical Sciences, College of Osteopathic Medicine, New York Institute of Technology, Northern Blvd., P.O. Box 8000, Old Westbury, NY 11568-8000, USA
Received 2015-3-14; Accepted 2015-8-2; Published 2015-9-3
Abstract
The subcellular localization, expression level, and activity of anti-cancer proteins alter in response to intrinsic and extrinsic cellular stresses to reverse tumor progression. The purpose of this study is to determine whether UBXN2A, an activator of the p53 tumor suppressor protein, has different subcellular compartmentalization in response to the stress of DNA damage. We measured trafficking of the UBXN2A protein in response to two different DNA damage stresses, UVB irradiation and the genotoxic agent Etoposide, in colon cancer cell lines. Using a cytosol-nuclear fractionation technique followed by western blot and immunofluorescence staining, we monitored and quantitated UBXN2A and p53 proteins as well as p53's downstream apoptotic pathway.
We showed that the anti-cancer protein UBXN2A acts in the early phase of cell response to two different DNA damage stresses, being induced to translocate into the cytoplasm in a dose- and time-dependent manner. UVB-induced cytoplasmic UBXN2A binds to mortalin-2 (mot-2), a known oncoprotein in colon tumors. UVB-dependent upregulation of UBXN2A in the cytoplasm decreases p53 binding to mot-2 and activates apoptotic events in colon cancer cells. In contrast, the shRNA-mediated depletion of UBXN2A leads to significant reduction in apoptosis in colon cancer cells exposed to UVB and Etoposide. Leptomycin B (LMB), which was able to block UBXN2A nuclear export following Etoposide treatment, sustained p53-mot-2 interaction and had partially antagonistic effects with Etoposide on cell apoptosis. The present study shows that nucleocytoplasmic translocation of UBXN2A in response to stresses is necessary for its anti-cancer function in the cytoplasm. In addition, LMB-dependent suppression of UBXN2A's translocation to the cytoplasm upon stress allows the presence of an active mot-2 oncoprotein in the cytoplasm, resulting in p53 sequestration as well as activation of other mot-2-dependent growth promoting pathways.
Keywords: DNA damage stress, UBXN2A, p53, mortalin-2, colon cancer cells
Introduction
The p53 protein suppresses the formation of tumor cells by arresting the cell cycle and by activating apoptosis in response to an array of cellular stressors [1]. Primarily, p53 is a cytoplasmic protein in normal cells and, upon genotoxic stresses, it translocates into the nucleus in a tightly regulated manner [2, 3]. However, this subcellular localization and subsequent function of p53 in nuclear and cytoplasmic compartments of a cancerous cell is compromised by p53's cytoplasmic sequestration. The Oncoprotein mortalin-2 (mot-2) [4] is the major player in this sequestration processes which leads to the development and progression of tumors. It is noteworthy that mot-2 binds and sequesters p53 particularly after exogenous stress [5], making mot-2 protein a potential candidate for targeted cancer therapies.
While cancer cells hijack the cell's signaling network to promote their proliferation, several proteins can interact with and inhibit the p53 negative-regulators and consequently reactivate p53 in a home-defense mechanism [6-9]. We have previously shown that a ubiquitin-like (UBX)-domain-containing protein, UBXN2A, binds to the oncoprotein mot-2 and unsequesters p53 from mot-2 in the cytoplasm, resulting in translocation of p53 to the nucleus where p53 proteins activate their downstream biological effects, including apoptosis [10]. Whether UBXN2A's response to stresses is similar to that of other anti-cancer defense proteins [9, 11, 12] remains obscure, as do its underlying anti-mot-2 mechanisms.
In this study, we have shown that induction of stresses, including exposure to Ultraviolet B radiation (UVB) or the genotoxic agent Etoposide, enhances UBXN2A translocation from the nucleus to the cytoplasm. Nucleocytoplasmic translocation of UBXN2A and its consequent binding to mot-2 in the cytoplasm is coordinated with p53 unsequestration and apoptosis events. The UBXN2A nucleocytoplasmic translocation mechanism and its interactions with oncoprotein mot-2 in response to genotoxic stresses may contribute to or enhance the anti-cancer pathways [13, 14] activated during the progression of cancer.
Material and methods
Cell culture
HCT-116 and LoVo cells were purchased from ATCC (American Type Culture Collection) and were grown in appropriate media, supplemented with 10% fetal bovine serum and penicillin/streptomycin. Leptomycin B (25ng/mL) was obtained from Sigma-Aldrich (St. Louis, MO, USA) and was used to block nuclear export of proteins [9]. The list of the primary antibodies and the titers used for western blotting (WB) and immunofluorescence staining is shown in Supplementary Table S1.
UVB treatment
UVB treatment of cells was carried out with a custom-made Spectroline XL-1000B UV chamber with a UVB unit that has a peak UV of 312 nm (Fisher Scientific, Pittsburgh, PA, USA). The energy-set (0-999,990 μJ/cm2) and time-set (0-9,900 seconds) modes of Spectroline XL-1000, based on a formula of “UV Dosage (W-Sec/cm2) = UV intensity (W/cm2) X Time (second)”, were used to calculate UVB irradiation for cell culture. Plates were exposed to UVB for designated doses without lids or media and were immediately returned to the CO2 incubator or subjected to cell lysates.
Etoposide treatment
Etoposide (ETO) was purchased from Sigma-Aldrich. We used Etoposide in this study as a DNA damaging agent in the cells. Cells were cultured as per ATCC instructions and allowed to grow to 70% confluence before treatment with ETO. HCT-116 and LoVo cells were treated with different concentrations of ETO for different periods of time. Cells were prepared for downstream analysis.
Protein Extraction
Cells were harvested via mechanical scraping and split into nuclear and cytoplasmic fractions utilizing the NE-PER nuclear and cytoplasmic protein extraction kit (Thermo Fisher Scientific, Waltham, MA, USA). Total protein concentration was assessed using a BCA assay technique (Life Technologies, Grand Island, NY).
Immunoprecipitation
To investigate the interaction between UBXN2A, mot-2, and p53 proteins under genotoxic stress, HCT-116 cells were exposed to UVB or Etoposide, and cytoplasmic and nuclear fractions were prepared after 24 hours. Immunoprecipitation (IP) was performed with Dynabeads magnetic beads (Life Technologies). Briefly, 50μl beads were pelleted by the magnet, washed, and incubated for 15mins with anti-mot-2 or anti-p53 antibodies at room temperature by gentle rotation. Mouse or rabbit serum IgG were used as negative controls. Cytoplasmic lysates were added to the beads and incubated for 1-2 hours at 4° C by gentle rotation. Beads were washed and the immunoprecipitated proteins released with the sample buffer, followed by WB analysis.
Western blot, immunofluorescence staining, and flow cytometry analysis
Proteins in cellular lysates or following IP experiments were separated on 4-20% SDS-PAGE followed by transfer of proteins to nitrocellulose membrane for WB analysis. Immunoblotting was carried out using antibodies against UBXN2A, p53, mot-2, p47, p97, HSC70 and cleaved-PARP, followed by secondary antibodies for chemiluminescent WB or infrared (LI-COR Odyssey, Lincoln, NE) imaging. Equal protein loading for cytoplasmic and nuclear fractions was confirmed by blotting against GAPDH (cytoplasmic marker) and ORC-2 (nuclear marker), respectively. Immunofluorescence staining with anti-UBXN2A and nuclear marker DAPI was conducted as previously described [10]. The rates of cell apoptosis were analyzed by a BD Accuri C6 flow cytometer following PerCP-Cy™5.5 Annexin V (BD Pharmingen, San Diego, CA) staining as previously described [15].
Densitometric quantification of western blot bands
Digitalized WB bands were quantified by measuring pixel numbers with an automated digitizing system (Image Studio, version 3.1). The total measured pixels of each band were normalized to the corresponding loading control.
Statistical analysis
Comparisons for differences in means (± SEM) were assessed by unpaired t-test or one-way ANOVA and post hoc Bonferroni's multiple-comparison t-test using GraphPad Prism 6.0. A p value of ≤ 0.05 was considered significant.
Results
UVB irradiation induces nucleocytoplasmic translocation of UBXN2A
We reported previously that UBXN2A facilitates the nuclear localization of the p53 tumor suppressor by binding to mot-2 in the cytoplasm [10]. To further elucidate the mechanisms of action of UBXN2A in colon cancer cells, we first examined the presence of subcellular localization signals within UBXN2A protein using ESLpred software developed by Bhasin and Raghava [16]. The software predicted potential nuclear export signals (NES) at amino acids 224-233 of the UBXN2A protein, with prediction accuracies of 95.3% (Fig. S1A). This protein structure analysis showed UBXN2A's anti-cancer function can be partly mediated through its subcellular trafficking pathway besides its functional SEP domain [10] and the ubiquitin regulatory X domain (UBX domain) [17]. Based on the information provided by the prediction server, ESLpred, cell lysates of HCT-116 poorly differentiated colon cancer cells were separated into cytoplasmic and nuclear fractions and were subjected to WB analysis. WB results showed UBXN2A proteins were dominantly located in the nuclear fraction in the absence of exogenous stresses (Fig. S1B-C). As previously described for other p53 regulators [9, 18], we hypothesized that genotoxic stresses induce UBXN2A nucleocytoplasmic translocation in colon cancer cells where UBXN2A acts as a negative regulator of mot-2, resulting in activation of p53. To examine whether the subcellular localization of UBXN2A can alter upon genotoxic stresses, we chose UVB as the most biologically relevant mutagenic lesion that were also well characterized in the p53 pathways [19-21]. Furthermore, UVB irradiation was found to induce apoptosis through both intrinsic and extrinsic pathways, whereas UVC induces apoptosis only via the intrinsic pathway [22].
Our results showed that UVB irradiation of HCT-116 cells caused an increase in UBXN2A levels in the cytoplasm in a dose-dependent manner, with maximum expression of UBXN2A 24 hours after exposure to 1, 2, or 4 KJ/m2 UVB (Fig. 1A). Simultaneously, expression of UBXN2A in the nucleus decreased as doses of UVB radiation were increased (Fig. 1B). These results indicate that the induction of DNA damage by biological doses of UVB [19, 21] triggers nucleocytoplasmic translocation of UBXN2A in colon cancer cells. Expectedly, we observed two individual peaks of p53 in the cytoplasm and nucleus in response to low or high DNA damages induced by different levels of UVB treatment [23]. p53 accumulates in the nucleus and cytoplasmic fractions before and after upregulation of cytoplasmic UBXN2A upon DNA damage. We observed that the second p53 peak, which can activate the apoptosis pathways, is overlapped with UBXN2A translocation from the nucleus to the cytoplasm of cells (UVB=1, 2, and 4KJ/m2; Fig. 1A-B). Due to p53-induced apoptosis in these higher concentrations of UVB, cells were not able to recover and undergo apoptosis, and the p53 level reduced significantly when cells received 4KJ/m2. As previously described [24], we observed comparable levels of p53 upregulation in the nuclear compartment in response to DNA damage (Fig. 1B).
UVB induces nucleocytoplasmic translocation of UBXN2A. (A-B) HCT-116 colon cancer cells were UVB irradiated with doses of 0.05, 0.125, 0.250, 0.5, 1, 2, and 4 KJ/m2, followed by 24 hours recovery time. Proteins were extracted from cytoplasmic and nuclear fractions followed by a BCA assay to measure the total level of proteins. Proteins were loaded into 4-20% gradient SDS-PAGE followed by WB analysis. (A) Results indicate that UVB irradiation upregulated UBXN2A protein in the cytoplasm and, simultaneously, the UBXN2A level decreased in the nucleus fraction in the corresponding dosages (0.5, 1, 2, and 4 KJ/m2). Upregulation of UBXN2A in the cytoplasm was accompanied by the second p53 peak in the cytoplasm fraction and accumulation of p53 in the nucleus. P47 (UBXN2C), another member of the UBXD family showed no significant change in response to low UVB, while it decreased in higher doses of UVB (0.5, 1, 2, and 4 KJ/m2) when UBXN2A is increased in the cytoplasm. UBXN2A's protein partner, p97 showed no changes in the cytoplasm. Upregulation of UBXN2A in the cytoplasm and consequent accumulation of p53 in the nucleus were associated with the second peak of cleaved PARP at 1, 2, and 4 KJ/m2. Anti-GAPDH and ORC-2 antibodies were used as subcellular and loading control markers in Panels A and B. (C-D) HCT-116 colon cancer cells were UVB irradiated with biologically relevant doses of 1 and 2 KJ/m2. The total proteins from cytoplasmic and nuclear fractions were measured by BCA assay followed by WB analysis. The band density of UBXN2A was quantified and normalized with GAPDH and ORC-2 loading controls for the cytoplasm and nucleus, respectively. The data is shown as mean ± SEM of three different experiments where *p<0.05 using Bonferroni's t-test. UVB irradiation significantly increases UBXN2A protein in the cytoplasm, whereas in the nucleus UVB significantly decreased the level of UBXN2A protein.
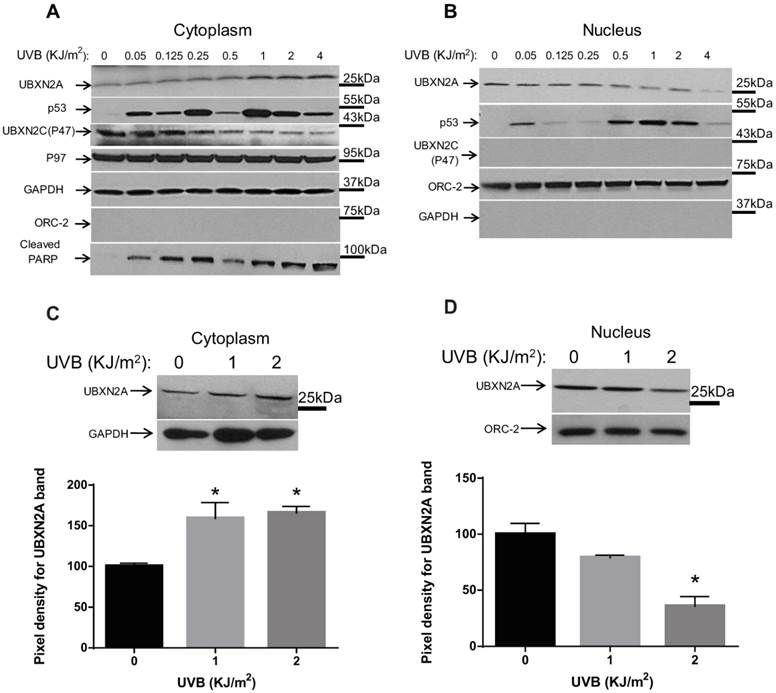
It has been shown that cleaved poly (ADP-ribose) polymerase (PARP) can be a reliable apoptosis marker activated downstream of p53 in HCT-116 colon cancer. Indeed, the silencing or knockout of p53 in HCT-116 can lead to a lower level of cleaved PARP [25]. Because cleaved PARP protein is one of the central steps in apoptotic cell death in colorectal cancer [26], we therefore decided to examine the effect of UBXN2A translocation from the nucleus to the cytoplasm on cleaved PARP. The WB data for cleaved PARP also showed two peaks of PARP in the cytoplasm (Fig. 1A), the second peak of which was coincident with UBXN2A upregulation in the cytoplasm.
To determine whether the upregulation of UBXN2A in response to UVB is a selective event, we also examined the expression of another UBX-domain-containing protein, p47. Interestingly, p47 (UBXN2C) was only detected in the cytoplasm (Fig. 1A). While the level of p47 (UBXN2C) remains unchanged following low doses of UVB (≤0.25 KJ/m2), p47 levels decrease in higher doses of UVB (≥0.5 KJ/m2). Due to their different co-factor roles [27, 28], members of the UBXD family can compete with each other (Fig. 1A) [29]. Therefore, downregulation of p47, which seems to concur with upregulation of UBXN2A in cytoplasm at higher doses of UVB, could be a compensatory mechanism between these two proteins during DNA damage response. Conceivably, p47 might be a target of proteasomal degradation during DNA damage response, as previously reported for other co-factors [30]. Future research investigations in our group will determine whether there is a cross-talk between UBXN2A and p47 following DNA damage response.
Because UBXN2A is also a co-factor of the p97 complex at the endoplasmic reticulum [31, 32], which responds to stresses, we decided to examine whether the p97 complex is involved in the UBXN2A-p53 axis during UVB exposure. The results showed that increased expression of UBXN2A in the cytoplasm has no effect on p97 expression (Fig. 1A). To further statistically confirm the above finding, we repeated the above experiments using 1 to 2 KJ/m2 UVB doses followed by WB analysis of the cytoplasm and nuclear lysates 24 hours after exposure. Quantification of signals after normalizing with the respective loading controls revealed that UBXN2A levels were significantly increased in the cytoplasm (Fig. 1C), whereas they were significantly decreased in the nucleus (Fig. 1D) of HCT-116 cells treated with 1 and 2 KJ/m2 of UVB, as compared to non-treated cells.
P53 has tight temporal regulation in response to genotoxic stress [33], and this regulation is coordinated with several adaptor proteins in a timely manner [34]. Based on the quantitative results illustrated in Panels C and D of Figure 1, we decided to determine the time interval of UBXN2A translocation in response to an intermediate dose of UVB. We therefore irradiated cells with 1KJ/m2 of UVB for different time periods (between 1 to 48 hours), followed by WB analysis. The results showed an increase in UBXN2A levels in the cytoplasm and subsequent decrease in levels of UBXN2A in the nucleus (Fig. 2A-B) is a time-dependent response event. This event starts 6 hours after UVB exposure and subsequently increases to maximum accumulation in the cytoplasm after 24 hours and is sustained until 48 hours after UVB exposure. Due to significant cell death, the WB of cells after 48 hours was not conclusive. Furthermore, we found the time frame of p53 accumulation in the nucleus and upregulation of the apoptotic marker, cleaved PARP, partially overlapped with UBXN2A's upregulation (Fig. 2A). This result resembles a positive feedback mechanism between UBXN2A and p53's downstream apoptotic pathway. Meanwhile, the levels of HSC70 proteins, a general heat shock protein in the cytoplasm, remained unchanged following UVB exposure (Fig. 2A). As previously described [35], the time required to recruit and upregulate the specific co-factors and regulators can contribute to the delayed accumulation of p53 in the nucleus (12 hours after UBXN2A peak, Fig. 2B), resulting in an intermediate-to-late induction of apoptotic genes.
Nucleocytoplasmic translocation of UBXN2A in response to UVB is an early event. (A-B) Cells were exposed to UVB radiation for different time intervals followed by WB analysis of cytoplasmic and nuclear fractions. (A) The nucleocytoplasmic translocation of UBXN2A in response to UVB started 6 hours after UVB exposure and reached a maximum 24 hours after 1KJ/m2 UVB exposure in the cytoplasm. Meanwhile, the levels of heat shock protein HSC70 showed no alteration in response to 1KJ/m2 UVB irradiation. Upregulation of p53 in the cytoplasm and its maximized accumulation in the nucleus were coincident with the cytoplasmic upregulation of UBXN2A. Anti-cleaved PARP showed a typical fluctuation of cleaved-PARP representing the level of apoptosis after UVB exposure. (C) HCT-116 cells were treated with three different doses of UVB followed by immunoprecipitation with anti-mot-2 antibody. Immunoblotting of the pulled down protein revealed UVB-induced UBXN2A binds to mot-2 in a dose-dependent manner. (D) Following UVB treatment, cell lysates were subjected to immunoprecipitation using anti-p53 antibodies followed by WB. Results showed reduction of p53-mot-2 binding at 1 and 2 KJ/m2 can coincide with increased association of UBXN2A with mot-2 (Panel D versus panel C). IgG immobilized on magnetic beads was used as control.
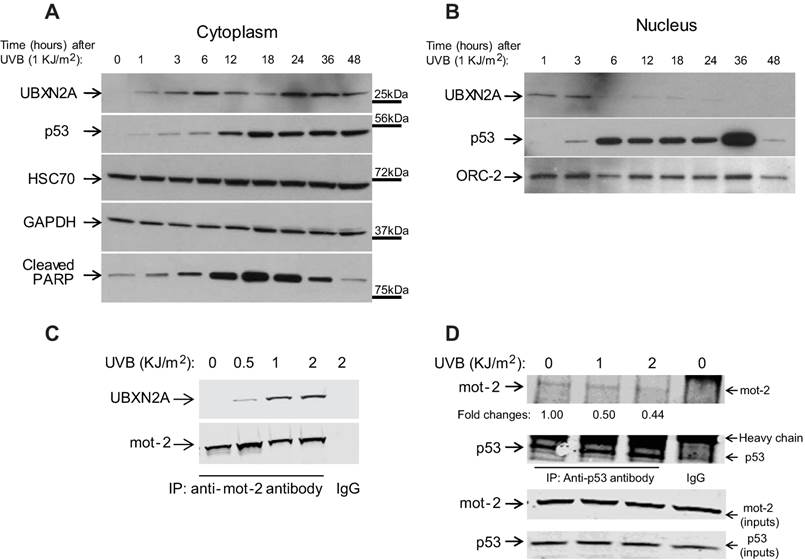
Because UBXN2A acts as a functional inhibitor of mot-2 in the cytoplasm [10], we decided to determine whether upregulated UBXN2A is functional in the cytoplasm following UVB treatment. Cytoplasmic cell lysates of HCT-116 cells treated with 0 to 2 KJ/m2 of UVB followed by 24 hours incubation were subjected to immunoprecipitation by anti-mot-2 antibodies. WB analysis in Fig. 2C shows that upregulated UBXN2A binds to mot-2 in the cytoplasm in a UVB-dose-dependent manner.
Binding UBXN2A to mot-2 leads to un-sequestration of p53 in the cytoplasm [10]. Based on the results shown in Figure 2C, we hypothesized that UBXN2A nuclear export following UVB ultimately displaces p53 from mot-2. HCT-116 cells were treated with UVB (0, 1, and 2 KJ/m2). After 24 hours, cytoplasmic fractions were subjected to immunoprecipitation using anti-p53 polyclonal antibodies or rabbit IgG (control). Pulled down proteins were analyzed with WB. Panel D in Figure 2 confirmed p53 binding to mot-2 protein attenuates following UV irradiation, and this event is synchronized with UBXN2A binding to mot-2 following 1 and 2 KJ/m2 UVB exposures (Fig. 2, Panel C).
These results indicate that nucleocytoplasmic translocation of UBXN2A is part of a positive regulatory element of DNA damage during UVB exposure. This can result in un-sequestration of p53 in the cytoplasm and consequent accumulation of p53 in the nucleus.
Etoposide induces nucleocytoplasmic translocation of UBXN2A
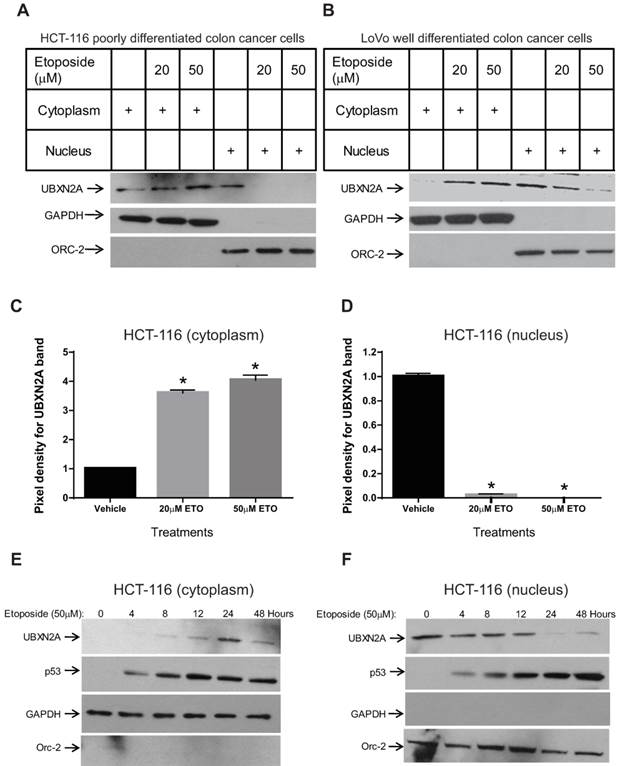
To further confirm that the nucleocytoplasmic translocation of UBXN2A is a general response to different types of DNA damage, we decided to use Etoposide, a genotoxic agent that induces DNA damage response in a p53-dependent manner [3] and is an alternative chemotherapeutic drug in colon cancer patients [36]. A poorly differentiated HCT-116 (Fig. 3A) colon cancer cell line and a well-differentiated LoVo (Fig. 3B) colon cancer cell line were treated with Etoposide (20μM and 50μM) [10] for 24 hours. We observed that, in both cell lines, Etoposide significantly increased UBXN2A levels in the cytoplasm while decreasing UBXN2A in the nucleus (Fig. 3A-3B). We then performed statistical analysis of the UBXN2A levels in the cytoplasm and the nucleus of HCT-116 after normalizing with the respective loading controls. A Bonferroni's modified t-test revealed that the Etoposide-induced DNA damage can significantly increase UBXN2A levels in the cytoplasm and decrease levels of UBXN2A in the nucleus, similar to UBXN2A's response to UVB (Fig. 3C-3D). With the same rationalization used for Figure 2A-B, we decided to determine the time line of UBXN2A's response to Etoposide. We treated HCT-116 cells with a clinical dosage of Etoposide (50μM) for different periods of time. WB analysis of the cytoplasm and nuclear cell lysates revealed that Etoposide increases cytoplasmic UBXN2A for up to 24 hours, and this event is coincident with a sustained decrease of nuclear UBXN2A (Fig. 3E-F). In addition, Etoposide induced a sustained increase of p53 levels in the cytoplasm and nucleus (Fig. 3E-F). Expectedly, we observed a decrease in the UBXN2A level after 48 hours incubation with Etoposide, which could be due to the late apoptosis and cell death induced by Etoposide-induced p53. Similar to UVB, we observed a coincident relationship between UBXN2A and p53 in response to Etoposide in the cytoplasm and nuclear compartments.
Etoposide induces nucleocytoplasmic translocation of UBXN2A in a dose- and time-dependent manner in poorly and well-differentiated colon cancer cells. (A-B) HCT-116 and LoVo cells were treated with Etoposide (20 and 50μM) for 24 hours. Nuclear and cytoplasm fractions were analyzed for UBXN2A localization. Etoposide induces UBXN2A nuclear export in both HCT-116 (poorly differentiated) and LoVo (well-differentiated) colon cancer cells. (C-D) The statistical analysis of HCT-116 data using Bonferroni's t-test revealed a significant upregulation and associated downregulation of UBXN2A levels in the cytoplasm and nucleus, respectively (n=3, *p<0.05). (E-F) HCT-116 cells were incubated with a clinical dose of Etoposide (50μM) for different time periods (4-48 hours). (E) DNA damage induced by Etoposide led to UBXN2A nuclear export following 4 hours incubation, and nucleocytoplasmic translocation was completed after 24 hours incubation. An anti-p53 antibody showed that upregulation of p53 started as early as 4 hours and reached maximum level at 48 hours, and it partially coincided with constitutively increased UBXN2A levels in the cytoplasm. Downregulation of UBXN2A after 48 hours may be due to the late-apoptosis/necrosis induced by p53.
The orchestrated UBXN2A nuclear export and its apoptosis events are only partially p53-dependent
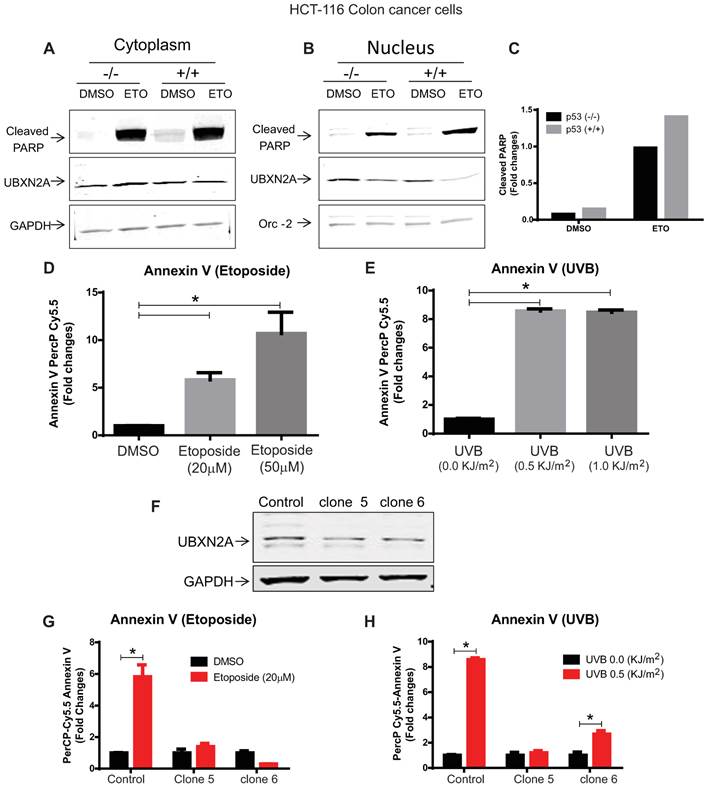
We decided to determine whether the presence of p53 is essential for UBXN2A nuclear export as well as apoptotic events (cleaved PARP) which occurs after UBXN2A nuclear export. We treated HCT-116 p53 +/+ and HCT-116 p53 -/- cells with the DNA damaging agent Etoposide (50μM) for 24 hours to evaluate the levels of UBXN2A and cleaved PARP in both cytoplasmic and nucleus compartments, as previously described [37, 38]. Cytoplasm and nuclear lysates were subjected to WB analysis using anti-UBXN2A, anti-cleaved PARP antibodies as well as cytoplasmic and nuclear markers and loading controls. Panels A and B in Figure 4 confirmed UBXN2A's nuclear exports in both HCT-116 p53 +/+ and p53 -/- cells. In addition, this set of experiments revealed a significant increase in cleaved PARP in both cytoplasmic and nuclear compartments in the presence of Etoposide in both p53 -/- and p53 +/+ cells. However, the sum of pixel intensities within the nucleus and cytoplasm of cleaved PARP revealed Etoposide generates more cleaved PARP in the presence of p53 protein (Fig. 4C). The experiment was reproduced two times with similar results. This set of results suggests UBXN2A nuclear export can be co-incident with apoptosis events (cleaved PARP) even in the absence p53, indicating p53 may have only a partial role in pre-apoptotic function of UBXN2A following DNA damage.
Silencing of UBXN2A decreases stress-induced apoptosis in colon cancer cells. (A-B) HCT-116 p53 -/- and p53 +/+ were treated with Etoposide (20μM) for 24 hours. Cytoplasmic and nuclear lysates were subjected to WB analysis using anti-cleaved PARP and anti-UBXN2A antibodies as well as anti-GAPDH and Orc-2 antibodies. (C) Signal intensities of cleaved PARP were quantified in both cytoplasmic and nuclear fractions. The sum of pixel densities of the cleaved PARP bands, normalized to the pixel densities of corresponding loading controls, was represented by bar graph. This experiment was conducted twice and generated comparable results. (D-E) Early apoptosis marker Annexin V significantly upregulated in HCT-116 p53 +/+, indicating the Annexin V marker can reliably indicate apoptosis following Etoposide and UVB stresses. (F) WB analysis verified efficient silencing of UBXN2A protein in HCT-116 cells (clones 5 and 6) stably expressing shRNA against UBXN2A. (G-H) HCT-116 expressing scrambled shRNA (control) and clones 5 and 6 were treated with Etoposide or UVB followed by flow-cytometry analysis using Annexin V marker. Statistical analysis revealed effective silencing of UBXN2A is associated with suppression of stress-induced early apoptosis in HCT-116 p53 +/+ cells (n=3, p<0.05).
UBXN2A is an essential factor in apoptosis events following DNA damage
To further understand the pre-apoptotic function of UBXN2A, we decided to employ an early apoptosis marker called Annexin V. Enns et al. showed both caspase-3 and phosphatidylserine translocation (Annexin V binding) represent apoptosis events in several cancer cells, including HCT-116 cells in the presence of WT-p53, while apoptosis was not detectable by Annexin V or caspase-3 assays in HCT-116 with inactivated p53 protein [39]. Based on this fact, we first decided to examine the level of Annexin V early apoptosis marker in HCT-116 exposed to Etoposide and UVB. Panels D and E in Figure 4 show that Annexin V showed a significant increase in response to Etoposide (20 and 50μM) and UVB (0.5 and 1 KJ/m2) in HCT-116 cells.
Using SilenciX technology (Tebu-bio, France), we stably silenced UBXN2A in the HCT-116 plus a control HCT-116 cell line expressing scrambled shRNA. A set of qRT-PCR (data not shown) and WB analysis (Fig. 4F) confirmed successful stable silencing of UBXN2A in HCT-116 cells. We then examined the levels of Annexin V in the presence of UBXN2A (scrambled shRNA) and shRNA against UBXN2A (clones 5 and 6). The data shown in Panels G and H of Figure 4 revealed the absence of UBXN2A significantly suppresses elevation of Annexin V apoptosis in HCT-116 cells exposed to Etoposide and UVB. The level of Annexin V showed a significant difference in clone 6 in response to UVB perhaps due to remaining UBXN2A protein. However, the level of Annexin V was significantly lower than control (Fig. 4H, column 6 versus column 2). Together, the results shown in Figure 4 suggest that UBXN2A plays important roles in apoptosis following DNA damage induced by Etoposide or UVB, and the presence of p53 can enhance the UBXN2A-induced apoptosis pathway.
In another set of experiments we decided to determine the kinetics of the nuclear and cytoplasmic p53 levels in HCT-116 UBXN2A-silenced. Control and silenced (clones 5 and 6) were treated with 1 and 2 KJ/m2 followed by WB analysis (Fig. S2A). Quantification of signals followed by normalization performed separately for treatment groups revealed the absence of UBXN2A can interfere with UVB-induced upregulation of p53 levels in the cytoplasm effectively at 1 KJ/m2 and moderately at 2 KJ/m2 (Fig. S2B). The absence of UBXN2A had no effects on upregulation of nuclear p53 (Fig. S2C). We repeated the same UVB-treatments (1 and 2 KJ/m2) in LoVo well-differentiated colon cancer cells and we achieved similar results (data not shown). One explanation for these contradictory results is that the nature of DNA damage induced by UVB is different from the DNA damage induced by Etoposide [40]. UVB induces double DNA breaks [41] as well as production of reactive oxygen species (ROS) [42] which can be more deleterious than Etoposide. An example is chromosome repositioning and altered nuclear organization, which are different in response to hydrogen peroxide versus UVB [43]. Therefore, while p53-mot-2 interaction sustains in the absence of UBXN2A, the upregulation of p53 in the nuclear compartment may in part be caused by other UVB-induced pathways in stressed cells.
Leptomycin B blocks UBXN2A nucleocytoplasmic translocation and its consequent apoptosis event in response to stress
To further understand the underlying mechanism of UBXN2A nucleocytoplasmic translocation, we decided to use Leptomycin B (LMB), a small molecule that inhibits nuclear export of proteins via inhibition of the CRM1 (chromosome region maintenance 1) pathway [9]. In this set of experiments, we first confirmed that Etoposide induces UBXN2A translocation from nucleus to cytoplasm in a dose-dependent manner following 24 hours incubation. When we blocked nuclear export with LMB, the level of UBXN2A showed no significant increase in the cytoplasm (Fig. 5A). In fact, UBXN2A remained in the nucleus regardless of the different doses of Etoposide (Fig. 5B). The level of p47, another member of the UBXD family in the cytoplasm, did not change in response to Etoposide with or without the presence of LMB treatment (Fig. 5A-B).
UBXN2A nucleocytoplasmic translocation is mediated via the CRM1 pathway. (A-B) HCT-116 cells were treated with Leptomycin B (LMB, an inhibitor of the CRM1 nuclear export pathway) and Etoposide for 24 hours. Prepared cytoplasmic and nuclear fractions were subjected to WB analysis. (A) In the cytoplasm, Etoposide-induced UBXN2A nuclear export was inhibited by LMB. (B) UBXN2A protein accumulated in the nucleus of cells following treatment with LMB in the presence of Etoposide. P47 remained unchanged in the presence and the absence of LMB. Etoposide-induced cleaved PARP proteins were observed in the presence and the absence of LMB. However, LMB caused less dramatic accumulation of cleaved-PARP following Etoposide treatment. (C-D) Quantification of the protein bands detected in WB further confirmed that the cytoplasmic UBXN2A protein levels (normalized with loading controls) were dramatically increased in the absence of LMB (Panel C: columns 2 and 3 versus 1) as compared to cells treated with LMB (Panel C: columns 5 and 6 versus 4). A similar phenomenon was observed in nuclei as analysis of protein bands (Panel D) showed the presence of LMB can neutralize UBXN2A nuclear export induced by Etoposide (Panel D, columns 4, 5, and 6 versus columns 1, 2, and 3). (E-F) Immunofluorescence analysis of UBXN2A in HCT-116 and LoVo cells treated with Etoposide for 24 hours confirmed the induction of DNA damage results in a significant translocation of UBXN2A from the nucleus to the cytoplasmic compartment. DAPI-stained cells show characteristic chromatin condensation and apoptotic bodies, particularly in LoVo well-differentiated cells treated with Etoposide. (E) The proposed working model suggests that, following genotoxic stress, UBXN2A proteins translocate from nucleus to cytoplasm. This leads to the binding of UBXN2A to mot-2 oncoprotein, which has several oncogenic functions in tumors, including cytoplasmic tethering of p53 protein.
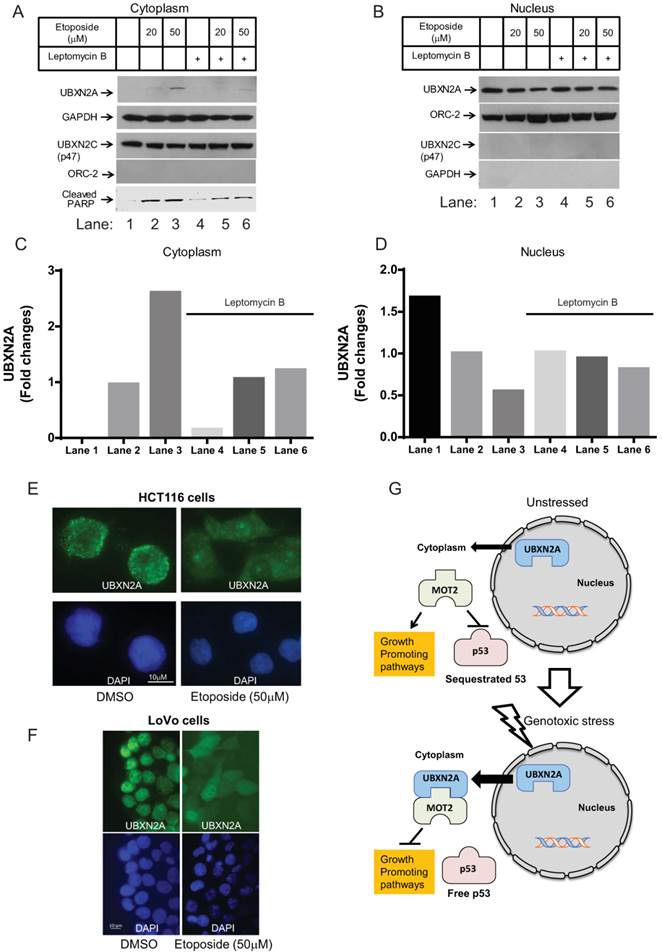
We quantified UBXN2A levels in Panels 5A (cytoplasmic fraction) and 5B (nucleus fraction). After normalization of UBXN2A signals with GAPDH (cytoplasmic marker) and Orc-2 (nuclear marker), we compared UBXN2A nuclear export in the presence and the absence of LMB. Quantification of UBXN2A signals confirmed LMB can suppress UBXN2A nuclear export following Etoposide (Fig. 5C-D). This experiment was conducted twice with similar results.
More importantly, we found upregulation of UBXN2A in the cytoplasm led to upregulation of cleaved PARP in the absence of LMB. However, in the presence of both Etoposide and LMB, we observed a lower level of apoptotic marker cleaved PARP, which was coincident with nuclear tethering of UBXN2A (Fig. 5A). Finally, we decided to determine whether LMB treatment can nullify the disrupted interaction between mot-2 and p53 by Etoposide-mediated UBXN2A cytoplasmic relocation. We conducted a set of immunoprecipitation (IP) experiments in HCT-116 cells treated with Etoposide (50mM), LMB (25ng/mL), and a combination of Etoposide and LMB for 24 hours. Cytoplasmic cell lysates were subjected to IP using anti-p53 polyclonal antibodies immobilized on IgA magnetic beads. Pulled down proteins were subjected to WB analysis using anti-mot-2 antibodies. We normalized the mot-2 signal after IP to mot-2 inputs. As illustrated in Figure S2D, while Etoposide can decrease mot-2-p53 binding (lane 3 versus lane 2), the presence of LMB increases mot-2 binding to p53 (lane 4 versus lane 3). In the reverse reaction, a combination of Etoposide and LMB decreases mot-2 binding to p53 in comparison to LMB alone (lane 5 versus lane 4). Together, these results suggest inhibition of nuclear export by LMB can partially nullify UBXN2A-dependent disruption between mot-2 and p53 upon Etoposide stress.
Using immunofluorescent staining followed by confocal microscopy of fixed HCT-116 cells, we examined the distribution of UBXN2A after 24 hours' incubation with DMSO or Etoposide. Figure 5E shows that the activated UBXN2A is indeed translocated from the nucleus stained with DAPI (blue) to the cytoplasm upon Etoposide DNA damage response. We observed similar translocation of UBXN2A from the nucleus to the cytoplasm when we stained UBXN2A and the nucleus in LoVo colon cancer cells treated with DMSO or Etoposide for 24 hours (Fig. 5F). As a well-differentiated colon cancer cell line, LoVo cells showed greater sensitivity to Etoposide, representing with condensed DNA and apoptotic bodies as reported previously with overexpression of UBXN2A [10].
Based on these current findings, we propose a model in which, under DNA-damaging stress conditions, UBXN2A can be trafficked to the cytoplasm to bind and inhibit mot-2 oncoprotein and consequently release p53. Suppression of mot-2 oncoprotein and the presence of free p53 can functionally activate the relevant downstream apoptotic pathways (Fig. 5G).
Discussion
To achieve an efficient and coordinated execution of cell death, signals generated in the nucleus following DNA damage need to propagate to all cellular compartments. The nucleocytoplasmic translocation of execution factors and their regulators is a vital part of the pro-apoptotic and apoptotic pathways, and this translocation is typically deregulated in cancer. Several proteins involved in the apoptosis pathway have been shown to traffic in and out of the nucleus in a timely manner following exogenous stresses [9, 33, 44]. Due to the vital role of p53 in the nucleus upon DNA damage, there is a set of specific regulators that translocate across the nuclear membrane to tightly regulate the nuclear import of p53 following genotoxic stresses [8, 9, 45]. Understanding the mechanisms of the nucleocytoplasmic transportation of these regulators provides a more detailed picture of their roles in cancer biology and their therapeutic potential.
UBXN2A disrupts tumor cell proliferation and survival in colon cancer cells largely by reactivation of wild-type p53 in the cytoplasm without affecting normal cells [10]. Understanding how UBXN2A interactions and functions are regulated in cancer cells remains a subject of intense interest. In this study, we examined the effects of the DNA damage stress induced by UVB and Etoposide on the subcellular distribution of UBXN2A. We found that increased cytoplasmic UBXN2A following exogenous stress is a functional protein and binds to mot-2 oncoprotein [46] depending on the severity of the stress [5] in a dose-dependent manner. As previously described, UBXN2A's binding to mot-2 protein results in unsequestration, stabilization, and upregulation of p53 in the cytoplasm and nucleus compartment [10]. A set of immunoprecipitation experiments confirmed UVB stress decreases p53-mot-2 interaction in a UVB dose-dependent manner. As previously described, upregulation of p53 in both nucleus and cytoplasmic fractions following DNA damage stress [47] were observed before and after UBXN2A nuclear export following DNA damage. The present results indicate that stress-induced elevation of UBXN2A in the cytoplasm is a critical modulator that exists alongside other p53 regulatory mechanisms activated during DNA damage response.
An example of a nuclear-positive regulation is the USP10 deubiquitinating enzyme, which translocates to the nucleus and stabilizes p53 upon DNA damage [9]. PUMA acts as a cytoplasmic positive regulator of p53 upon DNA damage [8], allowing p53 to induce mitochondrial membrane permeabilization and apoptosis [48]. UBXN2A increases in the cytoplasmic fraction upon DNA damage and displaces those p53 attached to mot-2 oncoprotein during the DNA damage response [5]. Further investigations are underway in our group to determine the fate of p53 in both cytoplasmic and nuclear fractions after releasing from mot-2 by UBXN2A.
Following the above experiments, three questions remained to be answered: 1) Does translocation of UBXN2A take place in the absence of p53? 2) Does UBXN2A induce apoptosis in the absence of p53 by inhibiting mot-2? 3) Is stress-induced apoptosis (UVB and Etoposide) in colon cancer cells suppressed in the absence of UBXN2A? The present study showed UBXN2A can translocate to the cytoplasm regardless of p53 status and induce apoptosis. These results indicate cytoplasmic UBXN2A can target other oncogenic functions of mot-2 [5, 49-52] beyond the p53 axis. However, as previously described, the presence of WT-p53 can further enhance apoptotic events following un-sequestration from mot-2 [46, 53].
Our results demonstrated that UBXN2A translocation to the cytoplasm is associated with induction of apoptosis determined by cleaved PARP protein. The fragmentation of PARP protein is an accepted protein marker for the activation of the p53/caspase-3 apoptotic pathway, and a high level of this cleaved PARP is correlated with a better prognosis in colon tumors [54]. In addition to cleaved PARP, we showed upregulation of cytoplasmic UBXN2A was also coincident with increased Annexin V, an early apoptosis marker following both UVB and Etoposide exposure. More importantly, we found silencing of UBXN2A suppresses elevation of Annexin V observed in the presence of DNA damage stress induced by UVB and Etoposide. These sets of results suggest the presence of UBXN2A and its mot-2 inhibitory functions is a critical step during the execution of DNA damage response in colon cancer cells.
HSC70 expression remained unchanged during the nucleocytoplasmic translocation of UBXN2A, implying that the induction of UBXN2A during DNA damage response is controlled by specific pathways and is not simply the consequence of the general heat shock stress following UVB exposure. Treatment of cancer cells with the CRM1-specific inhibitor LMB interrupted the cytoplasmic translocation of UBXN2A in response to stress and recovery of p53-mot-2 binding, confirming that nucleocytoplasmic trafficking of UBXN2A following DNA damage occurs through the CRM-1 nuclear export pathway and is essential in order to inhibit mot-2 and unsequester p53.
Posttranslational modification such as phosphorylation, ubiquitination, and sumoylation has been shown to regulate nuclear export of several proteins through the CRM-1 pathway [33, 55, 56]. UBXN2A can be posttranslationally modified, and the modification(s) of UBXN2A is likely to regulate UBXN2A nuclear export in response to DNA damage. As previously described for other proteins with an NES signal [56, 57], it is also possible that posttranslational modification of one or more UBXN2A nuclear protein partners in response to DNA damage may cause them to dissociate from UBXN2A, unmasking the NES signal and enabling UBXN2A to interact with the CRM-1 receptor for nuclear export. The specific role of these mechanisms in regulating the nuclear export of UBXN2A is under investigation by our group. Finally, it has been shown that APC (adenomatous polyposis coli), a dominant tumor suppressor protein in colon cancer, uses its NES signals to coordinate nuclear export of the oncogene product β-catenin for proteasomal degradation in the cytoplasm [58, 59]. Due to its ubiquitin-like activity [28], it is possible that UBXN2A, as part of its home-defense mechanism, also facilitates nuclear export of specific potential oncogenic factor(s) generated during the progression of cancer to be degraded by the 26S proteasome complex in the cytoplasm compartment. As a matter of fact, Ryu et al. recently reported that the oncogenic protein mot-2 localized in the nucleus in addition to its upregulation in the cytoplasm in cancer cells. Nuclear mot-2 promotes carcinogenesis and cancer cell metastasis by inactivation of p53 and by activation of telomerase and heterogeneous ribonucleoprotein K (hnRNP-K) proteins [52]. Ongoing investigation in our group will determine whether UBXN2A can interfere with oncogenic function of mot-2 in a similar mechanism as described for APC [58, 59].
In summary, our data show the DNA damage response is one of the underlying mechanisms that regulates UBXN2A in two colon cancer cell lines, one poorly- and one well-differentiated. This nuclear export would be expected to be necessary for the cytoplasmic apoptotic function of UBXN2A [10] but may also be important for its nuclear anti-cancer effect, which remains to be determined. A deeper understanding of how this process is initiated, regulated, and ended during the progression of colon tumors with and without genotoxic stress will help to resolve the relative importance of these activities to overall UBXN2A function. The cellular response to p53 activation following DNA damage is determined by cell type and stimuli [60]. The DNA damage stimuli (UVB and Etoposide) used in this study clarified the role of UBXN2A, mot-2 oncoprotein, p53 and its downstream biological cascade during DNA damage.
Supplementary Material
Figures S1-S2, and Table S1.
Acknowledgements
This project has been funded by the start-up package (Division of Basic Biomedical Sciences, University of South Dakota) and the National Institute of General Medical Sciences of the National Institutes of Health under award number 5P20GM103548 (Miskimins).
Conflict of interest
The authors disclose no potential conflicts of interest.
References
1. Appella E, Anderson CW. Post-translational modifications and activation of p53 by genotoxic stresses. Eur J Biochem. 2001;268:2764-72
2. Li Q, Feldman RA, Radhakrishnan VM, Carey S, Martinez JD. Hsf1 is required for the nuclear translocation of p53 tumor suppressor. Neoplasia. 2008;10:1138-45
3. Fritsche M, Haessler C, Brandner G. Induction of nuclear accumulation of the tumor-suppressor protein p53 by DNA-damaging agents. Oncogene. 1993;8:307-18
4. Wadhwa R, Takano S, Robert M, Yoshida A, Nomura H, Reddel RR. et al. Inactivation of tumor suppressor p53 by mot-2, a hsp70 family member. J Biol Chem. 1998;273:29586-91
5. Lu WJ, Lee NP, Kaul SC, Lan F, Poon RT, Wadhwa R. et al. Mortalin-p53 interaction in cancer cells is stress dependent and constitutes a selective target for cancer therapy. Cell Death Differ. 2011;6:1046-56
6. Yang HY, Wen YY, Chen CH, Lozano G, Lee MH. 14-3-3 sigma positively regulates p53 and suppresses tumor growth. Mol Cell Biol. 2003;23:7096-107
7. Zhang Y, Xiong Y, Yarbrough WG. ARF promotes MDM2 degradation and stabilizes p53: ARF-INK4a locus deletion impairs both the Rb and p53 tumor suppression pathways. Cell. 1998;92:725-34
8. Chipuk JE, Bouchier-Hayes L, Kuwana T, Newmeyer DD, Green DR. PUMA couples the nuclear and cytoplasmic proapoptotic function of p53. Science. 2005;309:1732-5
9. Yuan J, Luo K, Zhang L, Cheville JC, Lou Z. USP10 regulates p53 localization and stability by deubiquitinating p53. Cell. 2010;140:384-96
10. Sane S, Abdullah A, Boudreau DA, Autenried RK, Gupta BK, Wang X. et al. Ubiquitin-like (UBX)-domain-containing protein, UBXN2A, promotes cell death by interfering with the p53-Mortalin interactions in colon cancer cells. Cell Death Dis. 2014;5:e1118
11. Jauhiainen A, Thomsen C, Strombom L, Grundevik P, Andersson C, Danielsson A. et al. Distinct cytoplasmic and nuclear functions of the stress induced protein DDIT3/CHOP/GADD153. PLoS One. 2012;7:e33208
12. Zhang J, Cao M, Dong J, Li C, Xu W, Zhan Y. et al. ABRO1 suppresses tumourigenesis and regulates the DNA damage response by stabilizing p53. Nat Commun. 2014;5:5059
13. Hsu TI, Wang MC, Chen SY, Yeh YM, Su WC, Chang WC. et al. Sp1 expression regulates lung tumor progression. Oncogene. 2012;31:3973-88
14. Huang Q, Raya A, DeJesus P, Chao SH, Quon KC, Caldwell JS. et al. Identification of p53 regulators by genome-wide functional analysis. Proc Natl Acad Sci U S A. 2004;101:3456-61
15. Abdullah A, Sane S, Branick KA, Freeling JL, Wang H, Zhang D. et al. A plant alkaloid, veratridine, potentiates cancer chemosensitivity by UBXN2A-dependent inhibition of an oncoprotein, mortalin-2. Oncotarget. 2015
16. Bhasin M, Raghava GP. ESLpred: SVM-based method for subcellular localization of eukaryotic proteins using dipeptide composition and PSI-BLAST. Nucleic Acids Res. 2004;32:W414-9
17. Alexandru G. Exploring the role of p97 and its UBX-domain cofactors through identification of their interacting proteins. Methods Mol Biol. 2012;832:305-12
18. Tanaka T, Okada M, Hozumi Y, Tachibana K, Kitanaka C, Hamamoto Y. et al. Cytoplasmic localization of DGKzeta exerts a protective effect against p53-mediated cytotoxicity. J Cell Sci. 2013;126:2785-97
19. Latonen L, Taya Y, Laiho M. UV-radiation induces dose-dependent regulation of p53 response and modulates p53-HDM2 interaction in human fibroblasts. Oncogene. 2001;20:6784-93
20. Nicholls CD, Shields MA, Lee PW, Robbins SM, Beattie TL. UV-dependent alternative splicing uncouples p53 activity and PIG3 gene function through rapid proteolytic degradation. J Biol Chem. 2004;279:24171-8
21. Wang Y, Rosenstein B, Goldwyn S, Zhang X, Lebwohl M, Wei H. Differential regulation of P53 and Bcl-2 expression by ultraviolet A and B. J Invest Dermatol. 1998;111:380-4
22. Takasawa R, Nakamura H, Mori T, Tanuma S. Differential apoptotic pathways in human keratinocyte HaCaT cells exposed to UVB and UVC. Apoptosis. 2005;10:1121-30
23. Chen X, Chen J, Gan S, Guan H, Zhou Y, Ouyang Q. et al. DNA damage strength modulates a bimodal switch of p53 dynamics for cell-fate control. BMC Biol. 2013;11:73
24. Inoue T, Wu L, Stuart J, Maki CG. Control of p53 nuclear accumulation in stressed cells. FEBS Lett. 2005;579:4978-84
25. Lai CY, Tsai AC, Chen MC, Chang LH, Sun HL, Chang YL. et al. Aciculatin induces p53-dependent apoptosis via MDM2 depletion in human cancer cells in vitro and in vivo. PLoS One. 2012;7:e42192
26. Kim WH, Yeo M, Kim MS, Chun SB, Shin EC, Park JH. et al. Role of caspase-3 in apoptosis of colon cancer cells induced by nonsteroidal anti-inflammatory drugs. Int J Colorectal Dis. 2000;15:105-11
27. Kondo H, Rabouille C, Newman R, Levine TP, Pappin D, Freemont P. et al. p47 is a cofactor for p97-mediated membrane fusion. Nature. 1997;388:75-8
28. Rezvani K, Teng Y, Pan Y, Dani JA, Lindstrom J, Garcia Gras EA. et al. UBXD4, a UBX-containing protein, regulates the cell surface number and stability of alpha3-containing nicotinic acetylcholine receptors. J Neurosci. 2009;29:6883-96
29. Madsen L, Andersen KM, Prag S, Moos T, Semple CA, Seeger M. et al. Ubxd1 is a novel co-factor of the human p97 ATPase. Int J Biochem Cell Biol. 2008;40:2927-42
30. Yi J, Lu G, Li L, Wang X, Cao L, Lin M. et al. DNA damage-induced activation of CUL4B targets HUWE1 for proteasomal degradation. Nucleic Acids Res. 2015;43:4579-90
31. Alexandru G, Graumann J, Smith GT, Kolawa NJ, Fang R, Deshaies RJ. UBXD7 binds multiple ubiquitin ligases and implicates p97 in HIF1alpha turnover. Cell. 2008;134:804-16
32. Meyer H. p97 complexes as signal integration hubs. BMC Biol. 2012;10:48
33. Sen N, Kumari R, Singh MI, Das S. HDAC5, a key component in temporal regulation of p53-mediated transactivation in response to genotoxic stress. Mol Cell. 2013;52:406-20
34. Kurki S, Latonen L, Laiho M. Cellular stress and DNA damage invoke temporally distinct Mdm2, p53 and PML complexes and damage-specific nuclear relocalization. J Cell Sci. 2003;116:3917-25
35. Riley T, Sontag E, Chen P, Levine A. Transcriptional control of human p53-regulated genes. Nat Rev Mol Cell Biol. 2008;9:402-12
36. Amiri F, Zarnani AH, Zand H, Koohdani F, Jeddi-Tehrani M, Vafa M. Synergistic anti-proliferative effect of resveratrol and etoposide on human hepatocellular and colon cancer cell lines. Eur J Pharmacol. 2013;718:34-40
37. Soldani C, Bottone MG, Pellicciari C, Scovassi AI. Two-color fluorescence detection of Poly (ADP-Ribose) Polymerase-1 (PARP-1) cleavage and DNA strand breaks in etoposide-induced apoptotic cells. Eur J Histochem. 2001;45:389-92
38. Soldani C, Lazze MC, Bottone MG, Tognon G, Biggiogera M, Pellicciari CE. et al. Poly(ADP-ribose) polymerase cleavage during apoptosis: when and where? Exp Cell Res. 2001;269:193-201
39. Enns L, Bogen KT, Wizniak J, Murtha AD, Weinfeld M. Low-dose radiation hypersensitivity is associated with p53-dependent apoptosis. Mol Cancer Res. 2004;2:557-66
40. Long BH, Musial ST, Brattain MG. Single- and double-strand DNA breakage and repair in human lung adenocarcinoma cells exposed to etoposide and teniposide. Cancer Res. 1985;45:3106-12
41. Baumstark-Khan C, Hentschel U, Nikandrova Y, Krug J, Horneck G. Fluorometric analysis of DNA unwinding (FADU) as a method for detecting repair-induced DNA strand breaks in UV-irradiated mammalian cells. Photochem Photobiol. 2000;72:477-84
42. Rastogi RP, Singh SP, Hader DP, Sinha RP. Detection of reactive oxygen species (ROS) by the oxidant-sensing probe 2',7'-dichlorodihydrofluorescein diacetate in the cyanobacterium Anabaena variabilis PCC 7937. Biochem Biophys Res Commun. 2010;397:603-7
43. Ioannou D, Kandukuri L, Quadri A, Becerra V, Simpson JL, Tempest HG. Spatial positioning of all 24 chromosomes in the lymphocytes of six subjects: evidence of reproducible positioning and spatial repositioning following DNA damage with hydrogen peroxide and ultraviolet B. PLoS One. 2015;10:e0118886
44. Wilson AJ, Arango D, Mariadason JM, Heerdt BG, Augenlicht LH. TR3/Nur77 in colon cancer cell apoptosis. Cancer Res. 2003;63:5401-7
45. Marchenko ND, Hanel W, Li D, Becker K, Reich N, Moll UM. Stress-mediated nuclear stabilization of p53 is regulated by ubiquitination and importin-alpha3 binding. Cell Death Differ. 2010;17:255-67
46. Kaul SC, Aida S, Yaguchi T, Kaur K, Wadhwa R. Activation of wild type p53 function by its mortalin-binding, cytoplasmically localizing carboxyl terminus peptides. J Biol Chem. 2005;280:39373-9
47. Giaccia AJ, Kastan MB. The complexity of p53 modulation: emerging patterns from divergent signals. Genes Dev. 1998;12:2973-83
48. Chipuk JE, Kuwana T, Bouchier-Hayes L, Droin NM, Newmeyer DD, Schuler M. et al. Direct activation of Bax by p53 mediates mitochondrial membrane permeabilization and apoptosis. Science. 2004;303:1010-4
49. Wu PK, Hong SK, Veeranki S, Karkhanis M, Starenki D, Plaza JA. et al. A mortalin/HSPA9-mediated switch in tumor-suppressive signaling of Raf/MEK/extracellular signal-regulated kinase. Mol Cell Biol. 2013;33:4051-67
50. Yang L, Guo W, Zhang Q, Li H, Liu X, Yang Y. et al. Crosstalk between Raf/MEK/ERK and PI3K/AKT in suppression of Bax conformational change by Grp75 under glucose deprivation conditions. J Mol Biol. 2011;414:654-66
51. Saar Ray M, Moskovich O, Iosefson O, Fishelson Z. Mortalin/Grp75 Binds to Complement C9 and Plays a Role in Resistance to Complement-Dependent Cytotoxicity. J Biol Chem. 2014
52. Ryu J, Kaul Z, Yoon AR, Liu Y, Yaguchi T, Na Y. et al. Identification and functional characterization of nuclear mortalin in human carcinogenesis. J Biol Chem. 2014;289:24832-44
53. E. Oki KA, H. Saeki, T. Ohga, Y. Kakeji, S. Tsujitani, T. Kusumoto, Y. Maehara. Prognostic significance of mortalin expresion in gastric adenocarcinoma with wild-type p53. J Clin Oncol. 2011;29:abstr_e14662
54. Noble P, Vyas M, Al-Attar A, Durrant S, Scholefield J, Durrant L. High levels of cleaved caspase-3 in colorectal tumour stroma predict good survival. Br J Cancer. 2013;108:2097-105
55. Ishida N, Hara T, Kamura T, Yoshida M, Nakayama K, Nakayama KI. Phosphorylation of p27Kip1 on serine 10 is required for its binding to CRM1 and nuclear export. J Biol Chem. 2002;277:14355-8
56. Carter S, Bischof O, Dejean A, Vousden KH. C-terminal modifications regulate MDM2 dissociation and nuclear export of p53. Nat Cell Biol. 2007;9:428-35
57. Feng Z, Kachnic L, Zhang J, Powell SN, Xia F. DNA damage induces p53-dependent BRCA1 nuclear export. J Biol Chem. 2004;279:28574-84
58. Su Y, Fu C, Ishikawa S, Stella A, Kojima M, Shitoh K. et al. APC is essential for targeting phosphorylated beta-catenin to the SCFbeta-TrCP ubiquitin ligase. Mol Cell. 2008;32:652-61
59. Henderson BR. Nuclear-cytoplasmic shuttling of APC regulates beta-catenin subcellular localization and turnover. Nat Cell Biol. 2000;2:653-60
60. Li J, Tan J, Zhuang L, Banerjee B, Yang X, Chau JF. et al. Ribosomal protein S27-like, a p53-inducible modulator of cell fate in response to genotoxic stress. Cancer Res. 2007;67:11317-26
Author contact
![]() Corresponding author: Dr. Khosrow Rezvani, Division of Basic Biomedical Sciences, Sanford School of Medicine, The University of South Dakota, 414 E. Clark Street, Lee Medical Building, Vermillion, SD 57069, Telephone: 605-677-6898, Fax: 605-658-6383, E-mail: khosrow.rezvaniedu
Corresponding author: Dr. Khosrow Rezvani, Division of Basic Biomedical Sciences, Sanford School of Medicine, The University of South Dakota, 414 E. Clark Street, Lee Medical Building, Vermillion, SD 57069, Telephone: 605-677-6898, Fax: 605-658-6383, E-mail: khosrow.rezvaniedu