Impact Factor ISSN: 1837-9664

 Global reach, higher impact
Global reach, higher impactJ Cancer 2015; 6(12):1214-1221. doi:10.7150/jca.12191 This issue Cite
Research Paper
Expression Profiling Identifies Bezafibrate as Potential Therapeutic Drug for Lung Adenocarcinoma
Xinyan Liu1†, Xiaoqin Yang3†, Xinmei Chen4, Yantao Zhang2, Xuebin Pan2, Guiping Wang2 ![]() , Yun Ye5
, Yun Ye5 ![]()
1. Magazine office, Guangzhou Medical University, Guangzhou 510182, P.R. China
2. Department of Pharmacy, College of Health sciences, Guangzhou Medical University, Guangzhou 510180, P.R. China
3. Department of Bioinformatics, School of Life Science and Technology, Tongji University, Shanghai 200092, P.R. China
4. Department of Biochemistry, School of Basic Science, Guangzhou Medical University, Guangzhou 510182, P.R. China
5. College of Biological and Chemical Engineering, Guangxi University of Science and Technology, Liuzhou 545006, P.R. China
† These authors contributed equally to this work.
Received 2015-3-20; Accepted 2015-7-27; Published 2015-9-20
Abstract
Drug-induced gene expression patterns that invert disease profiles have recently been illustrated to be a new strategy for drug-repositioning. In the present study, we validated this approach and focused on prediction of novel drugs for lung adenocarcinoma (AC), for which there is a pressing need to find novel therapeutic compounds. Firstly, connectivity map (CMap) analysis computationally predicted bezafibrate as a putative compound against lung AC. Then this hypothesis was verified by in vitro assays of anti-proliferation and cell cycle arrest. In silico docking evidence indicated that bezafibrate could target cyclin dependent kinase 2(CDK2), which regulates progression through the cell cycle. Furthermore, we found that bezafibrate can significantly down-regulate the expression of CDK2 mRNA and p-CDK2. Using a nude mice xenograft model, we also found that bezafibrate could inhibit tumor growth of lung AC in vivo. In conclusion, this study proposed bezafibrate as a potential therapeutic option for lung AC patients, illustrating the potential of in silico drug screening.
Keywords: Lung adenocarcinoma, Connectivity map, Drug-repositioning, Bezafibrate
Introduction
Presently, lung cancer is one of the leading causes of cancer death around the world. Lung adenocarcinoma (AC), which usually originates in peripheral lung tissue, accounts for nearly 40% of primary lung cancers. Surgical resection remains the most effective therapy for early-stage lung AC patients. Unfortunately, survival rates for many patients are still disappointingly low due to late diagnosis. Though some targeting therapeutic agents, such as epithelial growth factor receptor (EGFR) tyrosine kinase inhibitors (TKIs), show survival advantages in some other types of lung cancer, survival improvement among patients with advanced lung AC is still unsatisfactory. Furthermore, some of these agents were confirmed to be ineffective or easy to gain drug resistance in several cases because of the genetic heterogeneity, including different gene mutations [1, 2]. Thus, gaining insight into the biological information such as expression profile and biological pathway in lung AC might facilitate the identification of novel and effective therapeutic agents.
According to conservative estimates, on average, a new drug approval takes about 10 years and costs around 1 billion dollars, and totally more than 90% of drugs would fail during different development stages. Recently, system and computational biology approaches have been used to screen approved drugs for new purposes, which provides valuable contributions in drug discovery and repositioning[3-5]. The differences in microarray expression profiles between disease and normal status can uncover the disease-gene-drug connections, which are often missing, hidden, or too scattered to find. Connectivity map (CMap, current version build 02) contains more than 7,000 expression profiles representing 1,309 compounds on cultured human cell lines, and has been regarded as one useful tool for discovering novel applications of existing drugs or compounds[6]. Recently, several studies have utilized connectivity mapping approach to predict candidate therapeutic compounds to prostate cancer[7], gastric cancer[8], neuroblastoma[9], etc.
In the present study, we set out to identify potential therapeutics for lung AC by using drug-induced gene-expression profiles to retrieve novel candidate drugs with expression profiles in normal and cancer tissues being inversely matched. Then we identified a list of compounds, whose treatment could reverse the expression direction in several cancer cells. One of the top-ranked candidates, bezafibrate was selected as a potential therapeutic agent for lung AC. Our subsequent experiments further validated that bezafibrate inhibited cell proliferation and induced G1 arrest in A549 and GLC-82 cells.
Materials and Methods
Analysis of gene expression and Connectivity Map
Lung AC microarray dataset used here for gene expression analysis was obtained from Gene Expression Omnibus (GEO) (accession number: GSE7670). Details of the dataset were summarized in Supplementary Table S1. The analysis workflow of microarray datasets and CMap was shown in Supplementary Figure S1. Firstly, dChip software (http://www.hsph.harvard.edu/cli/complab/dchip/) was used to normalize all chips together, and then derive fold changes for every single probe of lung AC samples relative to normal controls. Differentially expressed genes were identified with a two-fold change cutoff and corresponding probe IDs were obtained as the lung AC signatures for further CMap analysis. Then, those lung AC signatures consisting of up- and down-regulated genes were submitted simultaneously to CMap for compounds screening. Only records with negative score and p-value less than 0.05 were retained as potential therapeutic agents for lung AC.
Compounds and Cell culture
Bezafibrate (Sigma Aldrich) was dissolved in dimethylsulfoxide (DMSO) to a 0.5 M stock concentration and stored at -20°C. The concentration of DMSO was kept the same throughout each experiment, to a maximum of 0.5% v/v. Two human lung AC cell lines A549 and GLC-82 were obtained from Guangzhou Medical University cell repository and SUN YAT-SEN University cell repository, respectively. Cells were cultured in RPMI1640 medium supplemented with 10% fetal bovine serum at 37°C in a humidified atmosphere of 5% CO2.
Cell proliferation and viability assay
Cell viability assay was performed as previously described[10]. Briefly, A549 or GLC-82 cells were seeded into 96-well plates (Costar, Corning, NY) at a density of 4×104 cells/ml and exposed to the indicated concentrations of bezafibrate (BEZ) for 24, 48 and 74h, respectively. After drug exposure for the indicated concentrations and times, 10 μl of MTT reagent (0.5mg/ml) was added and incubated at 37°C for 4h. Then, the MTT solution was removed and DMSO was added to each well to dissolve the blue crystalline precipitate at 37°C for 20 min. The optical density values (OD value) were measured at 490 nm using a microplate reader (Tecan Sunrise, Switzerland). The cell viability was calculated and shown as the mean ± standard error of triplicate experiments.
Cell cycle analysis
A549 or GLC-82 cells were seeded into 6-well microplates at 5×106 cells/well, and incubated in medium containing 0, 100 or 200μM bezafibrate at 37°C for 24h. Cells were harvested and fixed with 70%(v/v) cold ethanol at -4°C for 12h. After fixed, cells were washed with cold phosphate-buffered saline (PBS), 100ug/ml RNase A and 50μg/ml propidium iodide staining solution were added and incubated for 30 minutes in dark. Finally, cells were analyzed by FACScan flow cytometer (Becton Dickinson, USA) and evaluated using the CellQuest program software.
Reverse docking using TarFisDock
TarFisDock (www.dddc.ac.cn/tarfisdock)[11], a webserver for identifying or seeking targets for a given ligand or a novel synthetic compound, could dock the given molecule into the protein targets in Potential Drug Target Database [12] (PDTD: http://www.dddc.ac.cn/pdtd/), and output candidates ranked by the corresponding energy scores. We performed TarFisDock analysis according to the reverse docking procedure. The 2D structure of bezafibrate was obtained from DrugBank (http://www.drugbank.ca), and gasteiger charges were assigned to the molecular structure. Then, the standard mol2 as query file was input into TarFisDock system for seeking bezafibrate-binding targets.
Real-Time reverse transcription-PCR (qRT-PCR)
qRT-PCR detection was done according to our previous experimental protocol[10]. Briefly, A549 or GLC-82 cells were treated with bezafibrate or DMSO for 24 h, and total RNAs were isolated with Trizol (Invitrogen, Karlsruhe, Germany) according to the manufacturer protocol. The qPCR amplification protocol was as follows: 95℃ for 5 min, followed by 40 cycles of 95℃ for 15s, 60℃for 15s and 72℃ for 30s. β-actin gene was used as the internal normalization control. The primers for CDK2 are 5-TCTGCCATTCTCATCGG-GTC-3 (forward) and 5-ATTTGCAGCCCAGGAGGATT-3 (reverse); the primers for β-actin are 5-GTTGCGTTACACCCTTTCTTG-3 (forward) and 5-GTCACCTTCACCGTTCCAGT-3 (reverse). The CDK2 relative expression was calculated using the formula 2-△△ct, where △△Ct is △Ct(treatment) -△Ct(control), △Ct is Ct(target gene) -Ct(β-actin) and Ct is the cycle at which the threshold is crossed.
Western blotting
A549 or GLC-82 cells (1×106/ml) seeded into 6-well plates (Costar, Corning, NY) were treated with 100 or 200μM bezafibrate for 24h. The cells were lysed with 100μl RIPA lysis buffer containing PMSF protease inhibitors. After the protein was extracted from each group, the total protein concentrations were determined using the BCA method according to the manufacturer's instructions. The protein were separated by 10% SDS-PAGE and then transferred onto polyvinylidene difluoride membranes. The membranes were blocked with 5% non-fat milk in Tris-buffered saline-Tween20 (TBST) at room temperature for 1h, then incubated with 1:1000 monoclonal rabbit anti-human antibody p-CDK2 (Thr-160) or CDK2 (ExCell Bio, Shanghai, China) at 4°C overnight. After washing, the membranes were incubated with corresponding horseradish peroxidase-conjugated secondary antibodies (1:10000) for 1 hour at room temperature. The proteins signals were then visualized by luminescent image analyzer (ImageQuant LAS4000 mini, GE Healthcare, USA) and β-actin was detected as a loading control.
In vivo tumor suppression test
All animal care and experimental protocols in this study were approved by the animal ethics committee of Guangzhou Medical University. The female BALB/c nude mice (4~6 weeks old) were purchased from Weitong Lihua Experimental Animal Technical Company (Beijing, China). About 5×107 A549 cells in 0.1 ml of PBS were subcutaneously injected at the right thigh of nude mice, and treatment was started when the tumors reached an average volume of 100 mm3. Mice bearing similar tumor volumes were chosen and randomly divided into 4 groups with 6 mice in each group: (a) control; (b) 200mg/kg of bezafibrate; (c) 2mg/kg of cisplatin; (d) 200mg/kg of bezafibrate+2 mg/kg of cisplatin. Cisplatin was injected intraperitoneally (i.p.), and bezafibrate was taken by intragastric administration once a day for two weeks. Mice were checked daily for toxicity/mortality relevant to treatment, and body weight and tumor size were measured every 3 days. Mice were sacrificed after three weeks and tumor volumes were calculated using the formula: tumor volume (mm3) = [length (mm)×width (mm)2]/2. Tumor growth inhibition rate (IR) was calculated according to the following formula: IR (%)=(1-mean weight experimental group/ mean weight control group)×100%.
Statistical analysis
The statistical significance of cell cycle distributions between the two groups was assessed with ANOVA followed by post-Hoc LSD and Dunnett T3 test using the SPSS software (version 17.0). A p-value<0.05 was considered statistically significant.
Results
CMap analysis identifies potential drugs targeting lung AC
To identify compounds exerting antitumor effects by altering the gene expression signature of lung AC to a favorable one, we performed CMap analysis by searching for negatively-correlated gene expression patterns associated with drug-treated cancer cells. For this analysis, we first defined the lung AC signature including 123 probes for up-regulated and 385 for down-regulated genes that were significantly differentially expressed between lung AC and normal lung tissue. Then, those signatures were used as input query items to compare with those produced from drug treatments in the CMap database. Several drugs were identified to reverse the signatures of lung AC. The top 10 drugs were summarized in Table 1. Among the top-ranked candidates, several drugs such as vorinostat[13], LY-294002[14], trichostatin A[15] and tanespimycin[10] have been reported to have anti-tumor effect for human cancers including lung cancer. Interestingly, bezafibrate, a PPARα agonist, showed significant negative enrichment, suggesting that this drug could potentially reverse the expression level of those lung AC signatures.
Top 10 candidate drugs of connectivity map analysis
| Rank | Cmap name | n | Enrich score | p | Drug category |
|---|---|---|---|---|---|
| 1 | vorinostat | 12 | -0.677 | 0 | HDAC inhibitor |
| 2 | LY-294002 | 61 | -0.347 | 0 | PI3K inhibitor |
| 3 | trichostatin A | 182 | -0.323 | 0 | HDAC inhibitor |
| 4 | tanespimycin | 62 | -0.304 | 0.00002 | HSP90 inhibitor |
| 5 | 15-delta prostaglandin J2 | 15 | -0.505 | 0.00042 | PPAR agonist |
| 6 | phenoxybenzamine | 4 | -0.852 | 0.00092 | alpha blocker |
| 7 | puromycin | 4 | -0.814 | 0.00223 | antibiotic |
| 8 | bepridil | 4 | -0.809 | 0.00261 | calcium blocker |
| 9 | resveratrol | 9 | -0.553 | 0.00376 | phytoalexin |
| 10 | bezafibrate | 4 | -0.772 | 0.00549 | PPAR agonist |
NOTE: All compounds listed here have at least four experiments
Bezafibrate inhibits cell growth and induces cell cycle arrest in lung AC cells in vitro
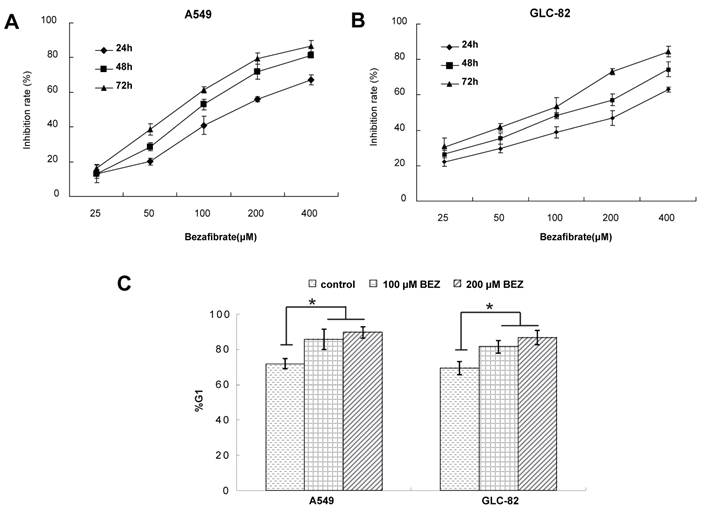
To experimentally validate the therapeutic efficacy of bezafibrate, we assessed the growth inhibitory effect of bezafibrate on two lung AC cell lines (A549 and GLC-82). Compared with untreated cells, bezafibrate significantly inhibited the growth of the two lung AC lines in a dose- and time-dependent manner (Figure 1A and B). To evaluate whether the anti-proliferative activity of bezafibrate was due to cell cycle arrest, we performed flow cytometry analysis on A549 and GLC-82 cells treated with 100 or 200μM bezafibrate for 24 hours. As compared to untreated cells, the G1 proportion of lung AC cells treated with bezafibrate increased significantly (P<0.05), indicating that bezafibrate induced G1 cell cycle arrest in those two lung AC cells (Figure 1C).
Cytotoxic effect and cell cycle arrest of bezafibrate in lung AC cell lines. A549 or GLC-82 cells were incubated with 25~400μM bezafibrate for 24, 48 and 72h. Cell viability was determined by the MTT assay and expressed as relative viability to control cells (A and B). Effect of bezafibrate on cell cycle was analyzed by flow cytometry after A549 or GLC-82 cells were exposed to 100 or 200μM bezafibrate (BEZ) for 24 h (C). * P<0.05 versus control group.
Potential protein targets for bezafibrate
Bezafibrate is a fibrate drug that lowers cholesterol and triglycerides, and is widely used for the treatment of hyperlipidaemia. Recent studies showed that bezafibrate might have anti-tumor effects[16, 17], however, the detailed mechanisms were not well understood. In our above experiments, bezafibrate was in silico screened out as a candidate compound for lung AC, and our subsequent experiments also validated that bezafibrate could inhibit cell proliferation and induce cell cycle arrest. To elucidate the anti-tumor mechanism of bezafibrate, virtual screening was carried out against a pool of potential targeting proteins by the online inverse docking pipeline TarFisDock. The top 2% candidate proteins ranked by binding energies were listed in Table 2. Amongst eighteen candidate targets, seven targets are related with cancers. Interestingly, cyclin dependent kinase 2(CDK2), a cell cycle kinase which might involve in several human cancers [18] was screened out as one candidate target of bezafibrate.
Protein target candidates of bezafibrate identified by TarFisDoc
| No | PDB_ID | Energy score (kcal/mol) | Target name | Related diseases |
|---|---|---|---|---|
| 1 | 2AGT | -38.21 | aldehyde reductase | diabetes |
| 2 | 1T41 | -38.09 | aldehyde reductase | diabetes |
| 3 | 1GAI | -38.01 | glucan 1,4-alpha-glucosidase | intestine disorder |
| 4 | 1FKN | -37.92 | memapsin 2 | Alzheimer's disease |
| 5 | 1HO4 | -37.82 | pyridoxine 5'-phosphate synthase | bacterial infections |
| 6 | 1LBV | -37.6 | inositol 1 phosphatase | affective disorder |
| 7 | 1J96 | -37.42 | aldo-keto reductase family 1 member c3 | cancers |
| 8 | 2ADA | -36.8 | adenosine deaminas | leukemia |
| 9 | 1ADD | -36.73 | adenosine deaminase | leukemia |
| 10 | 1HDT | -36.4 | serine proteinase alpha-thrombin | haemostatic disorders |
| 11 | 1GZ8 | -36.32 | cyclin dependent kinase 2(Cdk2) | cancers |
| 12 | 2ANY | -35.89 | kallikrein | rheumatoid arthritis |
| 13 | 1K3Y | -35.25 | glutathione transferase A1-1 | cancers |
| 14 | 1HDT | -35.12 | serine proteinase alpha-thrombin | haemostatic disorders |
| 15 | 1JP7 | -34.99 | methylthioadenosine phosphorylase | leukemia |
| 16 | 1D4P | -34.92 | serine proteinase alpha-thrombin | haemostatic disorders |
| 17 | 1M4H | -34.52 | memapsin 2 | Alzheimer's disease |
| 18 | 1I76 | -34.32 | neutrophil collagenase, MMP | tumor invasion |
Bezafibrate decreases CDK2 expression of lung AC cells
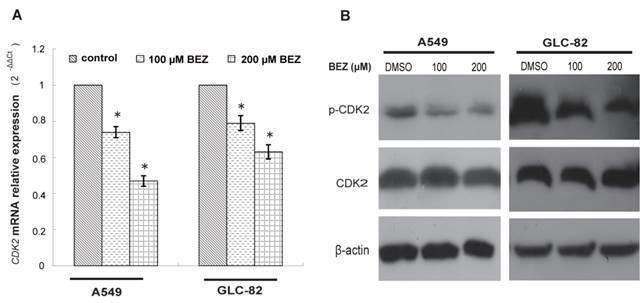
Based on the candidate list of bezafibrate-targets predicted by TarFisDock, we supposed that bezafibrate might induce G1 cell cycle arrest in lung AC cells through regulating the expression or activity of CDK2. To test our hypothesis, the expression level of CDK2 was evaluated by qRT-PCR and Western blotting in both A549 and GLC-82 cells after the treatment with bezafibrate for 24h. Our result (Figure 2A) showed that CDK2 mRNA in two lung AC cells was significantly inhibited by bezafibrate (P<0.05). Bezafibrate (100 or 200μM) down-regulated CDK2 mRNA expression in A549 and GLC-82 cells by 0.5~0.8 fold as compared to those in untreated cells. Furthermore, we also observed down-regulation of p-CDK2 (Thr-160) after incubation with 100 or 200μM bezafibrate without a significant change in total CDK2 protein level (Figure 2B), implying that bezafibrate might induce G1 cell cycle arrest by suppressing phosphorylation of CDK2 protein.
Bezafibrate under-regulates the expression of CDK2 mRNA and protein. A549 or GLC-82 cells were treated with 100 or 200μM bezafibrate (BEZ) or DMSO for 24h. Expression of CDK2 mRNA was determined by real time RT-PCR ( A ). Western blot was performed to analyse the expression of total CDK2 and p- CDK2 ((Thr-160) ( B ). * P<0.05 versus control group.
Bezafibrate suppressed the growth of A549 xenograft in vivo
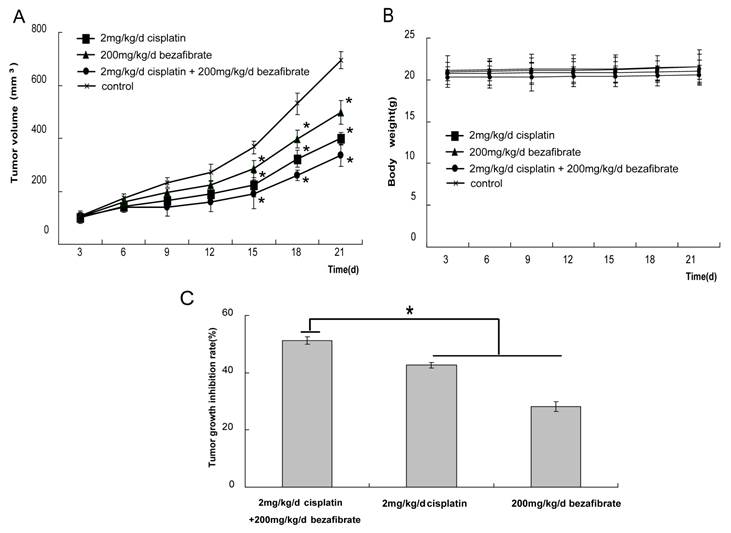
Finally, we evaluated the anti-cancer activity of bezafibrate in vivo using A549 cell xenograft model. After nude mice xenografts were treated with bezafibrate, cisplatin alone or together, the growth of tumor was inhibited to different extents. A statistically significant reduction in the speed of tumor progression was observed between treated and control groups (Figure 3A, p<0.05), suggesting bezafibrate significantly inhibited A549 xenograft growth in mice. Most importantly, the combination of bezafibrate and cisplatin strongly suppressed the growth of xenograft, the inhibition rate was up to 50%, which was significantly higher than that of cisplatin or bezafibate alone (Figure 3C, P<0.05). Whilst bezafibrate didn't significantly affect mice body weight, showing the relative safety of the agent (Figure 3B, P>0.05).
Antitumor activity of bezafibrate in A549 xenograft models. The BALB/c nude mice (female, 4∼6 weeks old) were randomized into 4 different groups (control, 200 mg/kg bezafibrate, 2mg/kg cisplaitn, 200 mg/kg bezafibrate+2mg/kg cisplaitn) with 6 mice each group. The detailed experimental procedures of establishted xenograft models were described in “Materials and Methods”. The nude mice were treated with indicated agents when the tumors reached an average volume of 100 mm3. Tumor volume (in mm3, recorded twice per week) (A), body weight (in grams, recorded twice per week) (B), and tumor growth inhibitory rate (percentage, at the end of the third week) (C) were shown. Each bar represents the means±SD (n = 6), *P< 0.05 vs group of control.
Discussion
The discovery of new compounds for medical conditions is generally time-consuming and very expensive, thus new strategies for drug discovery are very desperately needed. Comparing with the traditional development for new drug, the drug repurposing strategy, which finds new indications for existing drugs, could be not only time-saving, but also cost-effective. Connectivity mapping is an important method in drug repurposing by establishing connections among genes, drugs, and diseases. Recently, some studies successfully identified and validated potential therapeutic compounds for several different tumors by CMap analysis [8-10]. In present study, we identified some therapeutic candidate compounds for lung AC using the CMap, including HSP90 inhibitors, HDAC inhibitors, PPAR agonists, PI3K inhibitors, etc (Table 1), which were consistent with our previous study[10]. Some of the top-ranked candidates including vorinostat, tanespimycin, trichostatin A, and LY-294002, have been identified to have antitumor activity for many cancers including lung cancer[10, 13-15], suggesting that expression-based in silico screening with CMap is one effective approach to rapidly identifying novel potential applications for existing drugs.
In our preliminary study, the analysis of biological pathway with GSEA showed several pathways were dysfunctional in lung AC, such as down-regulated expression of PPARA pathway (data not shown). In accordance with results of GSEA, two PPAR agonists (15-delta prostaglandin J2 and bezafibrate) having negatively-correlated effects on expression of query lung AC signature, were in silico screened out as candidates using CMap. Peroxisomal proliferator-activated receptors (PPARs), PPARα, PPARγ, and PPARδ, could not only regulate cell proliferation, differentiation as well as survival, but also control carcinogenesis in different types of tissues [19]. Above all, it implied that PPARs might be potential targets for the therapy of lung AC. As a PPARγ agonist, the anticancer effects of 15-delta prostaglandin J2 have been extensively evaluated in different malignancies including lung cancer [20]. Clinically, the fibrates, such as fenofibrate, clofibrate, bezafibrate, could act as agonistic ligands of PPARα, and are widely used as lipid-lowering drugs with excellent tolerance and little side effects. Emerging evidences indicated that PPARα agonists exhibited anti-cancer effects on several human cancers including hepatoma[21], melanoma[22], as well as endometrial cancer[23]. In the present study, our expression-based in silico screening showed that bezafibrate can reverse the expression of lung AC signatures, hinting that bezafibrate may be a potential therapeutic agent for lung AC. Subsequent experiments further verified that bezafibrate inhibited cell proliferation and induced G1 cell cycle arrest in A549 and GLC-82 cells. Moreover, the antitumor effects of bezafibrate were evaluated in vivo by using transplanted tumor nude mice, and our results confirmed that bezafibrate has a notable antitumor effect on A549 xenograft (Figure 3). Most importantly, when combined with the commonly-prescribed cisplatin, bezafibrate enhenced antitumor effect of cisplatin. Of great interest, it was recently reported that PPARa activation with selective PPARa ligands could inhibit NSCLC primary and metastatic growth[16], which was consistent with our present study.
The precise mechanism of PPARα against cancers remains unclear and elusive. The evidences indicated that antitumor properties of PPARα activators were associated with proapoptosis and anti-inflammatory mechanisms [21, 24]. PPARα agonists also induce cell cycle arrest via diverse mechanisms. For example, fenofibrate inhibited G1/S phase progression in endometrial cancer by down-regulation of Cyclin D1 (CCND1)[23], and in glioblastoma cells by activation of FoxO3A/Bim apoptotic axis[25]. In head and neck squamous cell carcinoma, fenofibrate leaded to G2/M arrest via reducing the activity of the CDK1/cyclinB1 kinase complex[26]. To elucidate the anti-tumor mechanism of bezafibrate, we virtual screened out some bezafibrate-binding candidate proteins by TarFisDock (Table 2). Interestingly, cyclin dependent kinase 2 (CDK2), an especially critical regulator during the G1 to S phase transition, was screened out as a candidate target of bezafibrate. Our experiments validated that bezafibate can down-regulate the expression of CDK2 mRNA and down-regulation of p-CDK2 (Thr-160)(Figure 2), illustrating that bezafibrate might induce G1 arrest by suppressing phosphorylation of CDK2 protein.
In summary, our work attempted a new strategy in the medical therapy of lung cancer. Targeting PPARα may be a potential therapeutic strategy for lung AC, and fibrates such as bezafibrate may be promising anti-tumor drugs because of their excellent tolerance and little side effects. In addition to PPARα, bezafibrate could also target PPARδ and PPARγ[27], which are important molecular markers in lung cancer therapy as well[28, 29]. In spite of our observation indicated that bezafibrate attenuated the proliferation and induced cell cycle arrest in lung AC cells, the antitumor effect of bezafibrate still need to be verified in subsequent trials.
Supplementary Material
Figure S1 and Table S1.
Acknowledgements
Our special appreciation goes to Dr. Jie Gu from Jiangsu University and Dr. Ying Ge from Dalian Medical University for their careful revise and helpful suggestions. This work was supported by a grant from the Guangdong Natural Science Foundation (S2011010004147), a doctoral startup fund for Scientific Research at Guangzhou Medical University (2011C06), a grant from the National Natural Science Foundation of China (81301917), and a grant from Guangxi Natural Science Foundation (2012GXNSFBA053016).
Competing Interests
The authors have declared that no competing interest exists.
References
1. Massarelli E, Varella-Garcia M, Tang X, Xavier AC, Ozburn NC, Liu DD. et al. KRAS Mutation Is an Important Predictor of Resistance to Therapy with Epidermal Growth Factor Receptor Tyrosine Kinase Inhibitors in Non-Small-Cell Lung Cancer. Clinical Cancer Research. 2007;13:2890-6
2. Pao W, Miller VA, Politi KA, Riely GJ, Somwar R, Zakowski MF. et al. Acquired Resistance of Lung Adenocarcinomas to Gefitinib or Erlotinib Is Associated with a Second Mutation in the EGFR Kinase Domain. PLoS Med. 2005;2:e73
3. Bharadwaj U, Eckols TK, Kolosov M, Kasembeli MM, Adam A, Torres D. et al. Drug-repositioning screening identified piperlongumine as a direct STAT3 inhibitor with potent activity against breast cancer. Oncogene. 20140
4. Jahchan NS, Dudley JT, Mazur PK, Flores N, Yang D, Palmerton A. et al. A drug repositioning approach identifies tricyclic antidepressants as inhibitors of small cell lung cancer and other neuroendocrine tumors. Cancer Discov. 2013;3:1364-77
5. Zerbini LF, Bhasin MK, de Vasconcellos JF, Paccez JD, Gu X, Kung AL. et al. Computational repositioning and preclinical validation of pentamidine for renal cell cancer. Mol Cancer Ther. 2014;13:1929-41
6. Lamb J, Crawford ED, Peck D, Modell JW, Blat IC, Wrobel MJ. et al. The Connectivity Map: using gene-expression signatures to connect small molecules, genes, and disease. Science. 2006;313:1929-35
7. Li J, Xu YH, Lu Y, Ma XP, Chen P, Luo SW. et al. Identifying differentially expressed genes and small molecule drugs for prostate cancer by a bioinformatics strategy. Asian Pacific journal of cancer prevention: APJCP. 2013;14:5281-6
8. Claerhout S, Lim JY, Choi W, Park YY, Kim K, Kim SB. et al. Gene expression signature analysis identifies vorinostat as a candidate therapy for gastric cancer. PLoS One. 2011;6:e24662
9. De Preter K, De Brouwer S, Van Maerken T, Pattyn F, Schramm A, Eggert A. et al. Meta-mining of neuroblastoma and neuroblast gene expression profiles reveals candidate therapeutic compounds. Clin Cancer Res. 2009;15:3690-6
10. Wang G, Ye Y, Yang X, Liao H, Zhao C, Liang S. Expression-based in silico screening of candidate therapeutic compounds for lung adenocarcinoma. PLoS One. 2011;6:e14573
11. Li H, Gao Z, Kang L, Zhang H, Yang K, Yu K. et al. TarFisDock: a web server for identifying drug targets with docking approach. Nucleic Acids Res. 2006;34:W219-24
12. Gao Z, Li H, Zhang H, Liu X, Kang L, Luo X. et al. PDTD: a web-accessible protein database for drug target identification. BMC Bioinformatics. 2008;9:104
13. Owonikoko TK, Ramalingam SS, Kanterewicz B, Balius TE, Belani CP, Hershberger PA. Vorinostat increases carboplatin and paclitaxel activity in non-small-cell lung cancer cells. Int J Cancer. 2010;126:743-55
14. Kelly-Spratt KS, Philipp-Staheli J, Gurley KE, Hoon-Kim K, Knoblaugh S, Kemp CJ. Inhibition of PI-3K restores nuclear p27Kip1 expression in a mouse model of Kras-driven lung cancer. Oncogene. 2009;28:3652-62
15. Wang X, Li G, Wang A, Zhang Z, Merchan JR, Halmos B. Combined histone deacetylase and cyclooxygenase inhibition achieves enhanced antiangiogenic effects in lung cancer cells. Mol Carcinog. 2013;52:218-28
16. Skrypnyk N, Chen X, Hu W, Su Y, Mont S, Yang S. et al. PPARalpha activation can help prevent and treat non-small cell lung cancer. Cancer Res. 2014;74:621-31
17. Khanim FL, Hayden RE, Birtwistle J, Lodi A, Tiziani S, Davies NJ. et al. Combined bezafibrate and medroxyprogesterone acetate: potential novel therapy for acute myeloid leukaemia. PLoS One. 2009;4:e8147
18. Malumbres M, Barbacid M. Cell cycle, CDKs and cancer: a changing paradigm. Nat Rev Cancer. 2009;9:153-66
19. Michalik L, Desvergne B, Wahli W. Peroxisome-proliferator-activated receptors and cancers: complex stories. Nat Rev Cancer. 2004;4:61-70
20. Han S, Ritzenthaler JD, Rivera HN, Roman J. Peroxisome proliferator-activated receptor-γ ligands suppress fibronectin gene expression in human lung carcinoma cells: involvement of both CRE and Sp1. American Journal of Physiology - Lung Cellular and Molecular Physiology. 2005;289:L419-L28
21. Muzio G, Maggiora M, Oraldi M, Trombetta A, Canuto RA. PPARalpha and PP2A are involved in the proapoptotic effect of conjugated linoleic acid on human hepatoma cell line SK-HEP-1. Int J Cancer. 2007;121:2395-401
22. Grabacka M, Plonka PM, Urbanska K, Reiss K. Peroxisome proliferator-activated receptor alpha activation decreases metastatic potential of melanoma cells in vitro via down-regulation of Akt. Clin Cancer Res. 2006;12:3028-36
23. Saidi SA, Holland CM, Charnock-Jones DS, Smith SK. In vitro and in vivo effects of the PPAR-alpha agonists fenofibrate and retinoic acid in endometrial cancer. Mol Cancer. 2006;5:13
24. Grabacka M, Reiss K. Anticancer Properties of PPARalpha-Effects on Cellular Metabolism and Inflammation. PPAR Res. 2008;2008:930705
25. Wilk A, Urbanska K, Grabacka M, Mullinax J, Marcinkiewicz C, Impastato D. et al. Fenofibrate-induced nuclear translocation of FoxO3A triggers Bim-mediated apoptosis in glioblastoma cells in vitro. Cell Cycle. 2012;11:2660-71
26. Liu J, Ge YY, Zhu HC, Yang X, Cai J, Zhang C. et al. Fenofibrate Increases Radiosensitivity in Head and Neck Squamous Cell Carcinoma via Inducing G2/M Arrest and Apoptosis. Asian Pacific journal of cancer prevention: APJCP. 2014;15:6649-55
27. Willson TM, Brown PJ, Sternbach DD, Henke BR. The PPARs: from orphan receptors to drug discovery. Journal of medicinal chemistry. 2000;43:527-50
28. Fukumoto K, Yano Y, Virgona N, Hagiwara H, Sato H, Senba H. et al. Peroxisome proliferator-activated receptor delta as a molecular target to regulate lung cancer cell growth. FEBS Lett. 2005;579:3829-36
29. Jeong Y, Xie Y, Lee W, Bookout AL, Girard L, Raso G. et al. Research Resource: Diagnostic and Therapeutic Potential of Nuclear Receptor Expression in Lung Cancer. Molecular Endocrinology. 2012;26:1443-54
Author contact
![]() Corresponding authors: Gui-Ping Wang, Ph.D. Department of Pharmacy, College of Health sciences, Guangzhou Medical University, Guangzhou 510180, China. Tel: +86-20-86210012; E-mail: docgpwangcom. Yun Ye, Ph.D. College of Biological and Chemical Engineering, Guangxi University of Science and Technology, Liuzhou 545006, P.R. China. Tel: +86-0772-2687033; E-mail: yunyeedu.cn.
Corresponding authors: Gui-Ping Wang, Ph.D. Department of Pharmacy, College of Health sciences, Guangzhou Medical University, Guangzhou 510180, China. Tel: +86-20-86210012; E-mail: docgpwangcom. Yun Ye, Ph.D. College of Biological and Chemical Engineering, Guangxi University of Science and Technology, Liuzhou 545006, P.R. China. Tel: +86-0772-2687033; E-mail: yunyeedu.cn.