Impact Factor ISSN: 1837-9664

 Global reach, higher impact
Global reach, higher impactJ Cancer 2016; 7(2):214-224. doi:10.7150/jca.13150 This issue Cite
Research Paper
Genome-wide Analysis of Epstein-Barr Virus (EBV) Integration and Strain in C666-1 and Raji Cells
Kai Xiao1,2,3, Zhengyuan Yu2, Xiayu Li3, Xiaoling Li1,2,3, Ke Tang2, Chaofeng Tu2, Peng Qi2, Qianjin Liao1,2, Pan Chen1,2, Zhaoyang Zeng1,2,3, Guiyuan Li1,2,3, Wei Xiong1,2,3 ![]()
1. Hunan Key Laboratory of Translational Radiation Oncology, Hunan Cancer Hospital and the Affiliated Cancer Hospital of Xiangya School of Medicine, Central South University, Changsha, Hunan, 410013, China.
2. Key Laboratory of Carcinogenesis of Ministry of Health and Key Laboratory of Carcinogenesis and Cancer Invasion of Ministry of Education, Cancer Research Institute, Central South University, Changsha, Hunan, 410078, China.
3. Hunan Key Laboratory of Nonresolving Inflammation and Cancer, Disease Genome Research Center, The Third Xiangya Hospital, Central South University, Changsha, Hunan, 410013, China.
Received 2015-7-5; Accepted 2015-10-18; Published 2016-1-1
Abstract
EBV is a key risk factor for many malignancy diseases such as nasopharyngeal carcinoma (NPC) and Burkitt lymphoma (BL). EBV integration has been reported, but its scale and impact to cancer development is remains unclear. C666-1 (NPC cell line) and Raji (BL cell line) are commonly studied EBV-positive cancer cells. A rare few EBV integration sites in Raji were found in previous research by traditional methods. To deeply survey EBV integration, we sequenced C666-1 and Raji whole genomes by the next generation sequencing (NGS) technology and a total of 909 breakpoints were detected in the two cell lines. Moreover, we observed that the number of integration sites was positive correlated with the total amount of chromosome structural variations (SVs) and copy number structural variations (CNVs), and most breakpoints located inside or nearby genome structural variations regions. It suggested that host genome instability provided an opportunity for EBV integration on one hand and the integration aggravated host genome instability on the other hand. Then, we respectively assembled the C666-1 and Raji EBV strains which would be useful resources for EBV-relative studies. Thus, we report the most comprehensive characterization of EBV integration in NPC cell and BL cell, and EBV shows the wide range and random integration to increase the tumorigenesis. The NGS provides an incomparable level of resolution on EBV integration and a convenient approach to obtain viral strain compared to any research technology before.
Keywords: Epstein-Barr Virus (EBV), Whole genome sequencing, Raji, C666-1, Nasopharyngeal carcinoma (NPC), Burkitt lymphoma (BL)
Introduction
Epstein-Barr Virus (EBV) is a major etiologic agent for malignant diseases of epithelial and lymphoid cell origin [1-3]. Despite clear evidence supporting the correlation of EBV and many malignant tumors [4, 5], the underlying nature of EBV-host interaction remains elusive. The mode and influence of EBV integration is still not clear due to the lack of an impartial method to detect and survey genome-wide EBV integration sites, although EBV integration into host genome has been reported [6-11]. Recently, the advancement of sequencing technology [12-17] offers an opportunity to study the global extent and functional impact of EBV integration into the cancer genome. C666-1 [18] and Raji [19] are typical EBV positive cell lines. They are classic models to research viral-host interaction and used worldwide [20-28]. A rare few EBV integrations sites in Raji were reported in previous research by traditional methods such as genome library construction [8] and fluorescence in situ hybridization (FISH) [9]. Here we sequenced the C666-1 and Raji cell line genomes by deep (>60 coverage) whole genome sequencing to survey the entire viral integration distributions on virus and the host genome. Meanwhile, whole genome sequencing of C666-1 and Raji offered a chance to assemble their EBV genome strains, which should be useful resources for EBV-relative studies. To that end, sequencing the EBV-positive cancer cell genome provides a great solution to reveal the EBV integration distribution, the functional impact of viral integration on the host genome and the EBV genome characteristics.
Materials and Methods
Cell culture and sample preparation
C666-1 and Raji were grown in RPMI 1640 (HyClone, Logan, UT, USA) plus 10% fetal bovine serum (GIBCO, Grand Island, NY, USA) with 0.5% penicillin and streptomycin (GIBCO , Grand Island, NY, USA). Cells were grown at 37℃ in a humidified, 5% CO2 incubator. The genomic DNA of C666-1 and Raji cells was extracted and purified using Qiagen Gentra Puregene reagents according to the manufacturer's instructions.
Whole genome sequencing and analysis
The libraries of C666-1 and Raji DNA were prepared following the Illumina Truseq DNA sample preparation protocol and subjected to sequence with 90-base paired-end using an Illumina HiSeq instrument. Average fragment size used for sequencing was about 480 bp. Each library was subjected to 2 lanes, resulting in at least 60-fold haploid coverage for each sample. SOAPsnp [29] was used to detect single nucleotide polymorphisms (SNPs); SAMtools [30] was used to detect the InDels (small Insertion/Deletion); CREST [31] was used to detect structure variations (SVs, including Insertions, Deletions, Inversions and Translocations of chromosome bands or arms). Copy number variations (CNVs) were detected by an in-house program with an algorithm similar to software Segseq [32] of broad institute. EBV integrations were also detected by an in-house program. After the variations were detected, ANNOVAR [33] was used to do annotation and classification.
Analysis of EBV integration sites
First, we combine the human reference genome (hg19) and the wild type EBV reference genome (EBV-WT, GenBank accession number NC007605) [34] together to build a hybrid reference genome. Paired-end reads with fragment length of about 500bp were aligned to the hybrid reference genome with Burrows-Wheeler aligner (BWA) [35]. All mapped reads were subjected to a filtering process to remove possible PCR duplicates. Reads cover fuse boundaries of the human genome and the EBV genome are called split reads, which are partially mapped to the human genome (hg19) or the EBV genome (EBV-WT) in the original alignment result. Split reads are used to identify EBV integration positions. Extract split reads from the original alignment result and assemble the split reads into contigs. Match rate cutoff is set to 0.95. Split reads cover the same fuse boundary will be assembled into the same contig. Then realign the clipped part of the contigs to the hybrid reference genome. If the partially mapped part and the clipped part of a junction read respectively and uniquely mapped to the human reference genome (hg19) and the EBV genome (EBV-WT), the integration position is reported. After that, filter the integrations which length of sequence near the breakpoint are smaller than 30. The integrations need at least one split read to support them. Finally, pooled split reads come from the same integration position to get all integration events and its supporting split read numbers.
De novo assembly of EBV genomes
All sequencing reads were first aligned to the EBV-WT genome by BWA software [35], and the paired-end reads which at least one end mapped to the EBV-WT genome were collect to do the de novo assembly. We used SOAPdenovo [36] software which merged the overlapping reads based on de Bruijn graph algorithm to generate contigs. The paired-end information was then used to link contigs into scaffolds; subsequently, assembled scaffolds were aligned to the EBV-WT genome and the gaps were filled with corresponding reference sequence which was mostly located in the repeat region that make the sequence difficult to assemble.
The assembled full-length sequence of EBV strain derived from C666-1 was submitted to the GenBank database and assigned accession number AB828190. The sequence of Raji EBV strain was also submitted and assigned accession number AB828191.
Detection of SNVs
The C666-1 and Raji reads were aligned to the reference (EBV-WT) by BWA software [35]. Single nucleotide variations (SNVs) were detect by the SAMtools [30] software and applying the following criteria to filtering the raw variations result: (1) the minimal depth cover the variation site is not less than 4; (2) the minimal variation quality score is not less than 20; (3) the distance between two nearby variations is not less than 5; (4) the reads support the mutated allele is not less than 4.
Comparative and phylogenetic analyses
The MAFFT [37] software was used to align the EBV genomes of C666-1 (AB828190) and Raji (AB828191) with other 18 strains reported previously: EBV-WT (NC007605) [34], B95-8 (V01555) [34], AG876 (DQ279927) [38], Akata (KC207813) [39], Mutu (KC207814) [39], K4123-Mi (KC440851) [40], K4413-Mi (KC440852) [40], GD1 (AY961628) [41], GD2 (HQ020558) [42], HKNPC1(JQ009376) [43], and HKNPC2 ~ HKNPC9 (KF992564 ~ KF992571) [44]. Molecular Evolutionary Genetics Analysis (MEGA) [45] software, version 5.2 was used to perform the Phylogenetic analyses by Neighbor-joining (NJ) algorithm, based on multiple sequence alignments of the eight viral genomes and encoded genes. Protein sequences were aligned with Megalign (DNAStar, Lasergene, version 7.1).
Results
Detection of EBV integration
In this study, we processed whole-genome deep sequencing (> 60 coverage) on C666-1 and Raji cells to identifying of EBV insertion events and investigating impact of insertion on host genome. First, we aligned all short reads from whole-genome sequencing to the human (hg19) and EBV reference genome sequence (NC007605) (Figure 1). The total raw data of viral sequence in C666-1 is 117,172,040 bp and 114,444,102 bp in Raji. This approach yielded very high coverage depth (759 × in C666-1 and 1,312 × in Raji). Aligned to the reference human sequence, the total raw data of C666-1 is 182,230,285,487bp, and Raji is 183,866,611,697 bp, and average coverage depth of human genome in C666-1 and Raji are 62.90 × and 63.46 ×, respectively (Table 1). Based on the total number of viral and human sequencing data, we estimate that, on average, C666-1 contains 12 (759.39/62.9) copies of the viral genome per diploid human genome and Raji contains 20 (1312.05/63.46) copies, approximately. We identified EBV integration sites by searching for human-EBV chimeric reads, in which one end mapped to the human genome and the other mapped to the EBV genome. Chimeric reads supporting the same EBV integration event were then clustered, yielding 909 unique EBV insertion sites in the two cell line (350 in C666-1 and 559 in Raji). (Figure 2A & 2B), 26 of which are supported by at least 2 chimeric reads (9 in C666-1 and 17 in Raji, Additional File 1: Table S1). Compared to the > 60× sequencing coverage of human genome, integration event supported by relative less chimeric reads (maximum 33 reads) reveal heterogeneous, random viral integration events in both C666-1 and Raji cells.
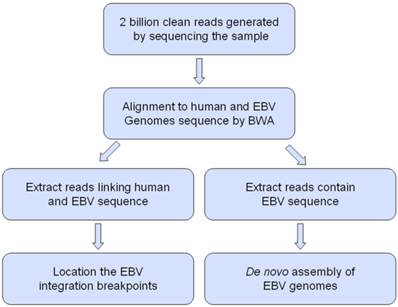
The workflow for analysis Epstein-Barr virus (EBV) integration and assembly of EBV genomes in C666-1 and Raji cells.
Summary of sequencing data of C666-1 and Raji.
| Categories | C666-1 | Raji |
|---|---|---|
| Raw reads | 2,328,493,430 | 2,298,076,610 |
| Raw bases (bp) | 209,564,408,700 | 206,826,894,900 |
| Clean reads | 2,200,200,436 | 2,175,753,790 |
| Clean bases (bp) | 198,018,039,240 | 195,817,841,100 |
| Rate of clean reads /raw reads | 94.49% | 94.68% |
| Total raw data mapped to human (bp) | 182,230,285,487 | 183,866,611,697 |
| Total raw data mapped to EBV (bp) | 117,172,040 | 114,444,102 |
| Effective sequencing depth in human (×) | 62.90 | 63.46 |
| Coverage in human (%) | 99.43 | 99.47 |
| Effective sequencing depth in EBV (×) | 759.39 | 1312.05 |
| Coverage in EBV (%) | 82.63 | 79.93 |
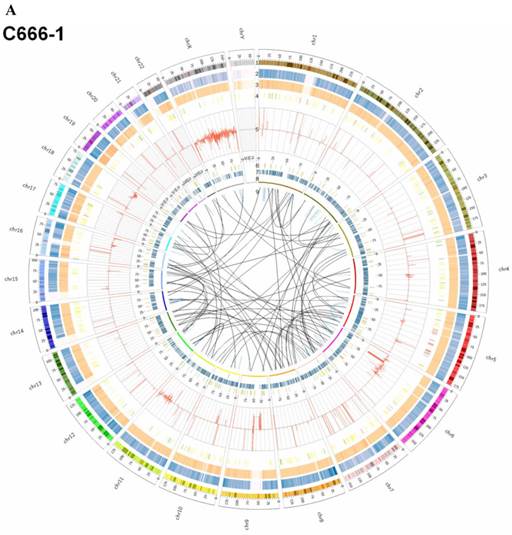
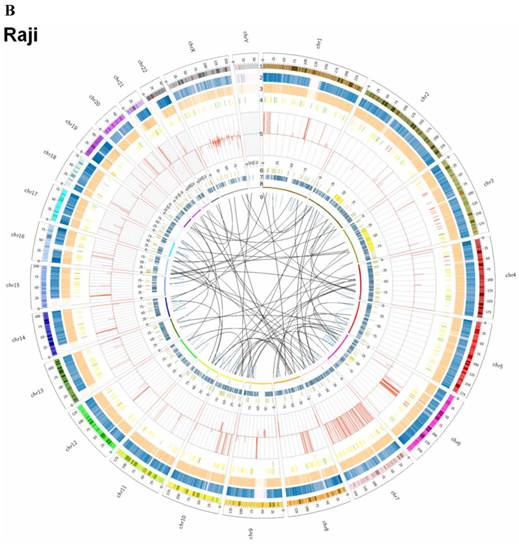
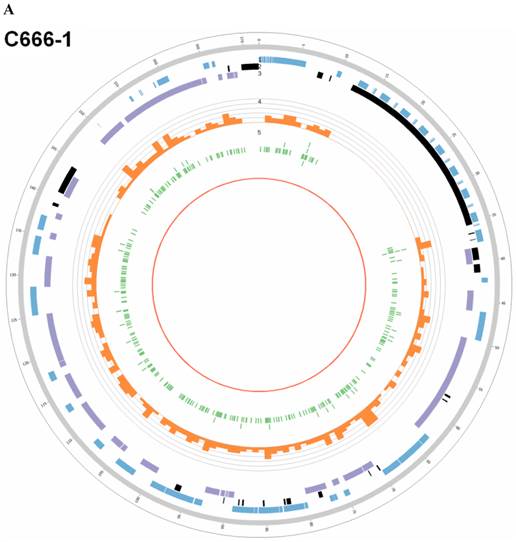
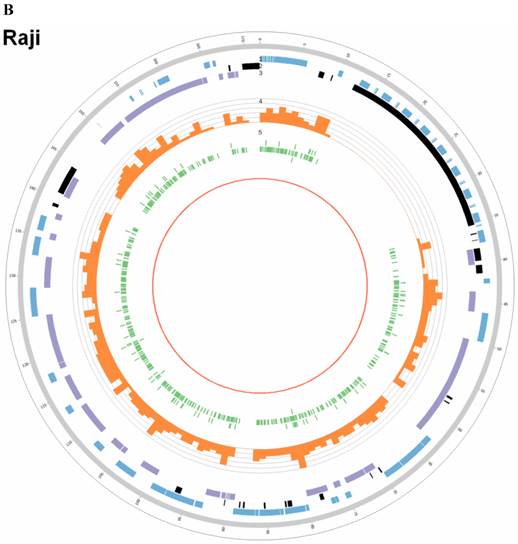
Summary of somatic genomic alterations in C666-1 and Raji cells. Various types of somatic alterations in C666-1(A) and Raji (B) genomes using circus plots. Tracks from outer to inner represent the following: (1) chromosome karyotype diagram; (2) SNP heatmap; (3) InDel heatmap; (4) virus integration sites (integrations within SV or CNV regions were represented in red, integrations near SV or CNV regions were represented in yellow, others were represented in green); (5) CNV diagram (red, outer of baseline: gain; inner of baseline: loss); (6) SV insertion (yellow); (7) SV deletion(blue); (8) SV invertion; (9) SV intra-chromosomal translocation (represent in blue) and inter-chromosomal translocation (represent in black).
EBV integrating sites were co-localized with chromosome instability sites
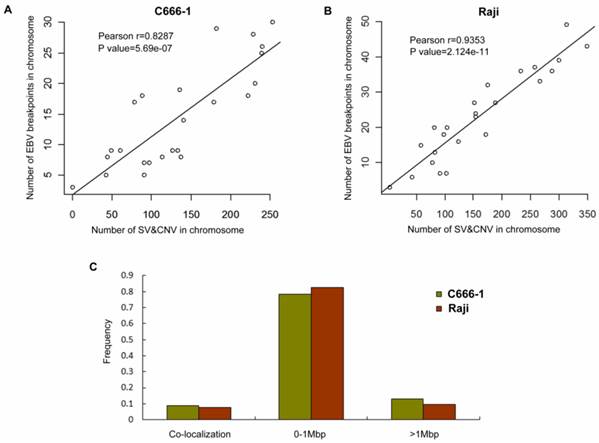
By whole genome sequencing, we found large number of SNPs, InDels, CNVs and SVs both in C666-1 (2,289,321 SNPs, 1,038,097 InDels, 1,510 CNVs and 5,820 SVs) and Raji (2,463,155 SNPs, 542,732 InDels, 426 CNVs and 6,996 SVs) cells (Figure 2A & 2B) compared to the reference human genome (hg19). The integration of virus DNA into the host chromosome could cause chromosome instability has been reported [46]. Here, we observed the number of viral integration sites was positive correlated with the total amount of the chromosome SVs and CNVs both in Raji (P = 2.124E-11) and C666-1 (P = 5.696E-07). Meanwhile, in C666-1, we found that 30 EBV breakpoints (8%) were located inside SV and CNV regions, 270 (77%) were located within 1 Mb of SV and CNV regions. Also in Raji, there are 43 breakpoints (7%) were located inside SV and CNV regions, 458 (81%) were located within 1 Mb of somatic SV and CNV regions (Figure 2 & Figure 3).
Correlation analysis of EBV integration and chromosome instability in C666-1 and Raji cells. A. The number of EBV integration sites versus the number of SVs and CNVs on C666-1 chromosomes. B. The number of EBV integration sites versus the number of SVs and CNVs on Raji chromosomes. C. Summary of distance between EBV integration sites and SVs and CNVs sites in C666-1 and Raji.
The highest frequency integration site (with 33 reads supported) in Raji was located in a CNV region (Additional File 2: Table S2). Based on the statistic analysis above, we concluded that most breakpoints located inside or nearby genome structural variation regions, which supporting that genome instability and chromosome structural variation was probably induced by EBV integration.
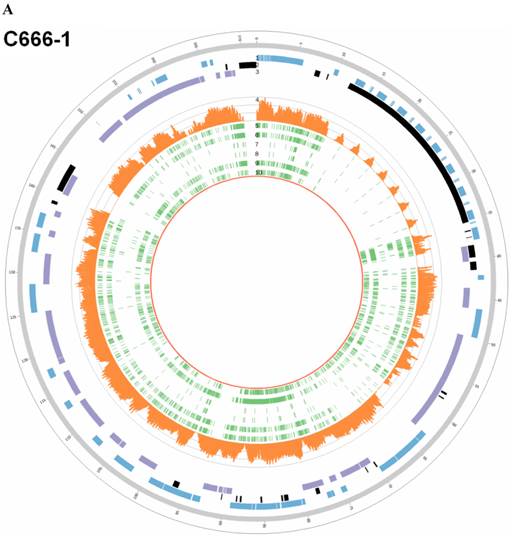
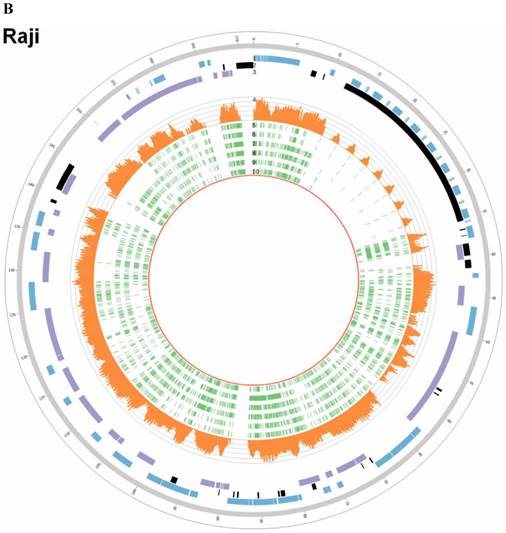
In EBV genome, we also surveyed the integration sites, which were proved to be randomly distributed in viral genome and almost covered the whole EBV genome both in C666-1 and Raji (Figure 4A & 4B).
EBV integration sites and breakpoints in the EBV genomes of C666-1 and Raji cells. The circus plots representing the EBV genomes from C666-1 (A) and Raji (B) cells. Tracks from outer to inner represent the following: (1) foward ORFs (blue); (2) repeat regions (black); (3) reward ORFs (purple); (4) number of integration sites per Kilobases of EBV genomes (up to 10 sites, orange); (5) integration sites (green), some of integration sites were identified two or more times in C666-1 or Raji genome.
EBV sequence and phylogenetic analyses
The C666-1 EBV draft genome is 171,021bp and Raji is 166,107bp. Multiple sequence alignments were carried out for the genome sequences of the newly assembled C666-1, Raji and other six EBV subtypes, including EBV-WT, AG876, B95-8, GD1, GD2 and HKNPC1 (Figure 5A & 5B). The sequence similarities between C666-1 and the other six EBV subtypes were 99.93% (GD1), 99.14% (EBV-WT), 99.14% (B95-8), 99.88% (GD2), 99.62% (HKNPC1) and 98.38% (AG876). The sequence similarities between Raji and the other six EBV subtypes were 99.17% (GD1), 99.33% (EBV-WT), 99.33% (B95-8), 99.00% (GD2), 98.77% (HKNPC1), and 98.51% (AG876). The result showed that Raji EBV genome was more similar to the subtypes derived from the lymphocyte (EBV-WT, B95-8) and C666-1 viral strain was more similar to the subtypes derived from the NPC (GD1, GD2, HKNPC1).
Sequencing depths and SNVs of EBV genome in C666-1 and Raji. Tracks from outer to inner represent the following: (1) foward ORFs(blue); (2) repeat regions(black); (3) reward ORFs (purple); (4) sequencing depths (up to 1800 reads, orange); single nucleotide variations (green) compared to B95-8 strain (5), reference sequence (NC_007605, 6), HKNPC1 strain (7), GD2 strain (8), AG876 strain (9) and GD1 strain (10). A. C666-1; B. Raji
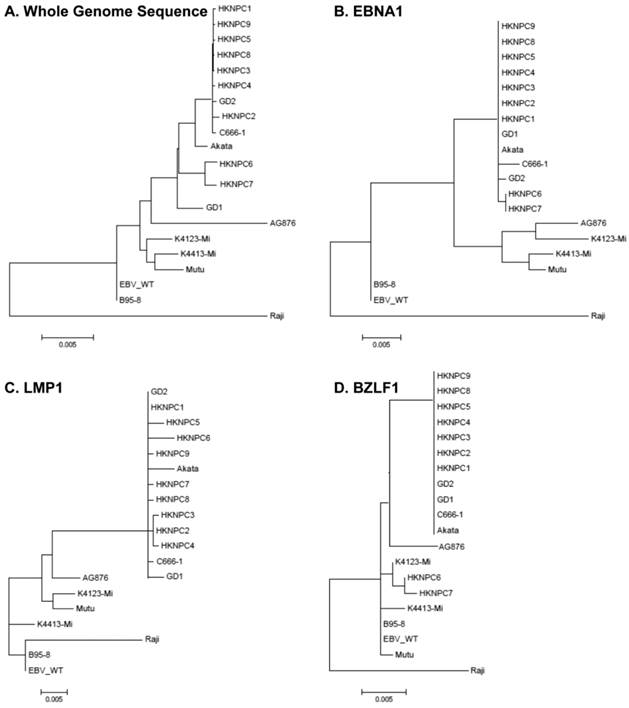
According to all published EBV genome and NPC-EBV genome reference sequence, the evolutionary trees of the EBV whole genome and its protein encoding genes, LMP1, EBNA-1 and BZLF1 were constructed. Raji could obviously separate from other EBV subtypes when the LMP1, EBNA-1, and BZLF1 gene were used to classify (Figure 6). The various subtypes of EBV showed a certain correlation with its location distribution. All the EBV subtypes from Asia could gather at a branch, including HKNPC1 to 9, GD1 and GD2, C666-1, and Japanese strain (Akata), which could separate from the EBV subtypes of non-Asian area, such as AG876, B95-8, Mutu, K4123-Mi, K4413-Mi, and K4123-Mi. Furthermore, by analyzing amino acid sequences, we found that LMP1 had two regions of insertions/deletions within CTAR2 and CTAR3 domains which presumably influenced LMP1's signaling functions (Figure 7). By comparison with the EBV-WT reference genome, a total of 891 SNVs (238 were nonsynonymous) were detected in the C666-1 EBV genome and 654 SNVs (224 were nonsynonymous) in Raji viral strain (Additional File 3: Table S3).
Phylogenetic analyses on whole EBV genomes and protein-encoding genes EBNA-1, LMP1, and BZLF1. A. Phylogenetic tree base on whole genome of 20 EBV strains, poorly aligned and highly divergent sequences were masked by Gblocks. Phylogenetic trees based on protein sequence alignment of EBNA1 (B), LMP1 (C), and BZLF1 (D) were generated. Phylogenetic analysis was performed using MEGA software (version 5), by Neighbor-joining (NJ) algorithm. Divergence scale, in numbers of substitution per site, is shown under each tree.
LMP1 protein sequences variation in different EBV strains. LMP1 protein sequences Alignment were generated using Megalign (DNAstar). Variant positions are marked in yellow. Functional domains of the gene are indicated above the consensus sequence.

Discussion
EBV plays a very important role in many kinds of cancer [47-54]. Our work provides the first synthetically analysis of genome variation in C666-1 and Raji, including EBV insertions, EBV genome alterations, and large human genomic structural variations. The traditional research methods such as genome library construction and FISH can be used to identify the presence of EBV integration, but rare integrations can be identified and only integrations near the targeted human or EBV sequences can be detected. In previous research of EBV integration on Raji, it detected a rare few breakpoints by library construction [8]. In addition, it was reported a number of probable regions by FISH [9], but not located exactly. The NGS technology provides an impartial and high-efficiency method for detecting viral integration, surveying their frequencies, analyzing EBV strain, and offering an opportunity to study the comprehensive impact of EBV integration on human genome.
We discovered 909 unique EBV insertion sites in the two cell lines by our detecting method. To reduce the possibility of false positive EBV integration identification, we chose two controls: one was negative genomic DNA (without EBV integration); the other was a mixture of free EBV DNA and human genomic DNA. We did not find any EBV integration events in either of the controls. Besides, we consider that the false negative viral integration rate would be decreased by increasing the sequencing depth. In Jiang's study [55], the number of HBV breakpoints which they detected added from 43 to 142 by improving the sequencing depth from 83× to 234×, and most of the newly detected sites were low frequency (supported by only one read).
The EBV genome size is much larger and more complicated than the other DNA viruses such as human papilloma virus (HPV) and hepatitis B virus (HBV), and its breakpoints almost covered the whole viral genome both in C666-1 and Raji (Figure 4 and Figure 5). This indicates EBV is more suitable for whole genome sequencing to identify integration sites. Based on the result that C666-1 contained about 12 EBV genome copies per diploid human genome and was detected 350 sites, and simultaneously Raji included nearly 20 EBV genome copies per diploid human genome and was detected 559 sites, it is reasonable to speculate that the more copies of EBV genome exist per diploid human genome, the more likely viral integrations occur.
Most of the integration sites were low frequency (supporting reads < = 2) and part of them could be detected by PCR. Our validation result was similar to Jiang's study of HBV integration [55]. Using whole genome sequencing, they found a lot of HBV low frequency integration sites and only validated 3 cases out of 24 sites (supporting reads = 2) by PCR. Hence, we could logically presume that EBV randomly integrates into human genome in most cases. Moreover, for detecting these low frequency sites, NGS is more sensitive than PCR. However, why most of integration sites are low frequency? There might be many potential reasons: Firstly, we observed the number of the chromosome viral integration sites was positive correlated with the total amount of the SVs and CNVs, and most breakpoints located inside or nearby genome structural variation regions, which is consistent with hit-and-run mechanism proposed by Jox [7]. He assumed the integration might constitute a chromosomal region prone to break events akin to the phenomenon of fragile sites, leading to the loss of viral DNA as well as chromosomal DNA. Therefore, the EBV integration sequences could easily be lost due to the genome instability. Secondly, the integration might cause fatal cell damage so that the EBV integration sequence could not be preserved during cell culture. Thirdly, the EBV integration sequences are probably eliminated by host defending and repairing mechanism. Fourthly, comparing to the reference genome (hg19, NC007605), the host and virus genome in the two cell lines have great sequence variations, so we might lose some potential integration sites by our detecting methods aligning to the reference genome.
By the whole genome sequencing, we obtain 117,172,040 bp raw data of EBV sequence in C666-1, and 114,444,102 bp in Raji. The sequencing depth of the EBV strain in C666-1 was nearly 760×, and in Raji was 1312×. We assembled about 171 kb EBV whole genome sequence in C666-1 and 166 kb in Raji by de novo assembly. Compared with EBV-WT (NC_007605), 891 SNVs were found in C666-1 and 654 SNVs in Raji. Then, we constructed phylogenetic trees (Figure 6) of 20 EBV subtypes (C666-1, Raji, EBV-WT, B95-8, AG876, Akata, Mutu, K4123-Mi, K4413-Mi, GD1, GD2, HKNPC1 ~ HKNPC9) and the result showed that subtypes derived from the same disease were closely clustered, which indicated the high-accuracy of NGS and the correlation between subtypes and diseases. Compared with traditional EBV sequencing methods, NGS technology showed immense advantage in EBV genome sequencing, viral genome variations analysis, viral genome comparison and phylogenetic analyses to other subtypes. Before NGS technique has been developed, it was very difficult to obtain EBV genomic sequence. We knew little about the association between viral subtypes and EBV-relative diseases due to the limitations of traditional sequencing methods. Our study also supports the idea that a genome-wide survey of EBV integration variation is feasible for correlation analysis of EBV subtypes and viral integration capability.
In summary, our study indicated that EBV showed random integration model to induce host genome instability and caused tumorigenesis in C666-1 and Raji. Using NGS, we acquired C666-1 and Raji host genome sequences and their EBV genome sequences, which would be widely used for EBV-relative researches. NGS improves our understanding on the essence of EBV-host interaction. In addition, our work could serve as a model for studies of other diseases with viral integration. In future study, a construction of cell line model before and after viral integration is helpful to observe transcriptional impact of integration. At present, most NGS platform can only sequence reads whose length is less than 400bp. With the advent of advanced sequencing technologies, such as single cell genome sequencing [56, 57] and the third generation sequencing technology which is able to sequence longer reads [58], we can reveal the mechanism of virus integration more deeply.
Supplementary Material
Additional File 1Table S1.
Table S2.
Table S3.
Acknowledgements
The authors greatly thank Minghui He, Zhibo Gao and Gang Chen at BGI-Shenzhen for their help on data analysis and valuable suggestions to our work.
Funding
This study was supported in part by grants from the National Natural Science Foundation of China (81172189, 81272298, 81372907, 81301757, 81472531, 81402009, 81572787, 81528019 and 91229122), the National High Technology Research and Development (863) Program of China (2012AA02A206) and the Natural Science Foundation of Hunan Province (14JJ1010 and 2015JJ1022).
Competing Interests
The authors have declared that no competing interest exists.
References
1. Rubicz R, Yolken R, Drigalenko E. et al. A genome-wide integrative genomic study localizes genetic factors influencing antibodies against Epstein-Barr virus nuclear antigen 1 (EBNA-1). PLoS genetics. 2013;9:e1003147
2. Meyer RM. EBV DNA: a Hodgkin lymphoma biomarker? Blood. 2013;121:3541-2
3. Zeng Z, Fan S, Zhang X. et al. Epstein-Barr virus-encoded small RNA 1 (EBER-1) could predict good prognosis in nasopharyngeal carcinoma. Clin Transl Oncol. 2015 [Epub ahead of print]
4. Zeng Z, Huang H, Huang L. et al. Regulation network and expression profiles of Epstein-Barr virus-encoded microRNAs and their potential target host genes in nasopharyngeal carcinomas. Sci China Life Sci. 2014;57:315-26
5. Yoshiyama H, Imai S, Shimizu N. et al. Epstein-Barr virus infection of human gastric carcinoma cells: implication of the existence of a new virus receptor different from CD21. Journal of virology. 1997;71:5688-91
6. Hurley EA, Agger S, McNeil JA. et al. When Epstein-Barr virus persistently infects B-cell lines, it frequently integrates. Journal of virology. 1991;65:1245-54
7. Jox A, Rohen C, Belge G. et al. Integration of Epstein-Barr virus in Burkitt's lymphoma cells leads to a region of enhanced chromosome instability. Annals of oncology: official journal of the European Society for Medical Oncology / ESMO. 1997;8(Suppl 2):131-5
8. Takakuwa T, Luo WJ, Ham MF. et al. Integration of Epstein-Barr virus into chromosome 6q15 of Burkitt lymphoma cell line (Raji) induces loss of BACH2 expression. The American journal of pathology. 2004;164:967-74
9. Gao J, Luo X, Tang K. et al. Epstein-Barr virus integrates frequently into chromosome 4q, 2q, 1q and 7q of Burkitt's lymphoma cell line (Raji). Journal of virological methods. 2006;136:193-9
10. Zhang HY, Qu G, Deng ZW. et al. Epstein-Barr virus DNA in nasopharyngeal biopsies. Virus research. 1989;12:53-9
11. Yu ZY, Song YL, Gong ZJ. et al. The Mechanism and Tumorigenesis of Oncogenic DNA Virus Integration. Prog Biochem Biophys. 2014;41:324-31
12. Cao X. Whole genome sequencing of cotton-a new chapter in cotton genomics. Sci China Life Sci. 2015;58:515-6
13. Chin FY, Leung HC, Yiu SM. Sequence assembly using next generation sequencing data-challenges and solutions. Sci China Life Sci. 2014;57:1140-8
14. Gao Y, Xie B, Liu R. Delivering noninvasive prenatal testing in a clinical setting using semiconductor sequencing platform. Sci China Life Sci. 2014;57:737-8
15. Hu W, Chen J. Whole-genome sequencing opens a new era for molecular breeding of grass carp (Ctenopharyngodon idellus). Sci China Life Sci. 2015;58:619-20
16. Wang Y, Ding Y, Yu D. et al. High-throughput sequencing-based genome-wide identification of microRNAs expressed in developing cotton seeds. Sci China Life Sci. 2015;58:778-86
17. Xie Q, Cao Y, Su J. et al. Genomic sequencing and analysis of the first imported Middle East Respiratory Syndrome Coronavirus (MERS CoV) in China. Sci China Life Sci. 2015;58:818-20
18. Cheung ST, Huang DP, Hui AB. et al. Nasopharyngeal carcinoma cell line (C666-1) consistently harbouring Epstein-Barr virus. International journal of cancer Journal international du cancer. 1999;83:121-6
19. Epstein MA, Achong BG, Barr YM. et al. Morphological and virological investigations on cultured Burkitt tumor lymphoblasts (strain Raji). Journal of the National Cancer Institute. 1966;37:547-59
20. Liao Q, Zeng Z, Guo X. et al. LPLUNC1 suppresses IL-6-induced nasopharyngeal carcinoma cell proliferation via inhibiting the Stat3 activation. Oncogene. 2014;33:2098-109
21. Zhang W, Zeng Z, Fan S. et al. Evaluation of the prognostic value of TGF-beta superfamily type I receptor and TGF-beta type II receptor expression in nasopharyngeal carcinoma using high-throughput tissue microarrays. J Mol Histol. 2012;43:297-306
22. Huang HB, Deng M, Zheng Y. et al. Innate immune protein lactotransferrin prevents initiation and arrests progression of nasopharyngeal carcinoma. Prog Biochem Biophys. 2013;40:319-24
23. Yang Y, Liao Q, Wei F. et al. LPLUNC1 inhibits nasopharyngeal carcinoma cell growth via down-regulation of the MAP kinase and cyclin D1/E2F pathways. PLoS One. 2013;8:e62869
24. Zhang W, Huang C, Gong Z. et al. Expression of LINC00312, a long intergenic non-coding RNA, is negatively correlated with tumor size but positively correlated with lymph node metastasis in nasopharyngeal carcinoma. J Mol Histol. 2013;44:545-54
25. Gong Z, Zhang S, Zeng Z. et al. LOC401317, a p53-Regulated Long Non-Coding RNA, Inhibits Cell Proliferation and Induces Apoptosis in the Nasopharyngeal Carcinoma Cell Line HNE2. PLoS One. 2014;9:e110674
26. Wei F, Li XY, Li XL. et al. The Effect and Mechanism of PLUNC Protein Family Against Inflammation and Carcinogenesis of Nasopharyngeal Carcinoma. Prog Biochem Biophys. 2014;41:24-31
27. Zhang W, Fan S, Zou G. et al. Lactotransferrin could be a novel independent molecular prognosticator of nasopharyngeal carcinoma. Tumour Biol. 2015;36:675-83
28. Bo H, Gong Z, Zhang W. et al. Upregulated long non-coding RNA AFAP1-AS1 expression is associated with progression and poor prognosis of nasopharyngeal carcinoma. Oncotarget. 2015;6:20404-18
29. Li R, Li Y, Fang X. et al. SNP detection for massively parallel whole-genome resequencing. Genome research. 2009;19:1124-32
30. Li H, Handsaker B, Wysoker A. et al. The Sequence Alignment/Map format and SAMtools. Bioinformatics. 2009;25:2078-9
31. Wang J, Mullighan CG, Easton J. et al. CREST maps somatic structural variation in cancer genomes with base-pair resolution. Nature methods. 2011;8:652-4
32. Chiang DY, Getz G, Jaffe DB. et al. High-resolution mapping of copy-number alterations with massively parallel sequencing. Nature methods. 2009;6:99-103
33. Wang K, Li M, Hakonarson H. ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic acids research. 2010;38:e164
34. Baer R, Bankier AT, Biggin MD. et al. DNA sequence and expression of the B95-8 Epstein-Barr virus genome. Nature. 1984;310:207-11
35. Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25:1754-60
36. Li R, Zhu H, Ruan J. et al. De novo assembly of human genomes with massively parallel short read sequencing. Genome research. 2010;20:265-72
37. Katoh K, Misawa K, Kuma K. et al. MAFFT: a novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic acids research. 2002;30:3059-66
38. Dolan A, Addison C, Gatherer D. et al. The genome of Epstein-Barr virus type 2 strain AG876. Virology. 2006;350:164-70
39. Lin Z, Wang X, Strong MJ. et al. Whole-genome sequencing of the Akata and Mutu Epstein-Barr virus strains. Journal of virology. 2013;87:1172-82
40. Lei H, Li T, Hung GC. et al. Identification and characterization of EBV genomes in spontaneously immortalized human peripheral blood B lymphocytes by NGS technology. BMC Genomics. 2013;14:804
41. Zeng MS, Li DJ, Liu QL. et al. Genomic sequence analysis of Epstein-Barr virus strain GD1 from a nasopharyngeal carcinoma patient. Journal of virology. 2005;79:15323-30
42. Liu P, Fang X, Feng Z. et al. Direct sequencing and characterization of a clinical isolate of Epstein-Barr virus from nasopharyngeal carcinoma tissue by using next-generation sequencing technology. Journal of virology. 2011;85:11291-9
43. Kwok H, Tong AH, Lin CH. et al. Genomic sequencing and comparative analysis of Epstein-Barr virus genome isolated from primary nasopharyngeal carcinoma biopsy. PLoS One. 2012;7:e36939
44. Kwok H, Wu CW, Palser AL. et al. Genomic diversity of Epstein-Barr virus genomes isolated from primary nasopharyngeal carcinoma biopsy samples. Journal of virology. 2014;88:10662-72
45. Tamura K, Peterson D, Peterson N. et al. MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Molecular biology and evolution. 2011;28:2731-9
46. Mooren JJ, Kremer B, Claessen SM. et al. Chromosome stability in tonsillar squamous cell carcinoma is associated with HPV16 integration and indicates a favorable prognosis. International journal of cancer Journal international du cancer. 2013;132:1781-9
47. Xiong W, Zeng ZY, Xia JH. et al. A susceptibility locus at chromosome 3p21 linked to familial nasopharyngeal carcinoma. Cancer Res. 2004;64:1972-4
48. Zeng Z, Zhou Y, Zhang W. et al. Family-based association analysis validates chromosome 3p21 as a putative nasopharyngeal carcinoma susceptibility locus. Genet Med. 2006;8:156-60
49. Zeng Z, Huang H, Zhang W. et al. Nasopharyngeal carcinoma: advances in genomics and molecular genetics. Sci China Life Sci. 2011;54:966-75
50. Gong Z, Yang Q, Zeng Z. et al. An integrative transcriptomic analysis reveals p53 regulated miRNA, mRNA, and lncRNA networks in nasopharyngeal carcinoma. Tumour Biol. 2015 [Epub ahead of print]
51. Xu K, Xiong W, Zhou M. et al. Integrating ChIP-sequencing and digital gene expression profiling to identify BRD7 downstream genes and construct their regulating network. Mol Cell Biochem. 2015 [Epub ahead of print]
52. Yan Q, Zeng Z, Gong Z. et al. EBV-miR-BART10-3p facilitates epithelial-mesenchymal transition and promotes metastasis of nasopharyngeal carcinoma by targeting BTRC. Oncotarget. 2015 [Epub ahead of print]
53. Zeng Z, Bo H, Gong Z. et al. AFAP1-AS1, a long noncoding RNA upregulated in lung cancer and promotes invasion and metastasis. Tumour Biol. 2015 [Epub ahead of print]
54. Zhang W, Zeng Z, Wei F. et al. SPLUNC1 is associated with nasopharyngeal carcinoma prognosis and plays an important role in all-trans-retinoic acid-induced growth inhibition and differentiation in nasopharyngeal cancer cells. FEBS J. 2014;281:4815-29
55. Jiang Z, Jhunjhunwala S, Liu J. et al. The effects of hepatitis B virus integration into the genomes of hepatocellular carcinoma patients. Genome research. 2012;22:593-601
56. Hou Y, Song L, Zhu P. et al. Single-cell exome sequencing and monoclonal evolution of a JAK2-negative myeloproliferative neoplasm. Cell. 2012;148:873-85
57. Wang J, Fan HC, Behr B. et al. Genome-wide single-cell analysis of recombination activity and de novo mutation rates in human sperm. Cell. 2012;150:402-12
58. Smith CC, Wang Q, Chin CS. et al. Validation of ITD mutations in FLT3 as a therapeutic target in human acute myeloid leukaemia. Nature. 2012;485:260-3
Author contact
![]() Corresponding author: Wei Xiong, Cancer Research Institute and the Affiliated Cancer Hospital of Xiangya School of Medicine, Central South University, 283 Tongzipo Road, Changsha, Hunan, 410013, China. Telephone: +86-731-8480-5446. Fax: +86-731-8480-5383. Email: xiongweiedu.cn
Corresponding author: Wei Xiong, Cancer Research Institute and the Affiliated Cancer Hospital of Xiangya School of Medicine, Central South University, 283 Tongzipo Road, Changsha, Hunan, 410013, China. Telephone: +86-731-8480-5446. Fax: +86-731-8480-5383. Email: xiongweiedu.cn