Impact Factor ISSN: 1837-9664

 Global reach, higher impact
Global reach, higher impactJ Cancer 2016; 7(4):353-363. doi:10.7150/jca.13754 This issue Cite
Research Paper
Ellagic Acid and Resveratrol Prevent the Development of Cisplatin Resistance in the Epithelial Ovarian Cancer Cell Line A2780
Laura H. Engelke1, Alexandra Hamacher1, Peter Proksch2, Matthias U. Kassack1 ![]()
1. Institut für Pharmazeutische und Medizinische Chemie, Heinrich-Heine-Universität Düsseldorf, Germany;
2. Institut für Pharmazeutische Biologie und Biotechnologie, Heinrich-Heine-Universität Düsseldorf, Germany.
Received 2015-9-4; Accepted 2015-12-8; Published 2016-1-10
Abstract
Purpose. Several studies have shown that natural compounds like resveratrol or ellagic acid have anticancer and antioxidant properties and can stimulate apoptosis in many cancer cell lines. The aim of this study was to elucidate if resveratrol or ellagic acid, respectively, could improve the efficacy of cisplatin in ovarian cancer.
Methods. As a cellular resistance model, the epithelial ovarian cancer cell line A2780 and its cisplatin-resistant subclone A2780CisR were used. A2780CisR was obtained by intermittent treatment of A2780 with cisplatin for 26 weekly cycles and showed a 4-6-fold increased resistance towards cisplatin compared to A2780.
Results. Pretreatment with resveratrol or ellagic acid 48 h prior to treatment with cisplatin showed a moderate enhancement of cisplatin cytotoxicity in A2780CisR cells (shift factors were 1.6 for ellagic acid and 2.5 for resveratrol). However, intermittent treatment of A2780 with cisplatin for 26 weekly cycles in permanent presence of resveratrol or ellagic acid, respectively, completely prevented the development of cisplatin resistance. The generated cell lines named A2780Resv and A2780Ellag displayed functional characteristics (migration, proliferation, apoptosis, activation of ErbB3, ROS generation) similar to the parental cell line A2780.
Conclusion. In conclusion, weekly intermittent treatment cycles of cisplatin-sensitive ovarian cancer cells with cisplatin retain cisplatin chemosensitivity in permanent presence of ellagic acid or resveratrol, respectively, whereas clinically relevant cisplatin chemoresistance develops in the absence of ellagic acid or resveratrol. Use of natural phenolic compounds may thus be a promising approach to prevent cisplatin resistance in ovarian cancer.
Keywords: ellagic acid, resveratrol, cisplatin, resistance development, ovarian cancer, A2780.
1. Introduction
Epithelial ovarian cancer is the fifth most lethal gynecological cancer due to its late diagnostic and asymptomatic behavior. Current treatment options are surgery followed by a platinum/taxane-based chemotherapy. Initially, patients show good response rates, but cancers relapse and develop a multifactorial drug resistance [1, 2]. Novel strategies to overcome chemoresistance are urgently needed.
Phytochemical products are a rich source for the treatment of various diseases like cancer. Nature-derived drugs can modulate molecular targets such as the EGFR or VEGFR family of receptor tyrosine kinases, established for therapy or prevention of cancer [3]. The natural compound resveratrol (RV) owns antitumorigenic and anti-inflammatory properties which are extensively described in the literature [4]. Its chemopreventive action is associated with arresting cellular proliferation. RV leads to cell cycle arrest in the S-phase and G2/M-phase [5-7]. In chemotherapy, RV is associated with inducing apoptosis, suppressing migration and sensitizing tumor cells to undergo chemotherapy-induced apoptosis [8, 9]. Ovarian cancer cell invasion is inhibited by RV through inhibition of EGFR phosphorylation [10, 11]. Generally, RV modulates many of those molecular targets in cancer stimulating cell migration, or regulating the ERK signaling pathway [3, 12]. In epithelial ovarian cancer cells, the overexpression of cyclin D1 is observed. This leads to an aggressive tumor phenotype and poor prognosis [12]. The expression of cyclin D1 is regulated by growth factor stimulation through the activation of a cascade of kinases such as MAPK/ERK kinase and PI3K signaling pathways [12]. The regulation of cyclin D1 is dependent on the inhibition of Akt-dependent signaling. RV influences the cyclin D1 level and decreases the phosphorylation of ERK and Akt in different ovarian cancer cells [12]. Furthermore, a direct substrate of Akt is mTOR. The phosphorylation of mTOR was decreased in response to RV treatment [12].
A phenolic phytochemical extensively described in literature is ellagic acid (EA) [13]. EA is known to quench reactive oxygen species (ROS) to reduce the toxic effects of ROS-generating chemotherapies [14]. Its chemopreventive actions are associated with regulating proliferation, inducing apoptosis, and cell cycle arrest [15-19]. EA like RV inhibits receptor tyrosine kinases such as VEGFR or IGFR [3, 16]. Furthermore, EA was shown to inhibit tumor growth in animal models induced by several chemical carcinogens, including aromatic amines, polycyclic aromatic hydrocarbons and N-nitrosamines [20-24]. Both, RV and EA, inhibit the tyrosine kinase activity of EGFR and ErbB2 through reduction of autophosphorylation [25]. A combination of EA and RV has been shown to interact synergistically in inducing apoptosis in human leukemia cells [26]. In different ovarian cancer, EA enhance the sensitivity of cytostatic drugs through regulation of various important pathways [3]. EA decreased cell survival in a dose- and time-dependent manner at which the growth of normal fibroblast cells was not inhibited [17]. In ovarian carcinoma cells, EA induces apoptosis through G1 phase arrest [17]. After treatment with EA, the level of p53 and p21 expression was increased, but the level of cyclin D1 and cyclin E was decreased [17].
Both, RV and EA, are rather nontoxic: subchronic oral toxicity studies for 90 days in rats of EA (up to 3 g/kg/day) or RV (up to 1000 mg/kg/day) showed a no observed adverse effect level (NOAEL) of around 3 g/kg/day for EA and NOAEL of around 200 mg/kg/day for RV [11, 27, 28].
In this study, the effects of EA and RV were studied in the ovarian cancer cell line A2780 and its 4.6-fold cisplatin-resistant subtype A2780CisR obtained by intermittent treatment with cisplatin (cDDP) in weekly cycles over 26 weeks, similar to a protocol previously published [29, 30]. Whereas it is known that short-term treatment of ovarian cancer cells with RV or EA leads to a moderate sensitization of ovarian cancer cells to undergo chemotherapy-induced apoptosis [8, 9, 17], nothing is known about long-term treatment effects of RV or EA on the development of cellular resistance. The aim of this study was thus to examine the effect of long-term treatment with RV or EA on the development of cDDP resistance next to the known short-term effects of the two natural compounds. Indeed, whereas short-term treatment showed only a moderate, partial reversal of cDDP-chemoresistance of A2780CisR, long-term treatment with RV or EA prevented the development of cDDP resistance in A2780 cells.
2. Materials and Methods
2.1 Materials
Roswell Park Memorial Institute (RPMI) media 1640, fetal bovine serum (FBS), penicillin/streptomycin (pen/strep) [10,000 U/ml; 10 mg/ml] and trypsin-EDTA (0.05% Trypsin, 0.02% EDTA in PBS) were purchased from PAN Biotech (Aidenbach, Germany). CDDP, RV and 2´-7´-dichlorodihydrofluorescein-diacetate (H2DCF-DA) were from Sigma-Aldrich (Steinheim, Germany). CDDP was dissolved and subsequently diluted in 0.9% saline. RV and H2DCF-DA were dissolved in DMSO and subsequently diluted with 1x PBS (10x PBS: 1.4 M NaCl, 30 mM KCl, 80 mM Na2HPO4 x 2 H2O, 15 mM KH2PO4). EA was purchased from Fluka Chemie AG (Buchs, Switzerland). The compound was dissolved in DMSO and subsequently diluted in PBS. Ascorbic acid was from Carl Roth (Karlsruhe, Germany) and was dissolved in 0.9% saline. 3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) was purchased from Serva (Heidelberg, Germany) and was dissolved in PBS at a concentration of 5 mg/ml. Propidium iodide and the annexin V apoptosis detection kit were purchased from Santa Cruz Biotechnology (Heidelberg, Germany). Propidium iodide was dissolved in PBS.
2.2 Cell lines
A2780 cells were obtained from ECACC (Salisbury, Wiltshire/UK) and cultivated in RPMI-1640 medium supplemented with 10% FBS, 120 µg/ml streptomycin and 120 U/ml penicillin. Cells were grown at 37°C in a humidified atmosphere containing 5% CO2. The cDDP-resistant subclone A2780CisR was obtained by intermittent treatment of A2780 cells with cDDP for 24 weekly cycles according to methods previously published [29, 30]. The cell types A2780Resv and A2780Ellag were generated by weekly cDDP treatment (IC50) of A2780 cells for 26 cycles in permanent presence of 10 µM RV or 3.2 µM EA. Furthermore, the cDDP-resistant cell line A2780CisR was permanently cultivated with 10 µM EA or 10 µM RV for 6 months, respectively. This treatment generated the cell lines termed A2780CisR+Ellag and A2780CisR+Resv. Cells were grown to 80-90% confluence, washed with 1x PBS, and treated with trypsin-EDTA before subculturing.
2.3 MTT assay
MTT Assays were performed as recently described [31]. Briefly, cells were plated into 96-well microtiter plates (approximately 9,000 cells/well) (Sarstedt, Nümbrecht, Germany) and pre-incubated with growth medium overnight. Then, cells were incubated with increasing concentrations of test compounds for 72 h. After 72 h, 25 µl of a solution of MTT were added to each well. After approximately 10 min, formazan crystals occurred, and medium was removed. Formazan crystals were then dissolved in 75 µl DMSO. Absorption was measured at 544 nm (test wavelength) and 690 nm (reference wavelength) using the BMG FLUOstar (BMG Labtechnologies, Offenburg, Germany). Absorption of the reference wavelength was subtracted from the absorption of the test wavelength.
2.4 Immunoblot analysis
For Immunoblotting, standard procedures were used as described in [30]. Briefly, samples were dissolved in 2X Laemmli-buffer with β-mercaptoethanol, heated to 95°C for 3 min, and loaded onto an 8% SDS-polyacrylamide gel. Proteins were transferred to a polyvinylidene difluoride membrane by semidry blotting at 40 mA for 1 h. The membrane was blocked with 3% milk in TBS-T (10X TBS-T: 200 mM Tris, 9% NaCl, 1% Tween 20, pH 7.4) for 1 h. Then PVDF membrane incubated overnight at 4°C with the primary antibody. Primary polyclonal goat antibodies and primary monoclonal rabbit antibodies were all from R&D Systems (Wiesbaden, Germany) except the primary anti-β-actin polyclonal antibody which was purchased from Santa Cruz Biotechnology and was used at a 1:2,000 dilution. ErbB3 and pErbB2 were used at a 1:100 dilution and phospho-ErbB3 used at a 1:200 dilution. ErbB2 and pEGFR were used at a 1:500 dilution and EGFR was used at a 1:250 dilution. Membrane was subsequently washed twice in TBS-T and once in TBS (10X TBS: 200 mM Tris, 9% NaCl, pH 7.4). Incubation with the corresponding HRP-conjugated secondary antibody (R&D Systems) followed for 1 h at room temperature. Then, the membrane was washed in TBS-T twice and in TBS once. Proteins were visualized using the Western Blotting Luminol Reagent (Santa Cruz Biotechnologies) and the INTAS Science Imaging Instrument (Gel iX Imager, Göttingen, Germany). β-actin was used as loading control at a 1:2,000 dilution.
2.5 Signal pathway analysis
The tyrosine-kinase phosphoproteom was investigated by a human phospho-receptor tyrosine-kinase antibody array (Cat# ARY001) from R&D Systems according to the manufacturer´s protocol.
2.6 Measurement of reactive oxygen species
A2780 or A2780CisR were plated in 24-well plates (approximately 150,000 cells/well) (Sarstedt) and incubated with grow medium overnight. Cells cultivated in phenolred-free RPMI-1640 without FBS and without antibiotics were exposed to EA dissolved in DMSO, RV in DMSO or ascorbic acid dissolved in 0.9% saline for 18 hours. Cells were then treated with 10 µM H2DCF-DA in the dark at 37°C for 30 min. After washing with PBS and cell detachment using trypsin-EDTA, cells were cultivated in phenolred-free RPMI-1640. Cells were stained with 5 µg/ml propidium iodide for 10 minutes in the dark. Then, fluorescence was measured by flow cytometry (CyFlow® space, Partec, Münster, Germany). The positive control was treated with 200 µM H2O2 in the dark at 37°C for 1 hour and then loaded with propidium iodide.
2.7 Doubling time
Each cell line was plated in 6-well plates (approximately 1,000 cells/well) (Sarstedt) and pre-incubated with growth medium for 72 h. Then, cells were harvested by trypsin-EDTA, washed with 1x PBS, and counted by flow cytometry.
2.8 Scratch assay
Scratch assay was performed according to standard procedures as described in [32]. Each cell line was plated in 6-well plates (approximately 3 million cells/well) and incubated in growth medium for approximately 6 hours at 37°C to create a confluent monolayer. A straight line was scraped through the cell monolayer with a 200 µl pipet tip. Cells were then washed with PBS and cultivated in serum-free RPMI-1640. Microscope images (Motic, Wetzlar, Germany) were acquired directly after scratching and after additional 24 h incubation at 37°C.
2.9 Cell cycle analysis
Cell cycle phases of the different cell lines were analyzed by flow cytometry using standard procedures [33]. After exposure to the various compounds, cells were washed in ice-cold PBS and approximately 2 million cells were fixed in 70% cold ethanol and stored for at least 24 h at -20°C. Fixed cells were washed with cold PBS and incubated in staining solution (0.1% (v/v) Triton X-100 in PBS, 200 µg/ml DNAse-free RNAse A (Fermentas/Thermo Fisher Scientific, Waltham, MA, USA), 20 µg/ml propidium iodide) in the dark at 37°C for 15 min. DNA content was then measured by flow cytometry.
2.10 Apoptosis analysis
To determine apoptotic cells, annexin V apoptosis detection kit from Santa Cruz Biotechnology was used according to the manufacturer´s protocol. Each cell line was exposed to 10 µM cDDP for 48 h. The supernatants and the cells released with trypsin-EDTA were centrifuged. After removing the supernatant, cells were washed with 1x PBS and resuspended in annexin V binding buffer (HEPES 10 mM, pH 7.4, NaCl 140 mM, CaCl2 2.5 mM). Approximately 100,000 cells were labeled with 2.5 µl of 50 µg Annexin V-FITC in 250 µl annexin V binding buffer and 10 µl of 50 µg/ml propidium iodide in PBS, and incubated in the dark at RT for 15 minutes. Apoptotic cells were then detected by flow cytometry. Apoptosis of untreated cells was subtracted from the cells treated with cDDP.
2.11 Statistical analysis
Assays were performed at least in three independent experiments. Concentration effect curves were then generated by nonlinear regression curve fitting using the 4-parameter logistic equation with variable hill slope (GraphPad Prism version 4, GraphPad Software Inc., San Diego, USA). IC50 is the concentration of the cytotoxic agent that led to a decrease of 50% of the recorded signal. The pIC50 values are -log IC50. Resistance factors are the ratios of the IC50 values determined in various treated cell lines (A2780CisR, A2780Resv, A2780Ellag) and the IC50 values in the A2780 cell line, and were determined by calculating and averaging the ratios of single pairs of IC50 values from individual experiments. Statistical significance was assessed by two-tailed Student´s t-test and considered significant if p < 0.05.
3. Results
The cytotoxic activity of EA and RV on A2780 and A2780CisR cells was assessed after 72 h incubation using MTT assay (Figure S1). RV gave a similar IC50 on both cell lines with 60.3 and 70.8 µM, respectively. For short- and long-term incubation with RV, a concentration of 10 µM was chosen at which RV has no cytotoxic effect. EA is however more potent at A2780 cells than at A2780CisR: the IC50 at A2780CisR is 36.3 µM and thus around 2-fold higher than the IC50 at A2780 with 17.0 µM. For short- and long-term incubation, A2780 cells were exposed to 3.2 µM and A2780CisR to 10 µM EA.
Cytotoxic activity of cDDP was assessed after 72 h incubation using MTT assay (Figure 1A, 1B, 1C, 1D).
Modulation of cDDP cytotoxicity and ROS induction by EA and RV (MTT and ROS assay). Incubation with 3.2 µM or 10 µM EA or 10 µM RV 48 h prior to addition of cDDP enhances cDDP cytotoxicity in A2780 and A2780CisR. In A2780 (A), the pIC50 value for cDDP was calculated as 5.73 ± 0.04 in the absence and 6.04 ± 0.04 in the presence of 3.2 µM EA (*: p < 0.05). In A2780CisR (B), the pIC50 value for cDDP was determined as 4.87 ± 0.02 in the absence and 4.99 ± 0.05 in the presence of 10 µM EA (not significant). Pre-incubation of 10 µM RV 48 h prior to addition of cDDP increased the pIC50 for cDDP in A2780 (C) to 6.14 ± 0.01 (*: p < 0.05). In A2780CisR (D), pIC50 value of cDDP in the presence of 10 µM RV was 5.05 ± 0.12 (not significant). Data shown (average ± SEM) are from a typical experiment out of a set of three independent experiments each performed with 3 replicates. Incubation of EA reduces the level of ROS similar to AA, also after hydrogen peroxide treatment. In contrast, RV promotes ROS in both cell lines (E, F). Additionally, EA inhibits ROS induction due to cDDP treatment. Combination of RV with cDDP has no further effects (G, H). Data shown are average from at least three independent experiments.
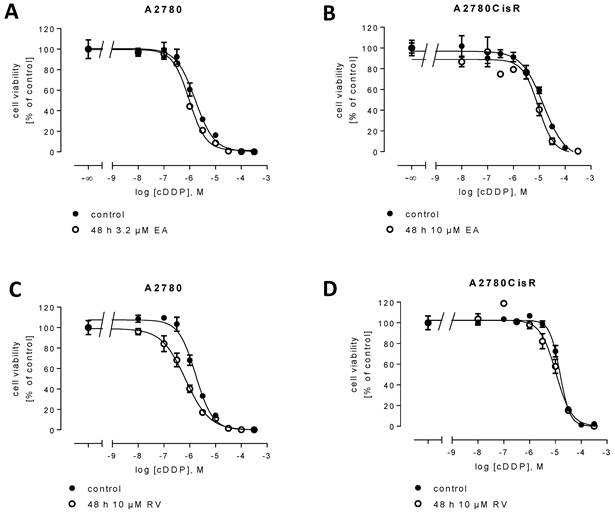
48 h pre-incubation of cells with EA or RV prior to cDDP addition increased the sensitivity of cDDP in A2780 and A2780CisR, respectively (shift factor EA: A2780: 2.4; A2780CisR: 1.3; shift factor RV: A2780: 3.1; A2780CisR: 1.4). Shift factors in A2780 cells are significant with p < 0.05. Next, we tested the effect of EA and RV on ROS in A2780 and A2780CisR. Similar to the antioxidant ascorbic acid (AA), EA reduced the level of ROS in H2O2-stressed cells, by about 12% in A2780 (Figure 1E) and by 19% in A2780CisR (Figure 1F). EA similar to AA had no effect on the basal level of ROS (untreated control). In contrast to EA, RV increased the level of ROS in A2780 and A2780CisR in untreated controls and after ROS-induction by H2O2 treatment (Figure 1E, 1F). RV increased ROS by about 32% in the native cell line and 18% in the cDDP-resistant cell line, significantly (*p < 0.05, **p < 0.01). Next, the effect of EA and RV on ROS in cDDP-treated cells was examined. EA reduced ROS in both cell lines significantly (Figure 1G, 1H). Interestingly, whereas cDDP induced ROS in A2780, it did not increase ROS in A2780CisR. RV alone increased ROS levels compared to the untreated control; however, the combination of RV with cDDP had no further effect.
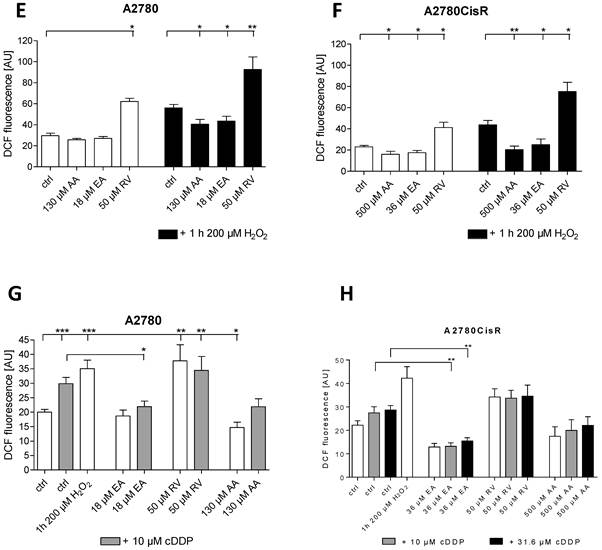
The small but significant effect of 48 h pre-incubation of RV and EA, respectively, on the chemosensitivity of cDDP in A2780 and the small effect in A2780CisR (Figure 1, Table 1) prompted us to examine the effect of RV and EA on the development of chemoresistance against cDDP. Thus, A2780 cells were exposed to weekly cycles (6 hours) of the cDDP IC50 under permanent presence of 10 µM RV or 3.2 µM EA, respectively, over a period of 26 cycles similar to the protocol previously published [30]. The resulting cell lines denoted A2780Resv and A2780Ellag did not develop cDDP resistance as observed in A2780CisR displaying a resistance factor of 4-6. The resistance factors remained constant up to the end of the 26 cDDP cycle treatments and were calculated as 1.1 for A2780Resv and 1.2 for A2780Ellag (not significant different from 1, Figure 2A, Table 1). Since permanent presence of EA and RV is able to prevent the development of chemoresistance induced by weekly stressing with an IC50 of cDDP, the effects of RV or EA on the cDDP cytotoxicity of A2780CisR were further investigated (Figure 2B). For 6 months A2780CisR cells were permanently cultivated with either 10 µM RV or 10 µM EA and were called A2780CisR+Resv and A2780CisR+Ellag. Then, the sensitivity against cDDP was tested by MTT. As shown in Figure 2B, IC50 values remained unchanged in A2780CisR+Resv and A2780CisR+Ellag compared to A2780CisR, indicating that permanent presence of RV and EA is not able to revert a fully established cDDP resistance (Figure 2B) but are able to prevent the development of resistance (Figure 2A). Table 1 summarizes all cDDP IC50 values and shift or resistance factors. Next, the level of ROS in A2780Ellag and A2780Resv compared to A2780 and A2780CisR was determined. Whereas RV and EA had opposite effects on ROS generation in short-term incubation experiments (Figure 1E, 1F, 1G, 1H) the basal level of ROS in the newly generated cell lines was significantly reduced for both, RV and EA compared to the parental cell line A2780 (Figure 2C). Notably, neither treatment with hydrogen peroxide nor with cDDP generated ROS in A2780Resv cells. In A2780Ellag cells, the level of ROS was higher than in A2780Resv cells but still significantly lower than in control A2780 (Figure 2C).
Summary of IC50 values of cDDP. Data shown are average ± SEM from three independent experiments.
| Cell line | pIC50 ± SEM | IC50 [µM] | Shift or resistance factor |
|---|---|---|---|
| A2780 | 5.73 ± 0.04 | 2.24 | |
| A2780 + 3.2 µM EA 48 h pre-incubation | 6.04 ± 0.04 | 0.92 | 2.4* |
| A2780 + 10 µM RV 48 h pre-incubation | 6.14 ± 0.01 | 0.72 | 3.1* |
| A2780CisR | 4.87 ± 0.02 | 13.9 | 6.2* |
| A2780CisR + 10 µM EA 48 h pre-incubation | 4.99 ± 0.05 | 10.8 | 1.3+ |
| A2780CisR + 10 µM RV 48 h pre-incubation | 5.05 ± 0.12 | 9.90 | 1.4+ |
| A2780Ellag | 5.64 ± 0.05 | 2.63 | 1.2# |
| A2780Resv | 5.67 ± 0.04 | 2.38 | 1.1# |
| A2780CisR+Ellag | 4.91 ± 0.02 | 12.6 | 1.1+ |
| A2780CisR+Resv | 4.81 ± 0.05 | 15.7 | 0.9+ |
*Resistance / shift factor significantly different from 1 compared to A2780; +resistance / shift factor not significantly different from 1 compared to A2780CisR; #resistance factor not significantly different from 1 compared to A2780.
EA and RV prevent the development of cDDP resistance in A2780 cells. A2780 cells were exposed to the IC50 of cDDP for 6h weekly over a period of 26 cycles under permanent presence of 10 µM RV or 3.2 µM EA. (A) Generated cell lines A2780Resv and A2780Ellag did not develop resistance as their calculated resistance factors were not significantly different from 1. (B) In contrast, treatment of A2780CisR cells with 10 µM RV or 10 µM EA over a period of 26 weeks did not alter chemoresistance. (C) Long-term incubation of EA or RV reduced the basic level of ROS. Both, RV and EA, completely inhibited ROS induction by hydrogen peroxide or cDDP. Data shown are average ± SEM from 3 independent experiments (**p < 0.01, ***p < 0.001).
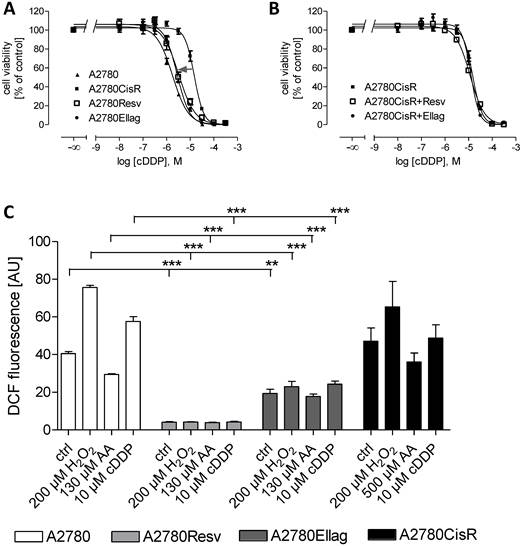
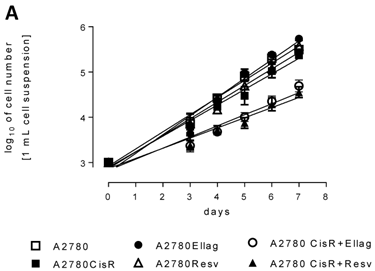
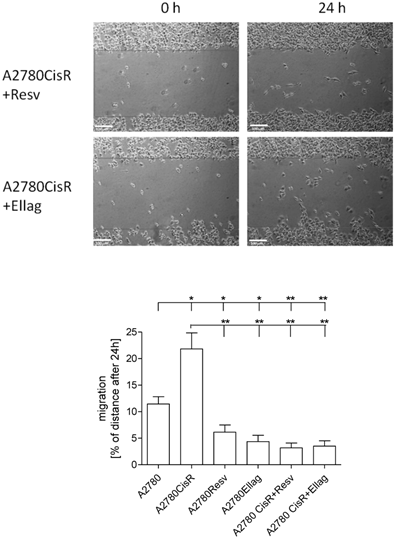
To obtain deeper insight into the mechanisms by which RV and EA prevent the development of resistance against cDDP, we analyzed cell biological and biochemical effects of RV and EA on A2780Resv and A2780Ellag as well as A2780CisR+Ellag and A2780CisR+Resv compared to A2780 and A2780CisR. A2780 and A2780CisR showed identical proliferation behavior (Figure 3A). The proliferation of A2780Resv and A2780Ellag was moderately enhanced compared to A2780 and A2780CisR. However, permanent presence (26 weeks) of RV or EA in A2780CisR cells resulting in A2780CisR+Resv and A2780CisR+Ellag cell lines led to a significantly decreased proliferation. Migration capacity was assessed by an in vitro scratch assay. Figure 3B displays images of the cell lines immediately and 24 h after applying a scratch. A2780CisR cells show significantly enhanced migration compared to A2780 (22% compared to 11%). A2780Resv and A2780Ellag displays even lower migration than A2780 although displaying slightly higher proliferation. Interestingly, the cell lines A2780CisR+Resv and A2780CisR+Ellag show much lower migration than A2780CisR (***p < 0.001) and even lower migration than A2780 (Figure 3B) although they retained cDDP chemoresistance similar to A2780CisR (Table 1). The effect of EA and RV in A2780CisR+Resv and A2780CisR+Ellag may however be due to strongly decreased proliferation as shown in Figure 3A.
Proliferation and migration behavior of A2780Resv, A2780Ellag, A2780CisR+Resv and A2780CisR+Ellag compared to A2780 and A2780CisR. (A) A2780 and A2780CisR showed identical proliferation, A2780Rev and A2780Ellag a slightly increased proliferation. However, A2780CisR+Resv and A2780CisR+Ellag showed a significantly reduced proliferation. Each cell line was seeded with the same number of cells (1,000 cells). Data shown are average ± SEM from 4 independent experiments. (B) Analysis of cell migration by in vitro scratch assay. The images were acquired at 0 and 24 h after applying a pipet tip-induced scratch using phase-contrast microscopy and represent one typical experiment of three independent experiments. A2780CisR showed the highest migratory potential whereas the cell lines A2780Resv, A2780Ellag, A2780+Resv and A2780CisR+Ellag migrated at a slower rate compared to parental cell line A2780 (*p < 0.05, **p < 0.01). The white bar represents a length of 100 µm. Data shown are average ± SEM of at least three independent experiments.

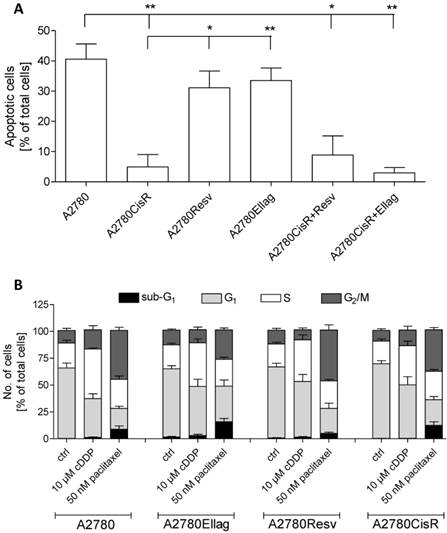
Next, 10 µM cDDP-induced effects on apoptosis (48 h incubation) and cell cycle (24 h incubation) were studied in the various cell lines (Figure 4). The number of apoptotic cells is similar in A2780Resv and A2780Ellag as in A2780 upon treatment with 10 µM cDDP (Figure 4A): in A2780, 43% of the cells were apoptotic, in A2780Resv 37%, and in A2780Ellag 40%, respectively. In contrast, A2780CisR showed almost no apoptosis induction (only 5%). Furthermore, A2780CisR+Resv and A2780CisR+Ellag cell lines behaved like A2780CisR in terms of apoptosis induction. Effects of 24 h cDDP or paclitaxel treatment on the cell cycle were then analyzed in A2780, A2780CisR, A2780Ellag and A2780Resv (Figure 4B). As control, paclitaxel induced a G2/M arrest in all cell lines whereas cDDP led to a S-phase arrest which was most pronounced in A2780 cells and the least in A2780CisR (number of cells in S-phase: A2780 46%; A2780Ellag 41%; A2780Resv 39%; A2780CisR 36%; Figure 4B). Taken together, cell cycle distribution upon paclitaxel or cDDP stress was not significantly affected by long term treatment of EA or RV. However, apoptosis data showed that A2780Resv and A2780Ellag cells retain the same cDDP-sensitive phenotype as the parental cell line A2780.
CDDP-induced apoptosis and cell cycle arrest in RV and EA treated versus untreated A2780 and A2780CisR. (A) All cell lines were exposed to 10 µM cDDP for 48 h. The amount of apoptosis induction in A2780Resv (37%) and A2780Ellag (40%) is similar to the amount of apoptotic cells in A2780 (43%). In A2780CisR+Resv and A2780CisR+Ellag, cDDP only induces 9% and 3%, respectively, similar to apoptosis induction in A2780CisR (5%). Data shown are average ± SEM from 3 independent experiments and are percentage of total cells analyzed by flow cytometry (*p < 0.05; **p < 0.01). (B) For cell cycle analysis all cell lines were exposed to test compounds for 24 h. The amount of cells arresting in S-phase in A2870Ellag (41%) and A2780Resv (39%) are similar to the ones in A2780 (46%). In contrast, the amount of A2780CisR cells arresting in S-Phase is smaller (36%). Data shown are average ± SEM from 4 independent experiments and are percentage of total cells analyzed by flow cytometry.
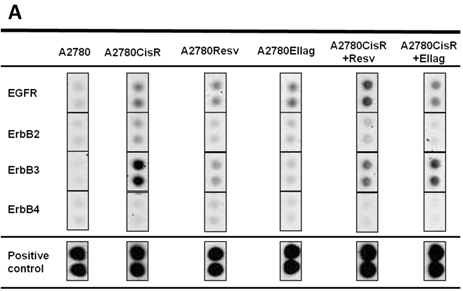
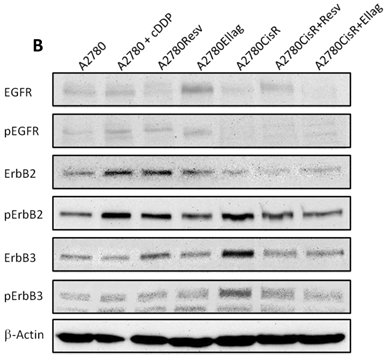
Previously, upregulation of IGF1, IGF1-R, and increased phosphorylation of IGF1-R has been assigned as a major reason for chemoresistance in A2780CisR [30]. However, subsequent approaches to reverse chemoresistance through inhibition of IGF1-R phosphorylation by NVP-AEW541 failed. 48 h pre-incubation of 1 µM NVP-AEW541 gave only an approximately 2-fold shift in A2780CisR and had no effect in A2780 cells (data not shown). Since receptor tyrosine kinases (RTK) such as the IGFR and EGFR family are involved in proliferation and cell invasiveness [34], the phosphorylation status of RTKs was measured in the various A2780 cell lines using proteome profiler arrays. Figure 5A shows results for the EGFR family, since besides EGF receptors and the previously published IGFR [30], no other RTKs were differently phosphorylated. Most prominent is the phosphorylation of ErbB3 in A2780CisR which is absent in sensitive A2780 and reduced in cell lines treated with EA or RV (A2780Resv, A2780Ellag, A2780CisR+Resv and A2780CisR+Ellag). To verify data from the proteome profiler, immunoblot analysis for expression and phosphorylation of EGFR, ErbB2 and ErbB3 was performed (Figure 5B). Furthermore, short-term effect of cDDP treatment in A2780 cells on RTK expression and phosphorylation was studied (“A2780 + cDDP”). Whereas EGFR and ErbB2 show similar expression in A2780 and A2780CisR, ErbB3 shows higher expression in A2780CisR. Phosphorylation in A2780CisR is however increased for ErbB2 and ErbB3. Short-term incubation of A2780 with cDDP (A2780 + cDDP) increased expression and phosphorylation of ErbB2 but did not affect ErbB3. Long-term incubation with RV or EA during cDDP resistance development (resulting in A2780Resv and A2780Ellag) gave reduced ErbB2 phosphorylation and an ERbB3 phosphorylation similar to A2780.
Immunoblot analysis of untreated and treated cell lines. (A) Human phospho-RTK arrays were used to determine the relative target phosphorylation. Only RTKs with detectable phosphorylation compared to control spots are displayed. Long term (26 weeks) permanent incubation with EA or RV inhibited phosphorylation of ErbB2 and ErbB3 induced by cDDP treatment. (B) Activation of ErbB2 and ErbB3 in the cDDP-resistant cell line was confirmed by western blot analysis. Short-time exposure of cDDP activates EGFR and ErbB2 in sensitive A2780 cells (A2780+cDDP). In A270CisR, ErbB2 and ErbB3 are activated. Long-term incubation of RV and EA inhibited phosphorylation of these receptors.
4. Discussion
Ovarian cancer is a malignancy with a worldwide incidence of more than 4% cases and almost every patient dies from short- or long-term consequences of this disease [2]. Currently established treatment is a combination of surgery and platinum/taxane-based chemotherapy [35]. Nevertheless, most cases relapse and develop multidrug resistance [35]. One approach in modern chemotherapy is the use of modulators of chemotherapy to increase the efficiency of cytostatic agents. Nature-derived compounds have emerged as modulators beneficial for prevention of cancer and reverting chemoresistance [3, 8, 25]. A plethora of investigations has been performed with the phenolic compounds EA and RV discovering complex and mostly beneficial effects on cancer development and cancer treatment [3, 36]. However, a systematic study on the effect of EA and RV on the development of resistance against cytostatic agents has not yet been performed. This study investigates effects of EA or RV, respectively, on resistance development during 26 weekly cycles of intermittent cDDP treatment in the epithelial ovarian cancer cell line A2780. 48 h pre-incubation of RV or EA enhanced cDDP cytotoxicity by a factor of 3.1 or 2.4 (Table 1), similar to the results from Nessa et al. using RV pre-incubation prior cDDP in A2780 cells [9]. Interestingly, these effects of RV and EA were not found in A2780CisR (Table 1), indicating that a fully established cDDP resistance as in A2780CisR cannot be reverted by short-term (up to 48 h) EA or RV treatment. Neither can a fully established cDDP resistance be reverted by long-term incubation (up to 26 weeks) as shown in A2780CisR+Resv and A2780CisR+Ellag cell lines. These cell lines showed decreased cellular proliferation compared to A2780 and A2780CisR (Figure 3A), but they were still as resistant towards cDDP as A2780CisR (Figure 2B, Table 1). However, A2780CisR+Ellag and A2780CisR+Resv cell lines showed reduced migration (Figure 3B) which may be attributed to reduced proliferation (Figure 3A) or reduced activation of EGF receptors (Figure 5) compared to A2780CisR.
In the next step, we tested if permanent presence of EA or RV in the cDDP-sensitive A2780 cell line prevents the development of cDDP resistance induced by 26 weekly cycles of intermittent cDDP treatments. This weekly intermittent cDDP treatment has previously been shown to result in a cDDP resistant cell line named A2780CisR [30]. Indeed, permanent EA or RV treatment, respectively, resulting in the cell lines A2780Ellag and A2780Resv, prevented the development of cDDP resistance in contrast to cDDP treatment in the absence of EA or RV (Figure 2A, Table 1). Both, A2780Ellag and A2780Resv resemble the cDDP-sensitive phenotype of A2780 even though they have undergone a 26 week intermittent treatment with cDDP. First of all, with a shift factor of 1.2 and 1.1, respectively, A2780Ellag and A2780Resv showed no significantly different chemosensitivity towards cDDP (Table 1). Their proliferation is slightly higher than in A2780 or A2780CisR cells (Figure 3A). However, long-term (26 weeks) EA or RV-treated A2780CisR cells show significantly reduced proliferation in Figure 3A which is similar to the effects of EA or RV published for leukemic, MCF-7 or Caco-2 cells [7, 37]. Second, the rate of apoptosis induction upon cDDP treatment is not different in A2780 and in A2780Ellag or A2780Resv whereas A2780CisR shows clearly reduced apoptosis induction (Figure 4A). EA and RV treatment in A2780CisR had no pro-apoptotic effect (A2780CisR+Ellag, A2780CisR+Resv, Figure 4A). Third, A2780Ellag and A2780Resv show even less migration than A2780 though owning a slightly higher proliferation rate (Figure 3). Again, this confirms maintaining a sensitive phenotype. Fourth, A2780Ellag and A2780Resv show lower activation of ErbB2 than A2780CisR and the same low phosphorylation of ErbB3 as A2780, thus confirming that long-term EA or RV treatment during cDDP resistance development retain the sensitive phenotype of A2780 (Figure 5). This is in accordance with literature data showing that inactivation of ErbB3 induces apoptosis in various carcinoma cell lines [34, 38]. However, reduction of ErbB receptor phosphorylation seems necessary for retaining a sensitive phenotype, but not sufficient as seen by long-term treatment with EA or RV of resistant A2780CisR resulting in A2780CisR+Ellag or A2780CisR+Resv cell lines displaying reduced phosphorylation but still resistance not different from A2780CisR.
Lastly, ROS modulation by EA and RV was different in short-term (48 h) and long-term (26 weeks) incubation experiments. 48 h RV increased ROS in the absence or presence of hydrogen peroxide in A2780 and A2780CisR, whereas EA had no effect or reduced ROS (Figure 1E, 1F). 48 h EA reduced cDDP-induced ROS in A2780 and A2780CisR whereas RV gave no further increase in ROS upon cDDP treatment (Figure 1G, 1H). Although RV and EA show different short-term effects in ROS production, both compounds are able to sensitize A2780 cells towards cDDP (Figure 1A, 1C, Table 1). Furthermore, both compounds inhibit the development of cDDP resistance in A2780 cells (Figure 2A). Whereas RV induced ROS in short-term treatment, in both long-term treatments, A2780Ellag and A2780Resv, ROS was significantly reduced compared to A2780 and neither cDDP or hydrogen peroxide were able to increase ROS in A2780Ellag and A2780A2780Resv in contrast to their effects in A2780 (Figure 2C). Thus, chemosensitivity in A2780 cells is not necessarily linked to increased oxidative stress after cDDP treatment since A2780Ellag and A2780Resv are as sensitive as A2780 but show reduced ROS.
In conclusion, permanent presence of the phytochemicals RV and EA, respectively, in non-toxic concentrations is able to prevent the development of cDDP resistance in the ovarian cancer cell line A2780. In the resistant sub-cell line A2780CisR, EA and RV were unable to reverse cDDP resistance even though reducing proliferation rate and migration.
Summary of the highlights of this study.
| 1) Short term treatment (48 h) with EA or RV of the chemosensitive ovarian cancer cell line A2780 leads to moderate (2-3 fold) increase in chemosensitivity. |
| 2) Short term treatment (48 h) with EA or RV of cDDP-resistant A2780CisR does not improve chemosensitivity. |
| 3) Permanent presence over a period of 26 weeks of EA or RV, respectively, does not revert cDDP-chemoresistance of A2780CisR. However, permanent presence of EA or RV inhibits proliferation and migration of A2780CisR. |
| 4) Permanent presence over a period of 26 weeks of EA or RV, respectively, prevents the development of chemoresistance upon weekly cDDP treatment in A2780 cells. A chemosensitive phenotype of A2780 is retained, and cell migration is inhibited. |
| 5) Phosphorylation of ErbB2 and ErbB3 is reduced in EA or RV treated cells and may account for reduced cell migration and prevention of chemoresistance. |
Supplementary Material
Figure S1.
Acknowledgements
This work was funded by grants from the Bundesministerium für Wirtschaft (BMWi) AiF/ZIM project KF2388801UL9 to MUK.
Competing Interests
None.
References
1. Mungenast F, Thalhammer T. Estrogen biosynthesis and action in ovarian cancer. Front Endocrinol. 2014;5:192
2. Siegel R, Ma J, Zou Z, Jemal A. Cancer Statistics. CA Cancer J Clin. 2014;64:9-29
3. Aggarwal BB, Shishodia S. Molecular targets of dietary agents for prevention and therapy of cancer. Biochem Pharmacol. 2006;71:1397-1421
4. Singh CK, George J, Ahmad N. Resveratrol-based combinatorial strategies for cancer management. Ann N Y Acad Sci. 2013;1290:113-21
5. Haider UG, Sorescu D, Griendling KK, Vollmar AM, Dirsch VM. Resveratrol increases serine15-phosphorylated but transcriptionally impaired p53 and induces a reversible DNA replication block in serum-activated vascular smooth muscle cells. Mol Pharmacol. 2003;63:925-32
6. Liang YC, Tsai SH, Chen L, Lin-Shiau SY, Lin JK. Resveratrol-induced G2 arrest through the inhibition of CDK7 and p34CDC2 kinases in colon carcinoma HT29 cells. Biochem Pharmacol. 2003;65:1053-60
7. Ferry-Dumazet H, Garnier O, Mamany-Matsuda M, Vercauteren J, Belloc F, Billiard C. et al. Resveratrol inhibits the growth and induces the apoptosis of both normal and leukemic hematopoietic cells. Carcinogenesis. 2002;23:1327-33
8. Kundu JK, Surh YJ. Cancer chemopreventive and therapeutic potential of resveratrol: Mechanistic perspectives. Cancer Lett. 2008;269:243-61
9. Nessa MU, Beale P, Chan C, Yu JQ, Huq F. Combinations of resveratrol, cisplatin and oxaliplatin applied to human ovarian cancer cells. Anticancer Res. 2012;32:53-60
10. Jeong KJ, Cho KH, Panupinthu N, Kin H, Kang J, Park CG. et al. EGFR mediates LPA-induced proteolytic enzyme expression and ovarian cancer invasion: Inhibition by resveratrol. Mol Oncol. 2013;7:121-29
11. Stakleff KS, Sloan T, Blanco D, Marcanthony S, Booth TD, Bishayee A. Resveratrol exerts differential effects in vitro and in vivo against ovarian cancer cells. Asian Pac J Cancer Prev. 2012;13:1333-40
12. Vergara D, Simeone P, Toraldo D, Del Boccio P, Vergaro V, Leporatti S. et al. Resveratrol downregulates Akt/GSK and ERK signalling pathways in OVCAR-3 ovarian cancer cells. Mol Biosyst. 2012;8:1078-87
13. Nambiar D, Rajamani P, Singh RP. Effects of phytochemicals on ionization radiation-mediated carcinogenesis and cancer therapy. Mut Res. 2011;728:139-57
14. Block KI, Koch AC, Mead MN, Tothy PK, Newman RA, Gyllenhaal C. Impact of antioxidant supplementation on chemotherapeutic toxicity: A systematic review of the evidence from randomized controlled trials. Int J Cancer. 2008;123:1227-1239
15. Narayanan BA, Geoffrey O, Willingham MC, Re GG, Nixon DW. p53/p21(WAF1/CIP1) expression and its possible role in G1 arrest and apoptosis in ellagic acid treated cancer cells. Cancer Lett. 1999;136:215-21
16. Narayanan BA, Re GG. IGF-II down regulation associated cell cycle arrest in colon cancer cells exposed to phenolic antioxidant ellagic acid. Anticancer Res. 2001;21:359-64
17. Chung YC, Lu LC, Tsai MH, Chen YJ, Chen YY, Yao SP, Hsu CP. The inhibitory effect of ellagic acid on cell growth of ovarian carcinoma cells. Evid Based Complement Alternat Med. 2013;2013:ArticleID 06705
18. Jurenka J. Therapeutic applications of promegranate (Punica granatum L.): a review. Altern Med Rev. 2008;13:128-44
19. Lansky EP, Newman RA. Punica granatum (pomegranate) and its potential for prevention and treatment of inflammation and cancer. J Ethnopharmacol. 2007;109:177-206
20. Bate-Smith EC. Detection and determination of ellagitannins. Phytochem. 1972;11:1153-6
21. Perchellet JP, Gali HU, Perchellet EM, Klish DS, Armbrust AD. Antitumor-promoting activities of tannic acid, ellagic acid, and several gallic acid derivatives in mouse skin. Basic Life Sci. 1992;59:783-801
22. Stoner GD, Morse MA. lsothiocyanates and plant polyphenols as inhibitors of lung and esophageal cancer. Cancer Lett. 1997;114:113-9
23. Dow RL, Chou TT, Bechle BM, Goddard C, Larson ER. Identification of tricyclic analogs related to ellagic acid as potent/selective tyrosine protein kinase inhibitors. J Med Chem. 1994;37:2224-31
24. Barch DH, Rundhaugen LM, Stoner GD, Pillay NS, Rosche WA. Structure-function relationships of the dietary anticarcinogen ellagic acid. Carcinogenesis. 1996;17:265-9
25. Aiyer HS, Warri AM, Woode DR, Hilakivi-Clarke L, Clarke R. Influence of berry polyphenols on receptor signaling and cell-death pathways: implications for breast cancer prevention. J Agric Food Chem. 2012;60:5693-708
26. Mertens-Talcott SU, Percival SS. Ellagic acid and quercetin interact synergistically with resveratrol in the induction of apoptosis and cause transient cell cycle arrest in human leukemia cells. Cancer Lett. 2005;218:141-51
27. Tasaki M, Umemura T, Maeda M, Ishii Y, Okamura T, Inoue T. et al. Safety assessment of ellagic acid, a food additive, in a subchronic toxicity study using F344 rats. Food Chem Toxicol. 2008;46:1119-24
28. Johnson WD, Morrissey RL, Usborne AL, Kapetanovic I, Crowell JA, Muzzio M, McCormick DL. Subchronic oral toxicity and cardiovascular safety pharmacology studies of resveratrol, a naturally occurring polyphenol with cancer preventive activity. Food Chem Toxicol. 2011;49:3319-27
29. Gosepath EM, Eckstein N, Hamacher A, Servan K, von Joquières G, Lage H. et al. Acquired cisplatin resistance in the head-neck cancer cell line Cal27 is associated with decreased DKK1 expression and can partially be reversed by overexpression of DKK1. Int J Cancer. 2008;123:2013-19
30. Eckstein N, Servan K, Hildebrandt B, Pölitz A, von Jonquières G, Wolf-Kümmeth S. et al. Hyperactivation of the insulin-like growth factor receptor I signaling pathway is an essential event for cisplatin resistance of ovarian cancer cells. Cancer Res. 2009;69:2996-3003
31. Mueller H, Kassack MU, Wiese M. Comparison of the usefulness of the MTT, ATP, and calcein assays to predict the potency of cytotoxic agents in various human cancer cell lines. J Biomol Screen. 2004;9:506-15
32. Liang CC, Park AY, Guan JL. In vitro scratch assay: a convenient and inexpensive method for analysis of cell migration in vitro. Nat Protoc. 2007;2:329-33
33. Darzynkiewicz Z. Overview of nucleic acid analysis. In: (ed.) Robinson JP. Current Protocols in Cytometry. New York: J Wiley & Sons. 2001 chapter7.1.1-7.1.4
34. Sithanandam G, Fornwald LW, Fields J, Anderson LM. Inactivation of ErbB3 by siRNA promotes apoptosis and attenuates growth and invasiveness of human lung adenocarcinoma cell line A549. Oncogene. 2005;24:1847-59
35. Coward JI, Middleton K, Murphy F. New perspectives on targeted therapy in ovarian cancer. Int J Womens Health. 2015;7:189-203
36. Núez-Sánchez MA, González-Sarrias A, Romo-Vaquero M, Garcia-Villalba R, Selma MV, Tomás-Barberán FA. et al. Dietary phenolics against colorectal cancer. From promising preclinical results to poor translation into clinical trials: pitfalls and future needs. Mol Nutr Food Res. 2015;59:1274-91
37. Losso JN, Bansode RR, Trappey A 2nd, Bawadi HA, Truax R. In vitro anti-proliferative activities of ellagic acid. J Nutr Biochem. 2004;15:672-8
38. Shun MC, Yu W, Park SK, Sanders BG, Kline K. Downregulation of Epidermal Growth Factor Receptor Expression Contributes to 𝛼-TEA's Proapoptotic Effects in Human Ovarian Cancer Cell Lines. J Oncol. 2010;2010:824571
Author contact
![]() Corresponding author: Matthias U. Kassack, Institut für Pharmazeutische und Medizinische Chemie, Heinrich-Heine-Universität Düsseldorf, Universitätsstraße 1, 40225 Düsseldorf, Germany. Phone: +49-211-8114587, Fax: +49-211-8110801, e-mail: Matthias.Kassackde.
Corresponding author: Matthias U. Kassack, Institut für Pharmazeutische und Medizinische Chemie, Heinrich-Heine-Universität Düsseldorf, Universitätsstraße 1, 40225 Düsseldorf, Germany. Phone: +49-211-8114587, Fax: +49-211-8110801, e-mail: Matthias.Kassackde.