Impact Factor ISSN: 1837-9664

 Global reach, higher impact
Global reach, higher impactJ Cancer 2016; 7(4):391-407. doi:10.7150/jca.13470 This issue Cite
Review
Calcitriol in Combination Therapy for Prostate Cancer: Pharmacokinetic and Pharmacodynamic Interactions
Mohamed Ben-Eltriki1, 2, Subrata Deb4, Emma S. Tomlinson Guns1, 3 ![]()
1. The Vancouver Prostate Centre at Vancouver General Hospital, Vancouver, BC, Canada
2. Department of Experimental Medicine, University of British Columbia, Vancouver, B.C, Canada
3. Department of Urologic Sciences, University of British Columbia, Vancouver, B.C, Canada
4. Department of Biopharmaceutical Sciences, College of Pharmacy at Roosevelt University, Schaumburg, IL, USA
Received 2015-8-5; Accepted 2015-10-18; Published 2016-1-15
Abstract
Epidemiological studies indicate that vitamin D insufficiency could have an etiological role in prostate cancer. In addition, calcitriol, used in combination with currently available drugs, has the potential to potentiate their anticancer effects or act synergistically by inhibiting distinct mechanisms involved in prostate cancer growth. Clinical data have not yet provided sufficient evidence to demonstrate benefit of vitamin D due to the limited and underpowered studies that have been published to date. Here, we review the preclinical and clinical studies that describe the activity of calcitriol, applied either alone or in combination and assessed the mechanistic basis of pharmacodynamic and pharmacokinetic interactions with calcitriol. Important considerations for calcitriol use in combination therapy with respect to safety and clinical outcomes have been discussed. Many of these combinations have therapeutic potential for the treatment of several cancer types and it is anticipated that future clinical research will put emphasis on well‑designed clinical trials to establish efficacy.
Keywords: Calcitriol, prostate cancer, pharmacokinetics, CYP enzymes, metabolism, pharmacodynamics, vitamin D receptor
Introduction
Vitamin D and Calcitriol
Two types of vitamin D molecules exist: vitamin D3 (cholecalciferol), which is animal-derived; and the plant derived vitamin D2. Vitamin D3 is a steroid-like molecule, which is considered to be a prohormone and is the predominant form of vitamin D in humans1. It can be endogenously synthesized from sterol 7-dehydrocholesterol in the skin through exposure to ultraviolet B radiation. Alternatively, vitamin D, in the form of either vitamin D3 or D2, can be acquired from the diet or dietary supplements. Vitamin D, either as D3 or D2, requires a two-step activation process to become biologically active1. Vitamin D3 is highly lipophilic that is transported in the blood stream only when bound to a specific plasma protein called vitamin D-binding globulin (VDBP). It is taken up within hours following synthesis or dietary uptake to be activated by liver and kidney2. In the liver, where multiple cytochrome P450 (CYP) enzymes are present, mitochondrial CYP27A1, microsomal CYP2J3 and CYP2R1 are readily available and capable of hydroxylating vitamin D3 at the C-25 position to form 25(OH)D3, which is then released into the blood stream for further activation, primarily in the kidney3,4. Serum 25(OH)D3 is often considered an acceptable measurement of vitamin D3 levels in the body5. It has a short plasma half-life and a long systemic half-life of ~6 hours and ~ 2 months, respectively2,6. When serum calcium levels are low, renal CYP27B1 converts 25(OH)D3 to 1, 25 dihydroxyvitamin D3 (1, 25(OH)2D3; calcitriol). Calcitriol is the most active form that exists as a small lipophilic molecule with superior cellular penetration5 (Figure 1).
Calcitriol activation pathways and biological functions in target tissues. In human, vitamin D3 is the predominant form of vitamin D, which is synthesized from 7- dehydrocholesterol upon sunlight exposure. Vitamin D may also be obtained from dietary sources or supplements as either vitamin D2 or D3. Vitamin D3 binds to vitamin D-binding protein (DBP) in the bloodstream and then taken up within hours following synthesis or dietary uptake to be activated by liver and kidney. In the liver it is first converted by mitochondrial cytochrome P450 (CYP) enzyme CYP27A1, microsomal CYP2J3 and CYP2R1 to 25-hydroxyvitamin D. This molecule is further converted by the renal enzyme 1-a hydroxylase (CYP27B1) to 1,25 dihydroxycholecalciferol (calcitriol), which is the active form of vitamin D. Finally, calcitriol binds to intracellular vitamin D receptor (VDR) in most cells in the body by functioning both as a paracrine and an autocrine agent.
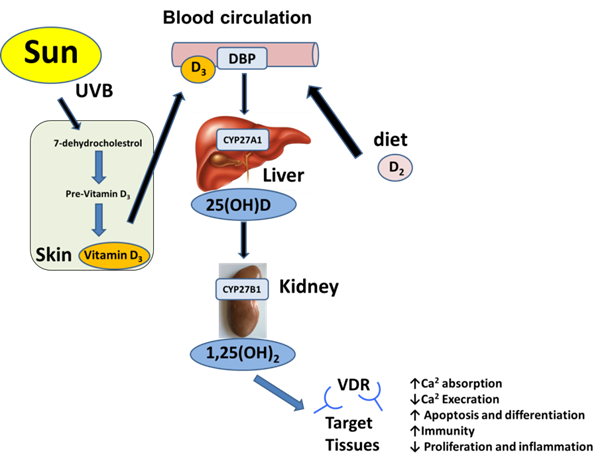
Traditionally, most of the randomized clinical trials (RCT) have focused on bone mineralisation and fracture risk that associated to serum vitamin D levels7. Numerous epidemiological studies recently have shown that low serum levels of vitamin D3 increases the risk of developing cancers of the colon, ovary, lung breast and prostate4,8-12. However, there have been mixed results from clinical studies with respect to their promise of cancer chemoprevention and/or treatment with vitamin D and we are yet to see compelling evidence that the numerous published preclinical studies may actually be translated to humans and cancer patients13,14. The most favorable benefits observed for vitamin D intake have been documented with respect to the inverse relationship between serum levels seen in patients with colon and breast cancer14. For prostate and skin cancer, however, inverse associations between cancer risk and serum vitamin D levels in clinical trial has been difficult to demonstrate so far14. Consequently, we are limited in our ability to provide guidelines for vitamin D intake, which serve as a model to define optimal levels of serum 25(OH)D3 that protect against the development of cancer4. Report from the U.S. Institute of Medicine (IOM) suggests that serum levels greater than 20 ng/mL are recommended that could offer optimal health benefits4,7,11,15,16. It is possible that, for individuals with limited sun exposure, these levels are achievable by taking vitamin D3 supplements daily in the range of 1000-4000 IU.
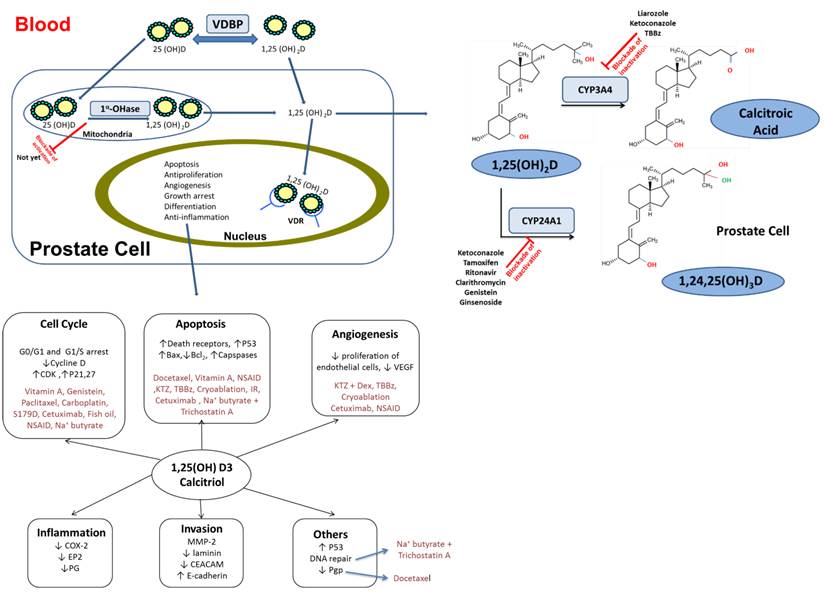
Calcitriol plays an important role in mineral homeostasis and bone growth by promoting the export of calcium from bone, absorption of calcium from the gastrointestinal tract and the stimulation of the absorption of phosphate and magnesium ions to support mineralization. It works as both a paracrine and an autocrine agent by binding to intracellular vitamin D receptor (VDR) to form a complex with the retinoid-X receptor. The resulting heterodimer binds to DNA to function as a transcription factor to either initiate or suppress gene expression17,18. The VDR has a higher affinity towards calcitriol than any other form of vitamin D, which explains its relatively high biological potency19. Besides calcitriol, various steroidal and non-steroidal synthetic analogs, non-vitamin D related endogenous compounds (e.g. lithocholic and arachidonic acid) and dietary ligands including curcumin and fatty acid derivatives, are also capable of activating VDR20,21. VDR is present in moderate levels in most cells in the body including the kidney, heart, muscle, breast, colon, prostate, brain and immune cells17. The gastrointestinal tract and bone are the two sites where VDR is highly expressed and consequently are the primary targets of calcitriol-mediated physiological effects4. Calcitriol has potent effects on factors involved in growth and differentiation of many types of cancer cells, as well as cell adhesion and apoptosis. This could be largely due to its role in regulating the cell cycle and number of genes that are implicated in cancer initiation4. Calcitriol has been shown to exhibit anti-proliferative, pro-differentiating, pro-apoptotic and anti-inflammatory actions in a variety of cancer cells in vitro including PCa cells 22-24(Figure 2).
Summary of calcitriol-drug combination exerting PK/PD interactions in distinct intracellular compartments of prostate cell. Inhibitors of CYP 27A1, CYP24A1 and CYP3A4 isoform can play a vital role in maintaining the active form of vitamin D3. PK interaction outcomes can lead to significant modification of calcitriol levels in both serum and tissues. Diagram also delineates the essential cell signaling pathways for prostate cancer development and crosstalk between them. PD interaction are mediated through alterations in these regulatory pathways that enhance overall anticancer effects. CYP: Cytochrome P450; PK: Pharmacokinetic; PD: pharmacodynamic; VDBP: vitamin D-binding globulin; CDK: Cyclin-dependent kinases; P21: cyclin-dependent kinase inhibitor; S179D:a molecular mimic of naturally phosphorylated human Prolactin; NSAID: Nonsteroidal anti-inflammatory drug; KTZ; Ketoconazole; DEX::Dexamethasone; IR: Ionizing radiation; P53:Tumor protein; COX-2: cyclooxygenase-2; TBBz :4, 5, 6, 7tetrabromobenzimidazole;MMP-2: matrix metalloproteinase-2; CEACAM; carcinoembryonic antigen-related cell adhesion molecules; Pgp: multidrug resistance protein 1;EP2: prostaglandin E receptor 2; PG: prostaglandin.
Calcitriol bioavailability in tissues is largely determined by its affinity to bind to the VDBP plasma transporter, rate of synthesis from 25(OH)D3 and rate of degradation by catabolizing enzymes. When calcitriol is administered orally, it is absorbed rapidly with approximately 99.9% bound to and transported via VDBP in the blood and a serum half-life of approximately 15 hours25,26. Calcitriol levels in the blood depend largely on its bioavailability, ratio of binding to plasma and tissue proteins, perhaps also its binding to lipoprotein as well as the equilibrium that exists for its biosynthesis from 25(OH)D3 and catabolic degradation steps in the kidney and liver, respectively. Within tissues, its levels are reflective of both blood 25(OH)D3 and 1, 25(OH)2D3 free levels as well as the balance between the local biosynthesis from 25(OH)D3 and its rate of degradation27.
Prostate cancer
PCa is the most commonly diagnosed cancer among old men worldwide and the third leading cause of cancer-related deaths in Canada after lung and colorectal cancers14,28-31. To effectively manage the disease and eventually develop novel cancer therapeutics that improve prognosis of PCa, better understanding of the different molecular mechanisms that occur during PCa initiation and progression is necessary. Prostate-specific antigen (PSA), which serves as a biomarker for PCa, has made it possible to detect the disease at the early stages and render better prediction of its behavior. However, there has been considerable debate about the benefits of screening patients using PSA due to limitations in its clinical use in predicting the outcome or response to therapy. PSA lacks specificity for PCa because as it is unable to distinguish well between benign prostatic hyperplasia and malignancy. This perhaps could lead to either over-treatment or unnecessary tissue biopsies32,33. There has been a great deal of efforts invested in searching for novels biomarkers in serum, urine and tissue but few presented with the ability to replace PSA33.
Initiation and progression of PCa are characterized by alterations in regulatory pathways of apoptosis, androgen receptor (AR) signaling, cell cycle regulation, signal transduction, cell adhesion and cohesion, and angiogenesis, which may occur within the prostate cells and/or the surrounding tissues. Disruption of these pathways leads to unregulated proliferation of the damaged cells and subsequent accumulation of mutations, resulting in malignancy. Examples of potential candidate markers of prognosis and therapeutic response involving these pathways include p53 and Bcl-2, AR, p27, p21 and CDK, Ki-67, retinoblastoma protein pRb and c-Myc, epidermal growth factor (EGF) receptor family and E-cadherin, vascular endothelial growth factor (VEGF), respectively32,34.
AR signaling pathways play a significant role in the early development of PCa, as well as in the advanced stages of the disease where the prostate is androgen-independent that fails to respond to hormone deprivation therapy. PCa is initially dependent on androgens, mainly testosterone or dihydrotestosterone (DHT), which activate the AR to transcriptionally regulate the expression of multiple genes including PSA. Androgens play a critical role in disease progression and are required for the growth and survival of castrate-sensitive as well as castration-resistant PCa (CRPC) cells. In addition, PCa cells are capable to grow even when the androgen levels are low due to de novo synthesis of androgen35. The majority of men with PCa eventually advance to an androgen-independent stage and become refractory after about two years with an increased PSA and/or symptomatic progression36. Nevertheless, chemotherapeutic agents seem to have little or no impact on the survival of such patients. Most patients who die from PCa have hormone refractory disease. While many PCas still express AR, only about 30% of the patients respond to anti-androgen therapy that prevents androgens from binding to AR to achieve complete androgen blockade37, thus indicating the development of CRPC. Several mechanisms of CRPC progression have been proposed: mutations in the AR resulting in amplification of the AR and emergence of AR splice variants, changes in the activity of AR coregulators or pathways other than AR-mediated pathways are involved38. Studies have also shown that CRPC development triggers a release from the arrest resulting in cell cycle progression, even in the absence of androgens. Furthermore, epidermal growth factor (EGF), insulin-like growth factor (IGF), keratinocyte growth factor (KGF) and cytokines interleukin-4 and -6 have also been demonstrated to activate AR39-42.
To date, therapeutic options for CRPC are limited. No particular class of chemotherapeutic drugs are considered highly effective. Among the existing therapeutics, docetaxel is currently considered to be the gold standard which demonstrates an increase in survival of only 3 to 4 months with considerable associated morbidity43-46. New therapeutic approaches are being explored. Agents with growth inhibitory properties that work independent of the androgen pathways are also of current interest.
Calcitriol in Prostate cancer
The vitamin D derivatives are currently being used in the treatment of many diseases such as psoriasis and vitiligo47. However, there is a limitation in the broader use of calcitriol as a single anticancer agent clinically due to its toxicity at higher doses. Calcitriol has limited bioavailability at the tumor site which is actively confounded by local CYP-mediated deactivation5. Since dose elevation constraints exist due to its hypercalcemic side effects, to achieve desired clinical outcomes in cancer patients, the pharmacological doses required often lead to an extreme elevated plasma levels of calcium and phosphorus and cause adverse hypercalcemia and hypercalcaemia effects5.
It has been reported that VDR is expressed in three human prostate carcinoma cell lines; LNCaP, PC-3, and DU-145 as well as in the primary culture of stromal and epithelial cells derived from normal and malignant prostate tissues48. Many preclinical studies have been quite consistent in their demonstration of exposure to high calcitriol concentrations resulting in inhibition of PCa cell growth in vitro and retardation or slow-down of tumor growth in animal models of PCa49. Pre-clinical studies demonstrated that 10 nM (4.2 ng/ml) concentrations of calcitriol are associated with anticancer activity in vitro and in vivo50,51. Pharmacokinetic (PK) studies in normal mice indicate that calcitriol, at a dose of 0.125 µg/mouse, is able to suppress tumor growth and results in a peak plasma calcitriol concentrations (Cmax) of 12 ng/ml with corresponding areas under the curve from time 0 to 24 hr of 47 ng•h/ml. Calcitriol anticancer activity is mainly dependent on its dose, and its serum concentration ranges escalate from 0.5 to 41 ng/ml, resulting in significant toxicity52,53. There have been many approaches taken to elevate tumor calcitriol levels that have been reported in the literature, making it feasible to see anticancer benefits while limiting its toxicity. Specifically, calcitriol may be administered on an intermittent schedule, analogues with low/no calcemic activity may be substituted, and using calcitriol in combination with other agents could enhance anticancer activity and allow for lower calcitriol doses to be optimised50.
Calcitriol in combination therapy for PCa
Monotherapy has limited efficacy in cancer prevention and is mostly ineffective in curing cancers because of the heterogeneous pathways which contribute to their occurrence. In recent years, new drugs and drug combinations have contributed to improvements in treatment outcomes of PCa54. Combination regimens of chemotherapeutic drugs were developed in the 1960s and early 1970s, based on observations that single drug administration at clinically tolerable dosages failed to cure cancer as normal tissue toxicity often limits the use of a single agent. Combination therapy offers significant benefits when compared to single-agent therapy by providing: (i) maximal cell kill within the range of tolerable toxicity by allowing lower drug doses; (ii) diverse interactions between the drugs procuring pleiotropic effects which may be optimal for treating a heterogeneous tumour; (iii) the potential to prevent or slow the subsequent development of cellular drug resistance as combined drugs work by different mechanisms; (iv) lower cost incurred as lower doses or/and less frequent drug administration is required; (v) reduced side effects of individual drugs because each drug can be used optimally at a lower dose; (vi) better clinical outcomes when the approach take is not curative but intended to reduce symptoms and prolong life.
Optimal drug combination effects are likely to be achieved when combined drugs have different mechanisms of action and no overlapping toxicities. Individual drugs which are used in combination should be partially effective against the same tumor type. In addition, PK and pharmacodynamic (PD) interactions could occur as a result of combining drugs and should therefore also be investigated at the onset. These interactions can be either beneficial by allowing a therapeutic effect to be achieved with low doses of component interventions, which often minimizes potential side effects. Alternatively, they can exist as problematic antagonistic interactions when combined pharmacological outcomes lead to unwanted, toxic and result in lower effectiveness than expected from the individual activities of the combined drugs. Drug combinations that interact in a manner that results in additive or synergistic effects, where the combined effects exceed that anticipated based on the effects of the individual agents are ideal and therefore of great interest.
Concurrent administration of a lower dose of calcitriol and other anticancer drugs may be the best option that helps to overcome this obstacle and lead to achieving the desired anticancer concentrations with low toxicity. The combination approach for calcitriol used recently with docetaxel in a clinical trial was successfully implemented and achieved the desired anticancer effect with no toxicity in PCa patients55. This study demonstrated that administration of calcitriol dose up to 45 μg/week combined with weekly i.v docetaxel led to Cmax values of 3.4 ± 0.8 nM (1.41 ± 0.33 ng/ml) and was not associated with any significant toxicity55. Other clinical studies suggest that the maximum tolerated dose (MTD) of i.v dose calcitriol in combination with gefitinib was 74 µg/week26,56. The Cmax at the MTD was 6.68 ± 1.42 ng/mL (16 ± 3.4 nM) and the area under the curve (AUC) was 35.65 ± 8.01 ng h/mL. However, this reported Cmax was much higher than calcitriol concentrations considered to be required for antitumor activity as demonstrated in vitro50,51,57.
The aim of this review is to summarize, the currently available preclinical and clinical studies with calcitriol used either alone or as part of a combination therapy approach to treat PCa. Articles for review were searched using PubMed using the keywords: vitamin D, calcitriol, 1, 25(OH)2D3 and PCa. In this review, we highlighted the major considerations for the use of calcitriol in combination therapy with respect to safety and factors that influence the PK/PD interactions and clinical outcomes.
Pharmacokinetic Based Interactions
When calcitriol is used in combination with other drugs, calcitriol-drug PK interactions have the potential to affect pharmacological outcome in several ways: (i) alteration of calcitriol serum and tissue concentrations, (ii) alteration of calcitriol hepatic metabolism due to induction or inhibition of hepatic enzymes, (iii) alteration of extrahepatic enzymes by induction or inhibition, and (iv) induction or inhibition of drug transporters that could interfere with calcitriol cellular uptake. Alterations in the synthesis (CYP27B1 mediated) and metabolism (mediated by CYP3A4 and CYP24A1) of calcitriol is modulated as part of the growth regulation of tumors; thus, compromising calcitriol potency and sensitivity. Overall, PK interaction outcomes can lead to the significant modification of calcitriol levels in both serum and tissues.
Calcitriol and CYP enzymes
CYP enzymes catalyze the synthesis and metabolism of a large number of endogenous substrates, including steroids, vitamins, fatty acids, prostaglandins and leukotrienes, as well as the detoxification of exogenous compounds, including drugs, environmental chemicals and pollutants, and natural plant products58-60. CYP monooxygenases are a major class of phase I metabolizing enzymes that acts either by adding or removing functional groups to/from substrates for further metabolism, thereby facilitating excretion of xenobiotics.
There are four CYP isoforms that participate in the synthesis and breakdown of calcitriol. CYP27A1 and CYP27B1, located in the mitochondria, are involved in the first steps of calcitriol synthesis from vitamin D3 and 25(OH)D3 is located in kidney and liver3,60. Mechanisms of vitamin D metabolism within the prostate remain poorly understood. Historically, mitochondrial CYP24A1 has been reported to be the main enzymes responsible for 25(OH)D3 and a calcitriol metabolism. However, recently our lab and others have shown that CYP3A4, which is located in the endoplasmic reticulum, is a contributor to calcitriol degradation in vivo and in vitro23,61-63. As shown in Figure 2, two metabolic pathways exist in the prostate; the first involves the 24-hydroxylase as the first-step in catabolism of calcitriol. There is strong data supporting 24-hydroxylase activity in the kidney; this enzyme is also present in many target tissues, such as the intestine, which possess vitamin D receptor. As depicted in Figure 2, the end product of this pathway is a side chain cleaved metabolite, calcitroic acid. The second pathway involves the conversion of calcitriol via stepwise hydroxylation of carbon-26 and carbon-23, and cyclization to ultimately yield 1α,25R(OH)2-26,23S-lactone D3 (Figure 2). In some instance mutations in certain CYP genes, have been suggested to play a role in PCa development and progression64. Specifically, mutations in CYP17 and SRD5A2 (encoding for 5α-reductase enzyme) genes have been reported to increase the risk of PCa and is found in some cases of hereditary PCa34,59,64,65. Over-expression and mutations in CYP24A1 induce idiopathic infantile hypercalcemia which is linked to PCa resistance66,67. Moreover, there is an association between polymorphisms in CYP3A4 and the increased risk of PCa in men with benign prostatic hyperplasia59,64,68. In general, changes in the DNA, RNA and/or protein levels, and the activities of the targets discussed above are found to significantly correlate with Gleason grade and may suggest PCa progression to an androgen-independent stage34,64. Therefore, it is reasonable to suggest that CYP enzymes, such as CYP24A1, may be used as novel drug targets and also perhaps allow accurate prediction of tumor progression.
Combinations that affect calcitriol synthesis
Role of CYP27B1
Circulating serum levels of 1,25(OH)2D3 are tightly regulated by the renal enzyme, 25-hydroxyvitamin D-1α-hydroxylase, CYP27B1, which synthesizes calcitriol from the prohormone, 25(OH)D3. The renal production of calcitriol is tightly regulated by plasma parathyroid hormone (PTH) levels and serum calcium and phosphorus levels69. In response to low calcium levels, PTH levels rise up and regulate the expression of CYP27B1 leading to the synthesis of calcitriol, which is then released into plasma to serve its endocrine function to maintain calcium homeostasis and bone metabolism.
Many cell types, including prostate cells, immune cells, cells of the gastrointestinal tract, brain cells, and importantly skin cells are capable of calcitriol production. Thus, they can provide hormone for local signaling which could also explain the wide distribution of the and VDR in these organs70. In prostate cells, since they contain VDR, an additional role of calcitriol as an autocrine/ paracrine regulator of cell functions could be expected5. It has been reported that primary cultures of normal prostate epithelial cells, as well as several PCa cell lines, express CYP27B1 and can synthesize the active hormone calcitriol intracellularly48,70. In addition, the CYP27B1 activity declines in BPH and PCa cells compared to the normal prostate cells71. In vitro studies using high performance liquid chromatography (HPLC) to measure calcitriol in the media have demonstrated that two PCa cell lines (DU145 and PC3, but not LNCaP) and normal prostate cells were able to produce calcitriol after cells were exposed to 25(OH)D3. In addition, in the presence of clotrimazole the ability of PCa cell lines, except LNCaP, to produce calcitriol was inhibited. These results correlated with an inhibition of CYP27B1 mRNA expression whereas no CYP27B1 mRNA or protein activity was detected in LNCaP cells, which may contribute to the resistance48,70.
It has been reported that the control of calcitriol synthesis in tissues like bone and prostate is distinct in which calcitriol levels potentially fall earlier in these tissues than in the plasma17. In vitro studies suggest that the prostate CYP27B1 enzyme is not regulated by serum levels of PTH and calcium compared to the renal enzyme. This supports the hypothesis that extrarenal administration of vitamin D may be useful as a chemopreventive agent, and that calcitriol should be synthesized from 25(OH)D3 locally within prostate cells72,73. However, a recent study in mouse prostate xenograft model has demonstrated that dietary vitamin D3 administration caused an increase in serum calcitriol levels without causing any alteration in kidney CYP27B1 mRNA74. In addition, both dietary vitamin D3 and calcitriol imparted equivalent reductions in tumor volume in this xenograft model. This hypothesis was also tested clinically and calcitriol prostate levels were shown to increase significantly after oral administration of vitamin D supplements27. These results suggest that extra renal sources of vitamin D and local synthesis within prostate do, in fact, contribute to the elevated circulating calcitriol. No studies have been reported to date which examine the effect of drugs combined with calcitriol and their impact on CYP27A1 expression or how it affects calcitriol levels within the prostate.
Combinations that inhibit calcitriol metabolism
Role of CYP24A1
CYP24A1 recognizes 25(OH)D3 as a substrate and can mediate its hydroxylation to the active metabolite, calcitriol 5. This enzyme also recognizes other vitamin D metabolites and analogs and actively converts them to their hydroxylated products5,75. CYP24A1 is also responsible for calcitriol metabolism, mainly in the kidney but also in a variety of other vitamin D target cells. It converts it to water-soluble calcitroic acid which is then conjugated and excreted in bile1,76. The cyp24a -knockout mice studies have confirmed the physiological role of CYP24A1 in 25(OH)D3 and calcitriol hemostasis as a build-up of vitamin D3 was observed in the knockout mouse phenotype76,77.
CYP24A1 is highly expressed in the kidney but is also present in other normal tissues such as the prostate78. Calcitriol can also induce CYP24A1 and its own metabolism, thus has the capacity to limit its physiological functions. The levels of CYP24A1 expression may reflect the endocrine and autocrine/paracrine effects of vitamin D3 in the human body. CYP24A1 overexpression is a common feature of several solid tumors and was detected in a wide range of cancers such as breast, prostate, skin, esophagus, and gastrointestinal tract cancers67,79. It can contribute to the pathology of diseases that otherwise would respond to endogenous or supplemented vitamin D sources and it is associated with poor prognosis due to rapid degradation of both 25(OH)D3 and calcitriol, limiting their levels in the tumor cells and thus abrogating local anti-cancer effects of calcitriol 27,80.
Administration of calcitriol in combination with CYP24A1 inhibitors slows its catabolism, thereby enhancing its antitumor activity and antiproliferative effect (Table 1). Following treatment of DU145 PCa cells for 4 days with 1 µM liarozole, which is an imidazole derivative known to inhibit CYP enzymes, in combination with 10 nM calcitriol, the inhibitory effect on cell viability was further enhanced almost by 65% compared with no significant effect seen with either treatment alone. It is likely that this effect was mediated by inhibiting CYP24A1 activity, which resulted in a significant increase in calcitriol half-life from 11 to 31 h, thus enhancing the calcitriol anticancer activity. The combination treatments also led to greater VDR upregulation than cells treated with either drug alone81. A recent study reported that protein kinase CK2 positively regulates CYP24A1 expression and mediates the regulation of its expression. In keeping with this, inhibition of CK2 activity has been shown to cause a reduction in PCa cell proliferation and enhance calcitriol -mediated antitumor effect82.
Summary of preclinical calcitriol combination studies for prostate cancer
| Drug/Agent combined | Calcitriol dose | Model used | Mechanisms PK/PD Based interaction | Pharmacological Outcomes & Comments | Ref. |
|---|---|---|---|---|---|
| Docetaxel | Pre-treatment 100 nM calcitriol | PC3 | ↑apoptosis ↓Bcl-2, ↑Bax, MRP mRNA and protein levels | Sensitization ↓ % Cell viability ↓ IC50 | 105 |
| Ketoconazole + Tetralone | Cotreatemt 10 nM calcitriol | PC3 DU145 | ↓CYP24A1 activity ↑p21 mRNA ↑Calcitriol t1/2 | Sensitization ↓% Cell viability | 114 |
| Vitamin A | Cotreatemt 10 nM calcitriol | PC3 LNCaP DU145 | ↑ Cyclin D1 mRNA and protein Bax mRNA and protein | Sensitization ↓ % Cell viability | 111 |
| Genistein | Cotreatemt 10 nM calcitriol | HPEC LNCaP | ↑G(2)M arrest and G(1/0) arrest | Synergistically ↓ % Cell viability | 109 |
| Ketoconazole + Dexamethasone | Pre-treatment 5 µM calcitriol for 24hr calcitriol alone IP (31.25μg/kg) (QD, twice, weekly) | PC3 PC3 prostate tumor xenograft mouse model | ↓CYP24A1 activity ↑apoptosis ↑Caspase 8 activation ↓CYP24A1 activity ↓PC3 tumor growth ↓Ki-67 ↑Apoptosis ↑, caspase-3 | Synergistically ↓ % Cell viability Synergistically ↓Tumor growth | 51 |
| Paclitaxel | Pre-treatment 5 µM calcitriol for 24 hr. Pre-treatment 0.75 μg I.P for 3 days | PC3 PC3 prostate tumor xenograft mouse model | ↑p21 protein levels ↓cdk1 protein levels ↑p21 protein levels ↓cdk1 protein levels | Synergistically ↓Tumor growth Synergistically ↓Tumor growth | 104 |
| Carboplatin | Drugs were combined at IC20 | LNCaP DU145 | ↑G(2)M arrest | Sensitization | 106 |
| TBBz | Pre-treatment 10 nM calcitriol | Transfected PC3 cells with siRNA-CK2 PC3 prostate tumor xenograft mouse model | ↓CYP24A1 mRNA ↓CYP24A1 activity ↓Tumor CYP24A1 mRNA ↓Ki-67 ↑apoptosis ↑caspase-3 cleavage | Sensitization ↓ % Cell viability ↓Tumor volume | 82 |
| Cryoablation | 4.0 µg/kg of calcitriol (intratumoral injection | Murine prostate tumors (RM-9) in male C57BL/6J mice | ↓Ki-67 ↑apoptosis ↓Procaspase-9 ↑Procaspase-9 cleavage ↑caspase-3 cleavage ↓AKT | Sensitization ↑Necrosis ↑Apoptosis ↓Proliferation ↓Tumor volume | 129 |
| Sodium valproate + Radiotherapy | Cotreatemt 10 nM calcitriol | DU145 | ↑DNA damage ↓DNA replication S-phase cell-cycle arrest ↑apoptosis | Sensitization | 103 |
| S179D | Cotreatemt 10 nM calcitriol | PC3 DU145 | ↑VDR ↓P21 | Sensitization ↓ % Cell viability | 130 |
| Cetuximab | Cotreatemt 10 nM calcitriol | DU145 | ↓cell proliferation ↑apoptosis | Sensitization | 131 |
| Fish oil | Cotreatemt 10 nM calcitriol | LNCaP-c115 | cycle arrest at G(1)/S-phase | Synergistically | 132 |
| Ibuprofen | Cotreatemt 10 nM calcitriol | LNCaP | ↓Proliferation cycle arrest ↑apoptosis | Sensitization | 102 |
| Radiotherapy | Antagonism at high IR dose Cotreatemt 10 nM calcitriol | LNCaP | ↑apoptosis | Synergistically ↓LNCaP growth | 113 |
| Sodium butyrate + Trichostatin A | Cotreatemt 10 nM calcitriol | LNCaP PC-3 DU-145 | ↑apoptosis ↓P21 | Synergistically, ↓inhibit the growth | 112 |
| Liarozole | Cotreatemt 10 nM calcitriol | DU 145 | ↓CYP24A1 activity ↑Calcitriol t1/2 (11 to 31 hr) ↑VDR activity | Synergistically ↓cell viability | 81 |
| Ginsenoside aPPD, aPPT | - | Human microsomal protein | CYP3A4 activity↓ | Unknown | 63 |
PK: Pharmacokinetic; PD: pharmacodynamic; CYP: Cytochrome P450; IC50: The half maximal inhibitory concentration; t1/2:half-life; VDR: vitamin D receptor; HPEC: Primary human prostate epithelial cells; CK2: Casein kinase 2; ki67: proliferation marker; BAX: pro-apoptotic protein; Bcl2: anti-apoptotic protein; VDBP: vitamin D-binding globulin; CDK: Cyclin-dependent kinases; P21: cyclin-dependent kinase inhibitor. TBBz: 4, 5, 6, 7-tetrabromobenzimidazole; S179D is a molecular mimic of naturally phosphorylated human Prolactin; aPPT: 20(S)-protopanaxatriol; aPPD: 20(S)-protopanaxadiol.
In vitro treatment of PC3 cells with calcitriol in combination with ketoconazole or RC2204 (a selective inhibitor of CYP24A1) acted synergistically by potentiating calcitriol-mediated antiproliferative effects, promoting the activation of caspase-independent apoptosis pathways. However, ketoconazole is a well-known non-selective inhibitor of CYP enzymes and dexamethasone (potent anti-inflammatory agent used routinely in the chemotherapeutic regimen) was added that has the ability to induce CYP and up-regulate VDR protein expression and activities in vitro83,84. Thus, conclusions deduced following this observation are limited as they are confounded by the contribution of other mechanisms. The authors proposed that potentially ketoconazole inhibit the CYP24A1-mediated oxidative metabolism of calcitriol leading to increase in half-life and systemic exposure previously observed in PC3 cells and C3H/HeJ mouse kidney tissues. These observations were confirmed in vivo when the calcitriol and ketoconazole-dexamethasone combination therapy suppressed the clonogenic survival and enhanced growth inhibition observed with calcitriol treatment alone in the PC3 human PCa xenograft mouse model51.
Role of CYP3A4
CYP3A4 is a major hepatic phase I oxidative drug-metabolizing enzyme. It has broad substrate specificity and is inducible after exposure to therapeutic, dietary, and environmental agents 85. Variability in CYP3A4 activity accounts for large interindividual differences in the disposition if endogenous, xenobiotic and therapeutic drugs68. CYP3A4 polymorphisms could contribute specifically to potential inter-individual and interethnic variation in steroid metabolism as it is characteristically involved in the oxidative metabolism of testosterone, thus mediating prostate cell growth. Consequently, CYP3A4 function may play a role in androgen-mediated prostate carcinogenesis if the bioavailability of testosterone is affected68. CYP3A4 is expressed in many organs including prostate, breast, gut, colon, and small intestine; however, its expression is most abundant in the human liver, accounting for 30 percent of the total hepatic CYP protein content86-88. CYP3A4 is also highly expressed in intestine but with remarkably different composition and abundance than the liver. The intestinal CYP system is mainly comprised of CYP3A enzymes (82%)87. However, the total amount of intestinal CYP3A protein is only approximately 1% of that in liver87. It exhibits a broad substrate specificity and is responsible for oxidation of many therapeutic drugs and a variety of structurally unrelated compounds, including steroids, fatty acids, and xenobiotics. It can also be inhibited by a variety compounds including drugs, nutrients, and carcinogens, resulting in low CYP3A4 activity in all of the tissues in which it is expressed, including the prostate.
It has been previously shown that in human liver and intestine CYP3A4 is responsible for the oxidative metabolism of calcitriol61,89. Specifically, CYP3A4 mediates 24- and 25-hydroxylation of calcitriol, but not vitamin D3, in human liver microsomes90. Using specific CYP chemical inhibitors we have reported that CYP3A isoforms are responsible for the microsomal biotransformation of calcitriol in liver and intestine in vitro23. Co-incubation of calcitriol with commonly used CYP3A4 substrates/drugs (e.g. ketoconazole, tamoxifen, ritonavir or clarithromycin) led to approximately 60-100% inhibition of CYP3A4-mediated inactivation of calcitriol in human liver and intestine microsomes61. In addition, recently we have shown that ginsenoside metabolites from ginseng herbs, mainly 20(S)-protopanaxadiol and 20(S)-protopanaxatriol, strongly inhibited CYP3A4 activity in vitro and could potentially therefore provide additional benefits to patients with cancer, neurodegenerative and metabolic diseases when used in combination with calcitriol63. Similarly, we have also reported that abiraterone, a CYP17A1-mediated steroidogenesis inhibitor used in late stage PCa treatment, inhibits the CYP3A4-mediated inactivation of calcitriol in human liver and intestine, potentially providing additional anti-cancer benefits to PCa patients62. In the context of PCa, the inhibition of CYP3A4 may lead to reduced ability of the enzyme to oxidize calcitriol in the liver and thus sparing calcitriol catabolism to yield higher intracellular levels. Therefore, the metabolism of calcitriol in the intestine and liver could contribute more to calcitriol bioavailability and tissue levels than CYP24A189. The combination of calcitriol with CYP3A4 inhibitors, therefore, could allow for the use of lower doses of calcitriol and still achieve significant anticancer effects.
Pharmacodynamic Based Interaction
VDR and AR cross talk
VDR is a nuclear receptor that binds calcitriol and regulates the transcription of target genes in the nucleus. VDR is universally expressed in both androgen-dependent and independent PCa cell lines which have differential sensitivity to calcitriol treatment91. Six PCa cell lines were reported to contain VDR mRNA and had high-affinity saturable binding sites for calcitriol78. Many factors regulate the amount of VDR in target cells that could potentially alter the magnitude of response to calcitriol exposure. Specific to the prostate, calcitriol response depends not only on absorption, transport into the circulation, plasma levels, ultimate prostate tissue levels and metabolism but also on intact VDR for signaling. The absence of intact VDR or loss of its function would decrease or eliminate the pharmacological responses to calcitriol in the prostate. A recent report that examined the expression of VDR protein in 841 PCa patients concluded that a high expression of VDR in prostate tumors is associated with a reduced risk of lethal cancer92. In addition, genetic predisposition is seen as a risk factor to prostate cancer, there are several studies suggesting the association between genetic variants in VDR and the development of PCa or likelihood of progression to an advanced stage93-95. Significant associations with five common genes (Fok1, Bsm1, Taq1 and ApaI), with high Gleason score have been reported with strongest evidence for Fok1 and Bsm194,96.The expression of VDR could, therefore, be used to help improve prognostic prediction of PCa progression and guide treatment decisions for men. In addition, VDR polymorphisms should also be integrated as a prediagnostic indicator of vitamin D status97.
PCa is known as an androgen-dependent malignancy and the fact that other hormones such as calcitriol are now recognized as modulators of PCa growth and differentiation, suggest a role for pharmacological agents, which might make use of this endocrine axis. There is now considerable evidence to suggest that cross-talk exists between VDR and AR that may vary between different PCa cell lines98. In addition, the phenomenon of sharing the same co-regulators which results in interdependence of AR and VDR signaling may contribute to the therapeutic activity of vitamin D in PCa98,99. The AR belongs to the superfamily of steroid nuclear receptors. AR coregulators, which were originally identified as AR-associated proteins, can modulate many other steroid receptors in this super-family. Co-regulators can act to enhance (co-activate) or decrease (co-repress) the AR and changes in its expression have been shown to correlate with poor prognosis in PCa patients38. It has been reported that a large number of coregulators are overexpressed in PCa100. Several AR regulators, including ARA54, ARA70, gelsolin, and supervillin, have been reported to promote VDR98. Thus, by targeting this cross talk new therapeutic strategies designed to treat PCa could be developed.
Summary of preclinical studies
Several studies carried out in cell culture and animal models, as well as clinical trials, have shown that calcitriol pre-treatment or combination with other therapeutic agents provides anticancer benefits which are superior to treatment with either agent alone101. Calcitriol apparently sensitizes cancer cells, enhancing their antitumor activity, and can act synergistically with other therapeutic agents. Targeting multiple pathways or the same pathway through a different mechanism maximizes the likelihood of a therapeutic effect while limiting tissue toxicity (Table 1 and 2).
Summary of clinical calcitriol combination studies for prostate cancer
| Drug combined | Patient Type & size | Calcitriol Dose or Formulation | Clinical Endpoint | Clinical Outcomes | Comments | Ref. |
|---|---|---|---|---|---|---|
| Docetaxel | AIPCa 37 | calcitriol (0.5 μg/kg) weekly (Rocaltrol, Roche Pharmaceuticals, Basel, Switzerland) | PSA, Safety & Efficacy | PSA ↓ n=30 No different in toxicity compared to docetaxel single treatment No PK interaction | This regimen was safe and well tolerated Phase I | 120 |
| Docetaxel | AIPCa 250 | 45 µg DN-101 weekly | PSA Survival time | PSA ↓ n=145 Survival time ↑ | There was an improvement in Patient survival with no an increase in toxicity | 55 |
| Docetaxel | Metastatic AIPCa 250 | 45 µg DN-101 weekly | PSA Clinical progress Safety & Efficacy | PSA ↓n=113 PSA ↔ n=113 Progress n=24 | Need further investigation | 116 |
| Docetaxel + Zoledronic acid | CRPC 30 | Calcitriol (0.5 µg/kg orally in 4 divided doses over 4 h ) | Safely& efficacy Toxicity | PSA ↓n=23 Survival time ↑ | This regimen shows a significant different, safe and well tolerated | 117 |
| Estramustine + Docetaxel | metastatic AIPCa 24 | High dose calcitriol 60 μg orally | Safety & Efficacy | Asymptomatic hypercalcemia n=4 | High dose calcitriol may be safely added to docetaxel and Estramustine administered | 123 |
| Naproxen open label | Relapse 21 | High dose calcitriol (DN101, Novacea) 45 µg /week | Safely& efficacy | PSA doubling time ↓ n=4 PSA doubling time ↑ n=14 | Well tolerated and safe 75% responded to the treatment regimen phase II | 133 |
| Dexamethasone | CRPCa 18 | i.v 74 μg weekly | PSA RECIST | No patient had response Progress n=14 Toxicity n=7 | Clinical trial response failed Phase II | 125 |
| Dexamethasone + Carboplatin | HRPC 34 | 0.5 µg daily | PSA | PSA ↓ n=13 Significant adverse events | This regimen has shown a good response with an acceptable side effect profile compare Phase II | 121 |
| Dexamethasone | 43 AIPCa | Intermittent high different doses of 8, 10 and 12 μg 3 times a week for a month (Rocaltrol, Roche Pharmaceuticals, Indianapolis, IN) | Toxicity | Partial PSA↓ n=8 Toxicity was low | No clear different and superior to dexamethasone treatment alone Calcitriol at high dose was safe, feasible, and had antitumor effects Calcitriol was administered a month prior to dexamethasone dosing Phase II | 124 |
| Dexamethasone + Prednisolone + Docetaxel | Metastatic CRPC 953 | 45 µg DN-101 | Overall survival (OS), assessed By the Kaplan-Meier method. | Shorter survival than the control | Clinical trial response failed. This failure might be due to either docetaxel regime or calcitriol dose Randomized, Open-Label Phase III Trial | 134 |
| Carboplatin | metastatic AIPCa patients 17 | (0.5 µg/kg) repeated dose every 4 weeks | PSA Palliative response | PSA↓ n=1 Partially PSA↓ n=4 Pain reduction n=3 | No different compare to single agent No dose related toxicity This might be due to low dose of calcitriol given | 122 |
| Mitoxantrone + Prednisone Phase II | 19 metastatic AIPCa patient | Calcitriol ((DN-101) 180 µg orally Every 3 weeks | PSA decline to half Quality of life was evaluated | PSA↓ n=5 No different in Toxicity physical functioning↓ fatigue, insomnia, and diarrhea↑ | This might be due to low dose of calcitriol given | 128 |
RCT: Randomized control trial; HRPC: Hormone-refractory prostate cancer; CRPS: AIPCa: Androgen independent prostate cancer; RECIST: Response Evaluation Criteria in Solid Tumors; PSA: Prostate cancer antigen; IV: Intravenous; PK: Pharmacokinetics; Ref: References; CRPCa: Castration-resistant prostate cancer; DN-101: a high-dose (15 μg) formulation of calcitriol.
Calcitriol combinations which sensitize PCa cells to anticancer drugs
Calcitriol has been shown to sensitize PCa to low temperature-induced, non-ice rupture-related cell death in vitro and in vivo. When calcitriol used with cryotherapy treatment, it significantly increased cell death and reducing cell viability via activation of apoptosis44. Calcitriol enhanced antitumor effect of cryoablation by increasing necrosis and apoptosis and reduced cell proliferation. Thus, calcitriol could potentially be an applicable reagent as a freeze sensitizer to cryoablation1. Radiotherapy, which is often a curative treatment option for PCa, however, also presents with a high risk of side effects at effective doses of ionizing radiation (IR). So when human androgen-insensitive DU145 PCa cells were pre-treated with a combination of 100 nM calcitriol and 1 mM sodium valproate, a well-tolerated histone deacetylase, led to sensitization of the PCa cells to IR102. This calcitriol pre-treatment with IR efficiently suppressed PCa cell proliferation and increased DNA double-strand breaks compared with non-pretreated cells. Combined pre-treatment of calcitriol with IR also resulted in 16 % enhancement in IR-induced activation of DNA damage CHEK2 compared to untreated cells. These molecular changes led to DNA replication blockade, S-phase cell cycle arrest and enhanced apoptosis. This combination approach could, therefore, be useful and allow for doses of radiation administered to cancer patients to be attenuated such that side effects are reduced22,103.
Calcitriol enhanced the anti-proliferative and the cytotoxic effects of taxanes chemotherapy (e.g., paclitaxel and docetaxel) in vitro when used in a combination treatment to treat PC3 cell lines104. This is thought to be due to its ability to reduce multidrug resistance-associated protein-1 expression and, therefore, inhibit transporter mediated efflux of docetaxel from cells105. Calcitriol also sensitizes PCa cells by inducing apoptosis and slowing cell cycle and cell proliferation. The growth of PCa cell lines (LNCaP and DU145) were inhibited when cells were treated with a platinum compound (cis- or carboplatin), and its synthetic analogue, Ro 25-6760. However, the growth inhibition was further enhanced by calcitriol. The enhancement of inhibitory effect was greater when lower concentrations of these compounds were used in combination with higher concentrations of calcitriol. At IC20 doses, calcitriol and a platinum compound acted in a synergistic manner to inhibit the growth of PC cells106.
Calcitriol Combinations which act additively or synergistically with other anticancer agents on PCa cells
Mitoxantrone combined with glucocorticoids has been widely used to treat androgen-independent PCa. Calcitriol synergistically increased mitoxantrone/dexamethasone mediated growth inhibition of PC-3 cells in vitro. The combination also caused significantly greater tumor regression in the PC-3 xenograft model system compared with treatment with mitoxantrone/dexamethasone or untreated controls107.
There is increasing evidence that the isolation of a single compound from complex foods may not be effective in preventing cancer even when administered at toxic doses, whereas combination therapy using lower doses with no or lower toxicity might work. Genistein is a dietary-derived isoflavonoid found in high concentrations in serum after ingestion of soy-rich meals108. Studies reported that low concentrations of 0.5µM genistein tested in vitro in combination with 0.1 or 0.5 nM calcitriol was synergistic in the inhibition of primary human prostatic epithelial and LNCaP PCa cell viability. However, treatment of cells with genistein or calcitriol alone had no significant effect on cell viability. In primary cells, the combination acted synergistically as both resulted in G2/M and G1/0 cell-cycle arrest, whereas in LNCaP cells the combination had similar effects compared to treatment alone109. When concentrations higher than 5 µM genistein were used in combination with 10 nM calcitriol, up-regulation of cell cycle inhibitor p21 levels were seen compared to treatment alone and genistein potentiated the effect of calcitriol regulation of VDR expression.
Calcitriol and 20% dietary soy protein were also examined in vivo as a combination diet in a mouse xenograft model of PCa110. The combination diet resulted in a more substantial inhibition of tumor growth than treatment with either agent alone. Upon examination of the tumor tissue, it was deduced that potential mechanisms affected by combination treatment included up-regulation of several genes involved in prostate cell regulation such as anti-proliferative (p21, IGFBP-3) and pro-apoptotic (Bax) genes, down-regulating the anti-apoptotic (Bcl-2), compared to treatment with either calcitriol or 20% dietary soy alone110. A significant enhancement was also observed in the combination treated group in the up-regulation of the expression of the prostaglandin G-degrading enzyme 15-PGDH. However, the combination had the potential to increase the risk of hypercalcemia as indicated by elevated expression of intestinal calcium absorption genes (TRPV6, calbindin-9k)110. Laboratory studies have also indicated that calcitriol and dietary omega 3-polyunsaturated fatty acids act synergistically to inhibit the growth of the high passage androgen-independent PCa cell line, LNCaP-c115 at the level of the G1/S-phase transition and cell division50. The combination of vitamin A and vitamin D synergistically reduced cell viability, expression of cyclin D1 and induced apoptosis by enhancing Bax protein expression111.
The combination of cetuximab, which is an anti-epidermal growth-factor receptor antibody, with calcitriol efficiently, suppressed hormone-resistant DU145 PCa cell growth. The combination inhibited DU145 cell proliferation, caused considerable cell-cycle arrest in the Go/Gal-phase and enhanced apoptosis49. The combined effect of calcitriol (10 nM) with ibuprofen, a well-known non-steroidal anti-inflammatory drug (NSAID) on LNCaP PCa cells was enhanced in vitro. This effect was found only to be additive. However, significant synergistic cell growth inhibition was achieved by combined treatment of calcitriol and ibuprofen in DHT-stimulated LNCaP cells. This combined treatment was effective in decreasing the cell transition from G1- to S-phase and enhanced apoptosis compared with the effect of single drugs102. Neither calcitriol (1-10 nM) nor liarozole (1-10 mM) had any effect on DU145 cell viability when used alone. However, 10 nM calcitriol plus 1 mM liarozole acted synergistically and significantly reduced cell viability. Liarozole induced considerable increase in calcitriol half-life from 11 to 31 h. It also enhanced calcitriol activity by potentiating its ability to up-regulate VDR protein compared with the effects of either drug alone. The mechanism of interaction is due to the ability of liarozole to inhibit CYP24A1activity, thus increasing calcitriol half-life and effect81.
Sodium butyrate and trichostatin A are inhibitors of histone deacetylases activity, which acted synergistically with calcitriol radio biologically combination on DU-145 PCa cells by enhancing apoptosis112. Moreover, in vitro calcitriol acted synergistically with IR to inhibit the growth of the LNCaP human PCa cell line by potentiating IR-induced apoptosis. At radiobiologically relevant doses of IR, calcitriol also showed synergistic inhibition of growth of LNCaP cells. However, at higher doses of IR, the combination resulted in moderate antagonism. The results suggested that the synergistic effect could permit a reduction in the dose of radiation given clinically and thus potentially reduce treatment-related morbidity113.
In general, these combinations acted on common pathways as well as on independent pathways, thus increasing overall, anticancer effect. The molecular mechanisms of this PD interaction are mediated through alterations in regulatory pathways within the prostate that change and enhance overall, anticancer effects such as the increase in cellular apoptosis demonstrated when calcitriol combined with IR therapy, paclitaxel, docetaxel, vitamin A, genistein, cetuximab, ibuprofen, sodium butyrate and trichostatin A. Moreover, the interaction was mediated via the inhibition of calcitriol metabolism, which increases the bioavailability of endogenous and exogenously administered calcitriol, thereby synergistically enhancing its anticancer effects. Up-regulation of VDR by genistein and liarozole was observed as a result of higher levels of calcitriol reaching the tumor site following increased half-life of calcitriol (Table 1).
Combinations which sensitize PCa cells to anticancer effects of calcitriol
Some compounds have been reported to enhance the antitumor activity of calcitriol. The combination of S179D, which is a molecular mimic of naturally phosphorylated human prolactin and calcitriol, was shown to inhibit the growth PC3 and DU145 PCa cell lines in vitro and vivo24. Beside that S179D sensitized the cells to calcitriol concentration that below the level that typically results in hypercalcemia. This effect was demonstrated to be in part mediated via the induction of cell death, increased protein expression of VDR and p2124. Androgen-independent PCa cells DU-145 and PC-3 are relatively insensitive to the anti-proliferative action of calcitriol. This is thought to be due to an increase in the calcitriol metabolism, as a result of CYP24 enzyme induction, which in turn leads to decreased anti-proliferative efficacy114. In vitro studies supporting this hypothesis include a rat kidney mitochondria assay which incorporates CYP24A1 inhibition using 2-(4-hydroxybenzyl)-6-methoxy-3, 4-dihydro-2H-naphthalen-1-one and calcitriol that led to produce a greater inhibition of proliferation in DU-145 cells compared to single agent treatments114. Examination of the regulation of VDR target gene mRNA in DU-1455 cells revealed that co-treatment of calcitriol plus inhibitor of CYP enzymes co-ordinately up-regulated CYP24, p21and GADD45alpha114. When calcitriol was combined with a CK2 inhibitor, it enhanced calcitriol-mediated antitumor effects82. The inhibition of CK2 by 5, 6, 7-tetrabromobenzimidazole, a protein kinase CK2 selective inhibitor, was also shown to inhibit CYP24A1 promoter activity induced by calcitriol in PC3 cells. Furthermore, the ability of calcitriol to induced CYP24A1 mRNA expression was reduced by using CK2 siRNA knockdown and this significantly enhanced calcitriol-mediated antiproliferative effects in vitro and vivo in a xenograft model. These observations suggested that protein kinase CK2 contributes to calcitriol mediated target gene expression and is involved in the regulation of CYP24A1 expression. To summarize, combining selected drugs with calcitriol sensitizes prostate cells to the actions of calcitriol by i) inhibiting its metabolism and ii) up-regulating VDR and its target genes, thus enhancing overall calcitriol antitumor activity.
Summary of Clinical Studies
Combining calcitriol with cytotoxic agents
Although all preclinical data published so far strongly support the significance of calcitriol as an anticancer agent for prevention and/or treatment of PCa, its use as an anticancer agent alone or in combination has not yet been fully established clinically (Table 2). A phase I clinical trial has shown that weekly dosing allows substantial dose-escalation of calcitriol, and in follow-up a phase II trial suggested that adding weekly high-dose calcitriol may enhance the activity of weekly docetaxel in patients with advance stage disease115. Adding DN-101, a high-dose oral formulation of calcitriol designed for cancer therapy, to docetaxel treatment significantly enhanced the reduction in serum PSA levels in response to docetaxel. The use of high oral calcitriol combined weekly with docetaxel for the treatment of metastatic androgen-independent PCa (AIPCa) patients was recognised as safe and generally well tolerated compared with the toxicity related complications experienced with single-agent docetaxel treatment55,115-122. Metastatic AIPCa patients were treated with 60 µg calcitriol orally in a combination with estramustine and docetaxel every 21 days for up to 12 cycles. High dose calcitriol (60 µg daily) appeared to be safe when added to this chemotherapeutic regimen and was well tolerated123. When CRPCa patients were given high weekly doses of calcitriol (0.5 µg/kg orally), in combination with docetaxel and zoledronic acid, half of the patients had a PSA response and this regimen was also well tolerated117. In addition, the pharmacokinetics of either calcitriol or docetaxel was not affected by the presence of its companion drug. In an exploratory substudy, PSA and measurable disease response rates as well as time to progression and survival were also promising when compared with phase II studies reported in the literature for single-agent docetaxel for the same patient population120. High dose oral calcitriol (0.5 µg/kg) used in combination with intravenous carboplatin in patients with metastatic AIPCa was not associated with an increase in the response rate compared with the reported activity for carboplatin alone122. The outcomes were similar to that expected with single-agent carboplatin with only one of seventeen patients in the study achieving a confirmed PSA declined response and no patient achieved the palliative response end point (2-point reduction or normalization of pain on the present pain intensity scale without increased analgesic consumption53 (Table 2).
Combining calcitriol with corticosteroids and NSAID
One year of dosing with weekly calcitriol and daily naproxen treatment to 21 patients with PCa relapse was well tolerated by most patients and effective in delaying PCa growth and progression according to a reduction in PSA doubling time which was achieved in 75% of patients. High-dose intermittent calcitriol plus dexamethasone appears to be safe, feasible, and also demonstrated antitumor activity26,124,125. The combination effect of the daily calcitriol, dexamethasone and carboplatin in thirty-four patients with HRPCa also produced a PSA response in 13 of 34 patients. PSA was decreased in 13 treated patients, and the median overall survival was 97.7 weeks. However, significant side effects were reported121. The combination of intermittent high doses (8, 10 and 12 µg) calcitriol plus dexamethasone in 43 patients with AIPCa caused slight PSA decline with minimum side effects. In this case, there was no clear evidence that combination treatment was better that dexamethasone treatment alone121. Calcitriol, dosed in a pulsatile manner, was safe and tolerated when its toxicity was examined with zoledronate and the addition of dexamethasone at the time of disease progression126. In patients with progressive PCa, adding dexamethasone to calcitriol at escalating doses from 4 µg up to 30 µg per day was well tolerated. In addition, when administered three times per week in combination with intravenous zoledronate (4 mg monthly) with or without dexamethasone. Patients tolerated therapy well, even in those patients who received higher calcitriol doses of 30 µg treatment group; a maximum tolerated dose was therefore not definable. The Cmax observed in the 24 µg and 30 µg cohorts ranged from 391 to 968 pg/mL with minimal antitumor effects observed. The Cmax in the 24 µg and 30 µg cohorts were greater than the preclinical levels associated with antitumor effects127. Conversely, adding calcitriol to mitoxantrone and prednisone in AIPCa patient did not cause any apparent enhancement of mitoxantrone toxicity128 (Table 2).
Conclusion
Calcitriol biosynthesis and metabolism pathways are catalyzed by multiple CYP enzymes. This provides the opportunity to the researchers and clinicians to either stimulate calcitriol biosynthesis or inhibit its metabolism in an organ-specific manner, leading to higher exposure to endogenous or exogenously administered calcitriol. Similarly, targeting different biological pathways with calcitriol and currently available drugs present pharmacodynamic interactions, where the pharmacological actions of the combination are synergized or potentiated. Combination therapy with calcitriol for the treatment of PCa appears to be efficacious in vitro and vivo PCa models. However, definitive evidence regarding treatment efficacy has not yet been established clinically. Most clinical trials have taken place in patients with recurrence or castration‑resistant disease; studies should be carried out in PCa patients on active surveillance and/or receiving adjuvant therapy. Other targets of calcitriol PK and PD need to be studied, and further research is required to better explain the relationships of PCa risk with low vitamin D exposure/supplementation. In addition, from a pre-clinical perspective, a better understanding of cross talk between AR and VDR and their role in PCa growth and progression will open new avenues to explore regarding therapeutic calcitriol combination regimens. In addition, combining other therapeutic agents with calcitriol lowers required calcitriol doses at which hypercalcemic effects are no longer a concern. Overall, combinations of calcitriol with selected drugs are promising and offer a contemporary approach to maximizing the multifaceted biological and therapeutic actions of vitamin D in the context of PCa.
Acknowledgements
M.B.E. is a recipient of a studentship award from the Libyan Ministry of Education and Scientific Research.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Jones G. Extrarenal vitamin D activation and interactions between vitamin D(2), vitamin D(3), and vitamin D analogs. Annu Rev Nutr. 2013;33:23-44
2. Mawer EB, Schaefer K, Lumb GA, Stanbury SW. The metabolism of isotopically labelled vitamin D3 in man: the influence of the state of vitamin D nutrition. Clin Sci. 1971;40(1):39-53
3. Prosser DE, Jones G. Enzymes involved in the activation and inactivation of vitamin D. Trends Biochem Sci. 2004;29(12):664-673
4. Ingraham BA, Bragdon B, Nohe A. Molecular basis of the potential of vitamin D to prevent cancer. Curr Med Res Opin. 2008;24(1):139-149
5. Schuster I. Cytochromes P450 are essential players in the vitamin D signaling system. Biochim Biophys Acta. 2011;1814(1):186-199
6. Jones G. Pharmacokinetics of vitamin D toxicity. Am J Clin Nutr. 2008;88(2):582S-586S
7. Ross AC, Taylor CL, Yaktine AL, Del Valle HB. In: Dietary Reference Intakes for Calcium and Vitamin D. Washington (DC). Institute of Medicine (2011).
8. Garland CF, Gorham ED, Mohr SB, Garland FC. Vitamin D for cancer prevention: global perspective. Ann Epidemiol. 2009;19(7):468-483
9. Garland CF, Gorham ED, Mohr SB. et al. Vitamin D and prevention of breast cancer: pooled analysis. J Steroid Biochem Mol Biol. 2007;103(3-5):708-711
10. Lin J, Manson JE, Lee IM, Cook NR, Buring JE, Zhang SM. Intakes of calcium and vitamin D and breast cancer risk in women. Arch Intern Med. 2007;167(10):1050-1059
11. Lappe JM, Travers-Gustafson D, Davies KM, Recker RR, Heaney RP. Vitamin D and calcium supplementation reduces cancer risk: results of a randomized trial. Am J Clin Nutr. 2007;85(6):1586-1591
12. Trump DL, Chadha MK, Sunga AY. et al. Vitamin D deficiency and insufficiency among patients with prostate cancer. BJU Int. 2009;104(7):909-914
13. Force UPST. Dietary Reference Intakes for Vitamin D and Calcium. 2012. (http://www.ahrq.gov/policymakers/case-studies/coe1301.html). Last accessed: Setpember 10. 2015
14. Bikle DD. Vitamin D and cancer: the promise not yet fulfilled. Endocrine. Jan 9. 2014
15. Vieth R, Bischoff-Ferrari H, Boucher BJ. et al. The urgent need to recommend an intake of vitamin D that is effective. Am J Clin Nutr. 2007;85(3):649-650
16. Balvers MG, Brouwer-Brolsma EM, Endenburg S, de Groot LC, Kok FJ, Gunnewiek JK. Recommended intakes of vitamin D to optimise health, associated circulating 25-hydroxyvitamin D concentrations, and dosing regimens to treat deficiency: workshop report and overview of current literature. Journal of nutritional science. 2015;4:e23
17. Morris HA, Anderson PH. Autocrine and paracrine actions of vitamin d. Clin Biochem Rev. 2010;31(4):129-138
18. Demay MB. Mechanism of vitamin D receptor action. Ann N Y Acad Sci. 2006;1068:204-213
19. Chawla A, Repa JJ, Evans RM, Mangelsdorf DJ. Nuclear receptors and lipid physiology: opening the X-files. Science. 2001;294(5548):1866-1870
20. Haussler MR, Haussler CA, Bartik L. et al. Vitamin D receptor: molecular signaling and actions of nutritional ligands in disease prevention. Nutr Rev. 2008;66(10 Suppl 2):S98-112
21. Rochel N, Wurtz JM, Mitschler A, Klaholz B, Moras D. The crystal structure of the nuclear receptor for vitamin D bound to its natural ligand. Mol Cell. 2000;5(1):173-179
22. Chen TC, Holick MF. Vitamin D and prostate cancer prevention and treatment. Trends Endocrinol Metab. 2003;14(9):423-430
23. Deeb KK, Trump DL, Johnson CS. Vitamin D signalling pathways in cancer: potential for anticancer therapeutics. Nat Rev Cancer. 2007;7(9):684-700
24. Zhuang SH, Burnstein KL. Antiproliferative effect of 1alpha,25-dihydroxyvitamin D3 in human prostate cancer cell line LNCaP involves reduction of cyclin-dependent kinase 2 activity and persistent G1 accumulation. Endocrinology. 1998;139(3):1197-1207
25. Levine BS, Singer FR, Bryce GF, Mallon JP, Miller ON, Coburn JW. Pharmacokinetics and biologic effects of calcitriol in normal humans. J Lab Clin Med. 1985;105(2):239-246
26. Fakih MG, Trump DL, Muindi JR. et al. A phase I pharmacokinetic and pharmacodynamic study of intravenous calcitriol in combination with oral gefitinib in patients with advanced solid tumors. Clin Cancer Res. 2007;13(4):1216-1223
27. Wagner D, Trudel D, Van der Kwast T. et al. Randomized clinical trial of vitamin D3 doses on prostatic vitamin D metabolite levels and ki67 labeling in prostate cancer patients. J Clin Endocrinol Metab. 2013;98(4):1498-1507
28. Ilic D, Neuberger MM, Djulbegovic M, Dahm P. Screening for prostate cancer. Cochrane Database Syst Rev. 2013;1:CD004720
29. Kachuri L, De P, Ellison LF, Semenciw R. Cancer incidence, mortality and survival trends in Canada, 1970-2007. Chronic Dis Inj Can. 2013;33(2):69-80
30. Society CC. Prostate cancer statistics 2013.
31. Society CC. Canadian Cancer Statistics, percentage of all estimated cancer death in mens 2014.
32. Romero Otero J, Garcia Gomez B, Campos Juanatey F, Touijer KA. Prostate cancer biomarkers: An update. Urol Oncol. Feb 1. 2014
33. Cary KC, Cooperberg MR. Biomarkers in prostate cancer surveillance and screening: past, present, and future. Ther Adv Urol. 2013;5(6):318-329
34. Quinn DI, Henshall SM, Sutherland RL. Molecular markers of prostate cancer outcome. Eur J Cancer. 2005;41(6):858-887
35. Sharifi N, Auchus RJ. Steroid biosynthesis and prostate cancer. Steroids. 2012;77(7):719-726
36. Bhandari MS, Crook J, Hussain M. Should intermittent androgen deprivation be used in routine clinical practice? J Clin Oncol. 2005;23(32):8212-8218
37. Hadaschik BA, Gleave ME. Therapeutic options for hormone-refractory prostate cancer in 2007. Urol Oncol. 2007;25(5):413-419
38. Zhong J, Ding L, Bohrer LR. et al. p300 Acetyltransferase Regulates Androgen Receptor Degradation and PTEN-Deficient Prostate Tumorigenesis. Cancer Res. 15. 2014;74(6):1870-1880
39. McMurtry J, Richards M, Brocht D. Serum corticosterone concentrations in developing shell-less and shelled turkey embryos. Comp Biochem Physiol A Comp Physiol. 1991;100(1):135-137
40. Wu Y, Zhao W, Zhao J. et al. Identification of androgen response elements in the insulin-like growth factor I upstream promoter. Endocrinology. 2007;148(6):2984-2993
41. Huang YW, Wang LS, Chang HL. et al. Effect of keratinocyte growth factor on cell viability in primary cultured human prostate cancer stromal cells. J Steroid Biochem Mol Biol. 2006;100(1-3):24-33
42. Culig Z, Bartsch G, Hobisch A. Interleukin-6 regulates androgen receptor activity and prostate cancer cell growth. Mol Cell Endocrinol. 2002;197(1-2):231-238
43. van Dodewaard-de Jong JM, Verheul HM, Bloemendal HJ, de Klerk JM, Carducci MA, van den Eertwegh AJ. New Treatment Options for Patients With Metastatic Prostate Cancer: What Is The Optimal Sequence? Clin Genitourin Cancer. Jan 21. 2015
44. Bauer JA, Thompson TA, Church DR, Ariazi EA, Wilding G. Growth inhibition and differentiation in human prostate carcinoma cells induced by the vitamin D analog 1alpha,24-dihydroxyvitamin D2. Prostate. 2003;55(3):159-167
45. Wolff JM. Chemotherapy in hormone-refractory prostate cancer. Front Radiat Ther Oncol. 2008;41:103-107
46. Sartor O, Gillessen S. Treatment sequencing in metastatic castrate-resistant prostate cancer. Asian J Androl. 2014;16(3):426-431
47. Sigmon JR, Yentzer BA, Feldman SR. Calcitriol ointment: a review of a topical vitamin D analog for psoriasis. J Dermatolog Treat. 2009;20(4):208-212
48. Skowronski RJ, Peehl DM, Feldman D. Actions of vitamin D3, analogs on human prostate cancer cell lines: comparison with 1,25-dihydroxyvitamin D3. Endocrinology. 1995;136(1):20-26
49. Skowronski RJ, Peehl DM, Feldman D. Vitamin D and prostate cancer: 1,25 dihydroxyvitamin D3 receptors and actions in human prostate cancer cell lines. Endocrinology. 1993;132(5):1952-1960
50. Trump DL, Hershberger PA, Bernardi RJ. et al. Anti-tumor activity of calcitriol: pre-clinical and clinical studies. J Steroid Biochem Mol Biol. 2004;89-90(1-5):519-526
51. Muindi JR, Yu WD, Ma Y. et al. CYP24A1 inhibition enhances the antitumor activity of calcitriol. Endocrinology. 2010;151(9):4301-4312
52. Smith DC, Johnson CS, Freeman CC, Muindi J, Wilson JW, Trump DL. A Phase I trial of calcitriol (1,25-dihydroxycholecalciferol) in patients with advanced malignancy. Clin Cancer Res. 1999;5(6):1339-1345
53. Gross C, Stamey T, Hancock S, Feldman D. Treatment of early recurrent prostate cancer with 1,25-dihydroxyvitamin D3 (calcitriol). J Urol. 1998;159(6):2035-2039 discussion 2039-2040
54. de Wit R. Chemotherapy in hormone-refractory prostate cancer. BJU Int. 2008;101(Suppl 2):11-15
55. Beer TM, Ryan CW, Venner PM. et al. Double-blinded randomized study of high-dose calcitriol plus docetaxel compared with placebo plus docetaxel in androgen-independent prostate cancer: a report from the ASCENT Investigators. J Clin Oncol. 2007;25(6):669-674
56. Muindi JR, Johnson CS, Trump DL, Christy R, Engler KL, Fakih MG. A phase I and pharmacokinetics study of intravenous calcitriol in combination with oral dexamethasone and gefitinib in patients with advanced solid tumors. Cancer Chemother Pharmacol. 2009;65(1):33-40
57. Muindi JR, Modzelewski RA, Peng Y, Trump DL, Johnson CS. Pharmacokinetics of 1alpha,25-dihydroxyvitamin D3 in normal mice after systemic exposure to effective and safe antitumor doses. Oncology. 2004;66(1):62-66
58. Gonzalez FJ, Nebert DW. Evolution of the P450 gene superfamily: animal-plant 'warfare', molecular drive and human genetic differences in drug oxidation. Trends Genet. 1990;6(6):182-186
59. Nebert DW, Russell DW. Clinical importance of the cytochromes P450. Lancet. 2002;360(9340):1155-1162
60. Chen TC, Sakaki T, Yamamoto K, Kittaka A. The roles of cytochrome P450 enzymes in prostate cancer development and treatment. Anticancer Res. 2012;32(1):291-298
61. Deb S, Pandey M, Adomat H, Guns ES. Cytochrome P450 3A-mediated microsomal biotransformation of 1alpha,25-dihydroxyvitamin D3 in mouse and human liver: drug-related induction and inhibition of catabolism. Drug Metab Dispos. 2012;40(5):907-918
62. Deb S, Chin MY, Adomat H, Guns ES. Abiraterone inhibits 1alpha,25-dihydroxyvitamin D3 metabolism by CYP3A4 in human liver and intestine in vitro. J Steroid Biochem Mol Biol. 2014;144(Pt A):50-58
63. Deb S, Chin MY, Adomat H, Guns ES. Ginsenoside-mediated blockade of 1alpha,25-dihydroxyvitamin D inactivation in human liver and intestine in vitro. J Steroid Biochem Mol Biol. 2014;141:94-103
64. Mazaris E, Tsiotras A. Molecular pathways in prostate cancer. Nephrourol Mon. 2013;5(3):792-800
65. Gonzalgo ML, Isaacs WB. Molecular pathways to prostate cancer. J Urol. 2003;170(6 Pt 1):2444-2452
66. Schlingmann KP, Kaufmann M, Weber S. et al. Mutations in CYP24A1 and idiopathic infantile hypercalcemia. N Engl J Med. 2011;365(5):410-421
67. Horvath HC, Lakatos P, Kosa JP. et al. The candidate oncogene CYP24A1: A potential biomarker for colorectal tumorigenesis. J Histochem Cytochem. 2010;58(3):277-285
68. Keshava C, McCanlies EC, Weston A. CYP3A4 polymorphisms-potential risk factors for breast and prostate cancer: a HuGE review. Am J Epidemiol. 2004;160(9):825-841
69. DeLuca HF. Overview of general physiologic features and functions of vitamin D. Am J Clin Nutr. 2004;80(6 Suppl):1689S-1696S
70. Schwartz GG, Whitlatch LW, Chen TC, Lokeshwar BL, Holick MF. Human prostate cells synthesize 1,25-dihydroxyvitamin D3 from 25-hydroxyvitamin D3. Cancer Epidemiol Biomarkers Prev. 1998;7(5):391-395
71. Whitlatch LW, Young MV, Schwartz GG. et al. 25-Hydroxyvitamin D-1alpha-hydroxylase activity is diminished in human prostate cancer cells and is enhanced by gene transfer. J Steroid Biochem Mol Biol. 2002;81(2):135-140
72. Sakaki T, Sawada N, Takeyama K, Kato S, Inouye K. Enzymatic properties of mouse 25-hydroxyvitamin D3 1 alpha-hydroxylase expressed in Escherichia coli. Eur J Biochem. 1999;259(3):731-738
73. Young MV, Schwartz GG, Wang L. et al. The prostate 25-hydroxyvitamin D-1 alpha-hydroxylase is not influenced by parathyroid hormone and calcium: implications for prostate cancer chemoprevention by vitamin D. Carcinogenesis. 2004;25(6):967-971
74. Swami S, Krishnan AV, Wang JY. et al. Dietary vitamin D(3) and 1,25-dihydroxyvitamin D(3) (calcitriol) exhibit equivalent anticancer activity in mouse xenograft models of breast and prostate cancer. Endocrinology. 2012;153(6):2576-2587
75. Sakaki T, Sawada N, Nonaka Y, Ohyama Y, Inouye K. Metabolic studies using recombinant escherichia coli cells producing rat mitochondrial CYP24 CYP24 can convert 1alpha,25-dihydroxyvitamin D3 to calcitroic acid. Eur J Biochem. 1999;262(1):43-48
76. St-Arnaud R. Targeted inactivation of vitamin D hydroxylases in mice. Bone. 1999;25(1):127-129
77. Masuda S, Byford V, Arabian A. et al. Altered pharmacokinetics of 1alpha,25-dihydroxyvitamin D3 and 25-hydroxyvitamin D3 in the blood and tissues of the 25-hydroxyvitamin D-24-hydroxylase (Cyp24a1) null mouse. Endocrinology. 2005;146(2):825-834
78. Miller GJ, Stapleton GE, Hedlund TE, Moffat KA. Vitamin D receptor expression, 24-hydroxylase activity, and inhibition of growth by 1alpha,25-dihydroxyvitamin D3 in seven human prostatic carcinoma cell lines. Clin Cancer Res. 1995;1(9):997-1003
79. Anderson MG, Nakane M, Ruan X, Kroeger PE, Wu-Wong JR. Expression of VDR and CYP24A1 mRNA in human tumors. Cancer Chemother Pharmacol. 2006;57(2):234-240
80. Holt SK, Kwon EM, Koopmeiners JS. et al. Vitamin D pathway gene variants and prostate cancer prognosis. Prostate. 2010;70(13):1448-1460
81. Ly LH, Zhao XY, Holloway L, Feldman D. Liarozole acts synergistically with 1alpha,25-dihydroxyvitamin D3 to inhibit growth of DU 145 human prostate cancer cells by blocking 24-hydroxylase activity. Endocrinology. 1999;140(5):2071-2076
82. Luo W, Yu WD, Ma Y, Chernov M, Trump DL, Johnson CS. Inhibition of protein kinase CK2 reduces Cyp24a1 expression and enhances 1,25-dihydroxyvitamin D(3) antitumor activity in human prostate cancer cells. Cancer Res. 2013;73(7):2289-2297
83. McCune JS, Hawke RL, LeCluyse EL. et al. In vivo and in vitro induction of human cytochrome P4503A4 by dexamethasone. Clin Pharmacol Ther. 2000;68(4):356-366
84. Pascussi JM, Drocourt L, Gerbal-Chaloin S, Fabre JM, Maurel P, Vilarem MJ. Dual effect of dexamethasone on CYP3A4 gene expression in human hepatocytes. Sequential role of glucocorticoid receptor and pregnane X receptor. Eur J Biochem. 2001;268(24):6346-6358
85. Thummel KE, Wilkinson GR. In vitro and in vivo drug interactions involving human CYP3A. Annu Rev Pharmacol Toxicol. 1998;38:389-430
86. Bieche I, Narjoz C, Asselah T. et al. Reverse transcriptase-PCR quantification of mRNA levels from cytochrome (CYP)1, CYP2 and CYP3 families in 22 different human tissues. Pharmacogenet Genomics. 2007;17(9):731-742
87. Paine MF, Hart HL, Ludington SS, Haining RL, Rettie AE, Zeldin DC. The human intestinal cytochrome P450 "pie". Drug Metab Dispos. 2006;34(5):880-886
88. Pavek P, Dvorak Z. Xenobiotic-induced transcriptional regulation of xenobiotic metabolizing enzymes of the cytochrome P450 superfamily in human extrahepatic tissues. Curr Drug Metab. 2008;9(2):129-143
89. Xu Y, Hashizume T, Shuhart MC. et al. Intestinal and hepatic CYP3A4 catalyze hydroxylation of 1alpha,25-dihydroxyvitamin D(3): implications for drug-induced osteomalacia. Mol Pharmacol. 2006;69(1):56-65
90. Gupta RP, He YA, Patrick KS, Halpert JR, Bell NH. CYP3A4 is a vitamin D-24- and 25-hydroxylase: analysis of structure function by site-directed mutagenesis. J Clin Endocrinol Metab. 2005;90(2):1210-1219
91. Zhuang SH, Schwartz GG, Cameron D, Burnstein KL. Vitamin D receptor content and transcriptional activity do not fully predict antiproliferative effects of vitamin D in human prostate cancer cell lines. Mol Cell Endocrinol. 1997;126(1):83-90
92. Hendrickson WK, Flavin R, Kasperzyk JL. et al. Vitamin D receptor protein expression in tumor tissue and prostate cancer progression. J Clin Oncol. 2011;29(17):2378-2385
93. Chen L, Davey Smith G, Evans DM. et al. Genetic variants in the vitamin d receptor are associated with advanced prostate cancer at diagnosis: findings from the prostate testing for cancer and treatment study and a systematic review. Cancer Epidemiol Biomarkers Prev. 2009;18(11):2874-2881
94. Raimondi S, Johansson H, Maisonneuve P, Gandini S. Review and meta-analysis on vitamin D receptor polymorphisms and cancer risk. Carcinogenesis. 2009;30(7):1170-1180
95. Mun MJ, Kim TH, Hwang JY, Jang WC. Vitamin D receptor gene polymorphisms and the risk for female reproductive cancers: A meta-analysis. Maturitas. 2015;81(2):256-265
96. Kostner K, Denzer N, Muller CS, Klein R, Tilgen W, Reichrath J. The relevance of vitamin D receptor (VDR) gene polymorphisms for cancer: a review of the literature. Anticancer Res. 2009;29(9):3511-3536
97. Gandini S, Gnagnarella P, Serrano D, Pasquali E, Raimondi S. Vitamin D receptor polymorphisms and cancer. Advances in experimental medicine and biology. 2014;810:69-105
98. Ting HJ, Bao BY, Hsu CL, Lee YF. Androgen-receptor coregulators mediate the suppressive effect of androgen signals on vitamin D receptor activity. Endocrine. 2005;26(1):1-9
99. Heinlein CA, Chang C. Androgen receptor (AR) coregulators: an overview. Endocr Rev. 2002;23(2):175-200
100. Culig Z, Santer FR. Molecular aspects of androgenic signaling and possible targets for therapeutic intervention in prostate cancer. Steroids. 2013;78(9):851-859
101. Uzoigwe J, Sauter ER. Cancer prevention and treatment using combination therapy with plant- and animal-derived compounds. Expert Rev Clin Pharmacol. 2012;5(6):701-709
102. Gavrilov V, Steiner M, Shany S. The combined treatment of 1,25-dihydroxyvitamin D3 and a non-steroid anti-inflammatory drug is highly effective in suppressing prostate cancer cell line (LNCaP) growth. Anticancer Res. 2005;25(5):3425-3429
103. Gavrilov V, Leibovich Y, Ariad S, Lavrenkov K, Shany S. A combined pretreatment of 1,25-dihydroxyvitamin D3 and sodium valproate enhances the damaging effect of ionizing radiation on prostate cancer cells. J Steroid Biochem Mol Biol. 2010;121(1-2):391-394
104. Hershberger PA, Yu WD, Modzelewski RA, Rueger RM, Johnson CS, Trump DL. Calcitriol (1,25-dihydroxycholecalciferol) enhances paclitaxel antitumor activity in vitro and in vivo and accelerates paclitaxel-induced apoptosis. Clin Cancer Res. 2001;7(4):1043-1051
105. Ting HJ, Hsu J, Bao BY, Lee YF. Docetaxel-induced growth inhibition and apoptosis in androgen independent prostate cancer cells are enhanced by 1alpha,25-dihydroxyvitamin D3. Cancer Lett. 2007;247(1):122-129
106. Moffatt KA, Johannes WU, Miller GJ. 1Alpha,25dihydroxyvitamin D3 and platinum drugs act synergistically to inhibit the growth of prostate cancer cell lines. Clin Cancer Res. 1999;5(3):695-703
107. Ahmed S, Johnson CS, Rueger RM, Trump DL. Calcitriol (1,25-dihydroxycholecalciferol) potentiates activity of mitoxantrone/dexamethasone in an androgen independent prostate cancer model. J Urol. 2002;168(2):756-761
108. Watanabe S, Yamaguchi M, Sobue T. et al. Pharmacokinetics of soybean isoflavones in plasma, urine and feces of men after ingestion of 60 g baked soybean powder (kinako). J Nutr. 1998;128(10):1710-1715
109. Rao A, Woodruff RD, Wade WN, Kute TE, Cramer SD. Genistein and vitamin D synergistically inhibit human prostatic epithelial cell growth. J Nutr. 2002;132(10):3191-3194
110. Wang JY, Swami S, Krishnan AV, Feldman D. Combination of calcitriol and dietary soy exhibits enhanced anticancer activity and increased hypercalcemic toxicity in a mouse xenograft model of prostate cancer. Prostate. 2012;72(15):1628-1637
111. Sha J, Pan J, Ping P. et al. Synergistic effect and mechanism of vitamin A and vitamin D on inducing apoptosis of prostate cancer cells. Mol Biol Rep. 2013;40(4):2763-2768
112. Rashid SF, Moore JS, Walker E. et al. Synergistic growth inhibition of prostate cancer cells by 1 alpha,25 Dihydroxyvitamin D(3) and its 19-nor-hexafluoride analogs in combination with either sodium butyrate or trichostatin A. Oncogene. 2001;20(15):1860-1872
113. Dunlap N, Schwartz GG, Eads D. et al. 1alpha,25-dihydroxyvitamin D(3) (calcitriol) and its analogue, 19-nor-1alpha,25(OH)(2)D(2), potentiate the effects of ionising radiation on human prostate cancer cells. Br J Cancer. 18. 2003;89(4):746-753
114. Yee SW, Campbell MJ, Simons C. Inhibition of Vitamin D3 metabolism enhances VDR signalling in androgen-independent prostate cancer cells. J Steroid Biochem Mol Biol. 2006;98(4-5):228-235
115. Beer TM. ASCENT: the androgen-independent prostate cancer study of calcitriol enhancing taxotere. BJU Int. 2005;96(4):508-513
116. Beer TM, Ryan CW, Venner PM. et al. Intermittent chemotherapy in patients with metastatic androgen-independent prostate cancer: results from ASCENT, a double-blinded, randomized comparison of high-dose calcitriol plus docetaxel with placebo plus docetaxel. Cancer. 2008;112(2):326-330
117. Shamseddine A, Farhat FS, Elias E, Khauli RB, Saleh A, Bulbul MA. High-dose calcitriol, docetaxel and zoledronic acid in patients with castration-resistant prostate cancer: a phase II study. Urol Int. 2013;90(1):56-61
118. Petrioli R, Pascucci A, Francini E. et al. Weekly high-dose calcitriol and docetaxel in patients with metastatic hormone-refractory prostate cancer previously exposed to docetaxel. BJU Int. 2007;100(4):775-779
119. Beer TM, Hough KM, Garzotto M, Lowe BA, Henner WD. Weekly high-dose calcitriol and docetaxel in advanced prostate cancer. Semin Oncol. 2001;28(4 Suppl 15):49-55
120. Beer TM, Eilers KM, Garzotto M, Egorin MJ, Lowe BA, Henner WD. Weekly high-dose calcitriol and docetaxel in metastatic androgen-independent prostate cancer. J Clin Oncol. 2003;21(1):123-128
121. Flaig TW, Barqawi A, Miller G. et al. A phase II trial of dexamethasone, vitamin D, and carboplatin in patients with hormone-refractory prostate cancer. Cancer. 2006;107(2):266-274
122. Beer TM, Garzotto M, Katovic NM. High-dose calcitriol and carboplatin in metastatic androgen-independent prostate cancer. Am J Clin Oncol. 2004;27(5):535-541
123. Tiffany NM, Ryan CW, Garzotto M, Wersinger EM, Beer TM. High dose pulse calcitriol, docetaxel and estramustine for androgen independent prostate cancer: a phase I/II study. J Urol. 2005;174(3):888-892
124. Trump DL, Potter DM, Muindi J, Brufsky A, Johnson CS. Phase II trial of high-dose, intermittent calcitriol (1,25 dihydroxyvitamin D3) and dexamethasone in androgen-independent prostate cancer. Cancer. 2006;106(10):2136-2142
125. Chadha MK, Tian L, Mashtare T. et al. Phase 2 trial of weekly intravenous 1,25 dihydroxy cholecalciferol (calcitriol) in combination with dexamethasone for castration-resistant prostate cancer. Cancer. 2010;116(9):2132-2139
126. Morris MJ, Smaletz O, Solit D. et al. High-dose calcitriol, zoledronate, and dexamethasone for the treatment of progressive prostate carcinoma. Cancer. 2004;100(9):1868-1875
127. Holick MF. Vitamin D deficiency. N Engl J Med. Jul 19. 2007;357(3):266-281
128. Chan JS, Beer TM, Quinn DI. et al. A phase II study of high-dose calcitriol combined with mitoxantrone and prednisone for androgen-independent prostate cancer. BJU Int. 2008;102(11):1601-1606
129. Kimura M, Rabbani Z, Mouraviev V. et al. Role of vitamin D(3) as a sensitizer to cryoablation in a murine prostate cancer model: preliminary in vivo study. Urology. 2010;76(3):764 e714-720
130. Wu W, Zanello L, Walker AM. S179D prolactin sensitizes human prostate cancer cells such that physiological concentrations of 1, 25 dihydroxy vitamin D3 result in growth inhibition and cell death. Prostate. 2007;67(14):1498-1506
131. Belochitski O, Ariad S, Shany S, Fridman V, Gavrilov V. Efficient dual treatment of the hormone-refractory prostate cancer cell line DU145 with cetuximab and 1,25-dihydroxyvitamin D3. In Vivo. 2007;21(2):371-376
132. Istfan NW, Person KS, Holick MF, Chen TC. 1alpha,25-Dihydroxyvitamin D and fish oil synergistically inhibit G1/S-phase transition in prostate cancer cells. J Steroid Biochem Mol Biol. 2007;103(3-5):726-730
133. Srinivas S, Feldman D. A phase II trial of calcitriol and naproxen in recurrent prostate cancer. Anticancer Res. 2009;29(9):3605-3610
134. Scher HI, Jia X, Chi K. et al. Randomized, open-label phase III trial of docetaxel plus high-dose calcitriol versus docetaxel plus prednisone for patients with castration-resistant prostate cancer. J Clin Oncol. 2011;29(16):2191-2198
Author Biography
 Mohamed Ben-Eltriki earned his Pharmacy degree from Benghazi University, Libya in 2007. He then received his Master's degree in Pharmaceutical Sciences, specializing in Pharmacokinetics, from University of Alberta, Canada in 2012. He is currently a PhD student in the Faculty of Medicine at University of British Columbia (BC, Canada) working under the supervision of Dr. Emma Guns. His current work focuses on characterization of the anticancer activity of Vitamin D and Ginsenoside metabolites in combination therapy for prostate cancer. His main research interests are in the area of pharmacokinetics/pharmacodynamics and prostate cancer chemoprevention. Mohamed has one review article and five research articles published on pharmacokinetics, and several posters/oral presentations at international and local conferences. Email: mbeneltrikicom
Mohamed Ben-Eltriki earned his Pharmacy degree from Benghazi University, Libya in 2007. He then received his Master's degree in Pharmaceutical Sciences, specializing in Pharmacokinetics, from University of Alberta, Canada in 2012. He is currently a PhD student in the Faculty of Medicine at University of British Columbia (BC, Canada) working under the supervision of Dr. Emma Guns. His current work focuses on characterization of the anticancer activity of Vitamin D and Ginsenoside metabolites in combination therapy for prostate cancer. His main research interests are in the area of pharmacokinetics/pharmacodynamics and prostate cancer chemoprevention. Mohamed has one review article and five research articles published on pharmacokinetics, and several posters/oral presentations at international and local conferences. Email: mbeneltrikicom
 Dr. Subrata Deb is an Assistant Professor in the Department of Biopharmaceutical Sciences, Roosevelt University College of Pharmacy (IL, USA). Previously, Dr. Deb received his Ph.D. from The University of British Columbia. His areas of research expertise and interests include vitamin D metabolism, prostate cancer chemoprevention, carcinogen metabolizing enzymes and cytochrome P450-related metabolic interactions. Email: sdebubc.ca
Dr. Subrata Deb is an Assistant Professor in the Department of Biopharmaceutical Sciences, Roosevelt University College of Pharmacy (IL, USA). Previously, Dr. Deb received his Ph.D. from The University of British Columbia. His areas of research expertise and interests include vitamin D metabolism, prostate cancer chemoprevention, carcinogen metabolizing enzymes and cytochrome P450-related metabolic interactions. Email: sdebubc.ca
 Dr. Emma S. Tomlinson Guns is an Associate Professor in the Department of Urologic Sciences, Faculty of Medicine at the University of British Columbia (BC, Canada). She obtained her PhD in steroid drug metabolism at the University of Liverpool, UK in 1997 and completed a postdoctoral fellowship in Experimental Therapeutics at the BC Cancer Agency, Vancouver BC (1997-1999). She currently co-directs the Pharmacology and Drug Design Core at the Vancouver Prostate Centre and a significant aspect of her research is orientated around the use of natural health products (NHPs) and dietary supplements by prostate cancer patients and she is co-founder and former President (2011-2013) of the Natural Health Products Research Society of Canada (www.nhprs.ca). The concurrent use of NHPs with conventional chemotherapeutics/treatment strategies have been an important focus and interest of her lab - to help patients and caregivers to discern both beneficial and adverse interactions with NHPs. Email: egunscom
Dr. Emma S. Tomlinson Guns is an Associate Professor in the Department of Urologic Sciences, Faculty of Medicine at the University of British Columbia (BC, Canada). She obtained her PhD in steroid drug metabolism at the University of Liverpool, UK in 1997 and completed a postdoctoral fellowship in Experimental Therapeutics at the BC Cancer Agency, Vancouver BC (1997-1999). She currently co-directs the Pharmacology and Drug Design Core at the Vancouver Prostate Centre and a significant aspect of her research is orientated around the use of natural health products (NHPs) and dietary supplements by prostate cancer patients and she is co-founder and former President (2011-2013) of the Natural Health Products Research Society of Canada (www.nhprs.ca). The concurrent use of NHPs with conventional chemotherapeutics/treatment strategies have been an important focus and interest of her lab - to help patients and caregivers to discern both beneficial and adverse interactions with NHPs. Email: egunscom
![]() Corresponding author: Emma Tomlinson Guns, PhD, The Vancouver Prostate Centre at Vancouver General Hospital, 2660 Oak Street, Vancouver, BC, Canada V6H 3Z6. Phone: 1-604-875-4111. E-mail: egunscom.
Corresponding author: Emma Tomlinson Guns, PhD, The Vancouver Prostate Centre at Vancouver General Hospital, 2660 Oak Street, Vancouver, BC, Canada V6H 3Z6. Phone: 1-604-875-4111. E-mail: egunscom.