Impact Factor ISSN: 1837-9664
- Issue 7; 2026
- Issue 6; 2026
- Issue 5; 2026
- Issue 4; 2026
- Issue 3; 2026
- Volume 17; 2026
- Past Issues
- Editorial Board
- Cover Images
- Index & Coverage
- Special Issues
Introduction
Why do some organs or tissues...
The normal progenitor cells of...
Normal cells and cancer cells...
Are invasive and metastatic...
Is lack of competition with...
In what way have the curable...
More mutations multiply the...
“Precision” is...
Prevention of new mutations is...
Concluding remarks
Acknowledgements
References

 Global reach, higher impact
Global reach, higher impactJ Cancer 2016; 7(4):436-445. doi:10.7150/jca.13832 This issue Cite
Review
Learning about the Importance of Mutation Prevention from Curable Cancers and Benign Tumors
Gangshi Wang1 ![]() , Lichan Chen2, Baofa Yu3, Lucas Zellmer2, Ningzhi Xu4
, Lichan Chen2, Baofa Yu3, Lucas Zellmer2, Ningzhi Xu4 ![]() , D. Joshua Liao5
, D. Joshua Liao5 ![]()
1. Department of Geriatric Gastroenterology, Chinese PLA General Hospital, Beijing 100853, P.R. China;
2. Hormel Institute, University of Minnesota, Austin, MN 55912, USA;
3. Beijing Baofa Cancer Hospital, Shahe Wangzhuang Gong Ye Yuan, Chang Pin Qu, Beijing 102206, P.R. China;
4. Laboratory of Cell and Molecular Biology, Cancer Institute, Chinese Academy of Medical Science, Beijing 100021, P.R. China;
5. D. Joshua Liao, Clinical Research Center, Guizhou Medical University Hospital, Guizhou, Guiyang 550004, P.R. China.
Received 2015-9-13; Accepted 2015-12-3; Published 2016-1-28
Abstract
Some cancers can be cured by chemotherapy or radiotherapy, presumably because they are derived from those cell types that not only can die easily but also have already been equipped with mobility and adaptability, which would later allow the cancers to metastasize without the acquisition of additional mutations. From a viewpoint of biological dispersal, invasive and metastatic cells may, among other possibilities, have been initial losers in the competition for resources with other cancer cells in the same primary tumor and thus have had to look for new habitats in order to survive. If this is really the case, manipulation of their ecosystems, such as by slightly ameliorating their hardship, may prevent metastasis. Since new mutations may occur, especially during and after therapy, to drive progression of cancer cells to metastasis and therapy-resistance, preventing new mutations from occurring should be a key principle for the development of new anticancer drugs. Such new drugs should be able to kill cancer cells very quickly without leaving the surviving cells enough time to develop new mutations and select resistant or metastatic clones. This principle questions the traditional use and the future development of genotoxic drugs for cancer therapy.
Keywords: invasive and metastatic cells
Introduction
Cancer research these days, for the most part, looks at incredible minutiae of very specific molecules and their interactions in cancer cells. We now have a great wealth of information on what happens at the DNA, RNA and protein levels and on the biochemical reactions of various metabolisms. However, occasionally it may be a good idea to step back and look at things from a greater distance or from a completely different angle, so as to refocus and refresh. For example, Dr. Robert Axelrod, although specializing in political science, has, by cooperating with biologists and oncologists, shaped an intriguing hypothesis as to why and how tumor cells cooperate with each other during progressive carcinogenesis [1;2]. We now and then refresh ourselves in not only the clinical manifestations of cancers but also the evolution, ecology and dispersal of different organisms [3-7], and then rethink these cancer behaviors and these basic biological phenomena from the “first principles”, and not only from what laboratory research has told us. By doing so, we sometimes come up with some new thoughts that are counterintuitive or challenge the mainstreams of cancer research [8-11]. This essay describes some of our musings.
Why do some organs or tissues find it much easier than others to develop malignancy?
Sporadic tumors, either benign or malignant, can only develop in those tissues or organs that retain regeneration ability, because tumorigenesis requires cell proliferation to fix mutations onto progeny cells [12]. Those cell types that are no longer capable of regeneration are usually incapable of developing tumors. This is the reason why tumors of neuron-origin only initiate during the embryonic stage and develop in childhood, but do not occur in adulthood when the neurons have lost replication ability. One may further infer that those cell types that have a quicker and more-massive cell turnover may have a higher chance of, and a shorter latent period for, developing neoplasia and thus are collectively referred by us to as “anabolic cell type”[13]. Indeed, tumors in the skin, gastrointestinal (GI) tract and lung as well as bone-marrow-derived lymphoma, leukemia and myeloma are among the most common malignancies. Of course, this conclusion needs to preclude specific etiological factors that appear only in some countries or during some specific time periods. For example, the hepatitis B virus infection was omnipresent decades ago in China and in turn made liver cancer also omnipresent there then, but both the infection and the cancer are much less common in the United States and Europe and are less now in China.
The reason behind the contribution of a frequent cell turnover to the easier formation of cancers goes beyond the requirement of cell proliferation to fix mutations onto progeny cells, when thought about from an evolutionary point of view: As we described before [11], multicellular organisms, unlike unicellular ones, have evolved cell specialization with the fitness of the organism as a whole, but not the fitness of individual cells, as the ultimate interest. This whole-body-interest requires some cell types to die for the sake of the whole body. For instance, white blood cells are required to fight against bacteria, viruses and other infectious pathogens. Skin keratinocytes are required to protect the body from many detrimental physical (e.g. ultraviolet light), chemical (e.g. acidic material), and biological (e.g. bacteria) factors in the environment. Mucosal epithelial cells inside the GI tract also need to protect the body from variant biological, chemical and physical harmful factors, besides needing to carry out their main functions of digestion and nutrient absorption. All these “body-guard” cells die easily in their actions. Evolution does not equip these cells, expect their stem-cell progenitors, with sophisticated survival mechanisms, not so sophisticated as in those long-lived cell types such as the cardiac muscle cells and neurons, since they can be sacrificed at any time and the body can quickly produce millions of replacement cells. Probably, DNA repair mechanisms in these cells, relative to those in long-lived ones, may in general be less sophisticated as well, making them more susceptible to being mutated and malignantly transformed, although this speculation requires experimental verification. Theoretically, all these body-guard cells should unselfishly sacrifice their lives when such sacrifice is needed for sustaining the body's life. However, sometimes some of them do become selfish and try to evade the body's surveillances, likely due to some epigenetic or genetic alterations that result in aberrant function of some oncogenes or tumor suppressor genes and ensuing sustained cell proliferation, i.e. immortalization.
The normal progenitor cells of several curable cancers share some common properties
Gestational choriocarcinoma and testicular cancer have a high chance of being cured. Gestational choriocarcinoma derives from a trophoblast, which in turn is one of the progeny cells of a fertilized egg with half of its chromosomes foreign, i.e. from the paternal contribution. Genes on these foreign chromosomes may make trophoblasts more immunogenic and thus more easily removed by the patient's immune system. Testicular cancer, including its metastases to the organs where the temperature is 37ºC, seems to be very sensitive to hyperthermia-caused cell death. As deduced before [10], this may be because male germ cells as testicular cancer progenitors have evolved to be highly sensitive to heat-stress and thus are confined in the scrotum, the temperature of which is 2-7°C lower than the core body temperature, varying among different mammals [14]. Moreover, compared with other somatic cells, male germ cells are under a much tighter surveillance of genomic alteration to ensure the normality of the sperm, in part because the blood-testis barrier keeps the cells in a hypoxic situation [15;16]. This surveillance makes it much easier for these cells to die from stress-induced-cell-death (SICD) that has properties of programmed events, as described before [8-10], because the body wants to eliminate mutation-containing sperm.
Many cases of leukemia and lymphoma are curable as well. Their normal counterparts and also their progenitors, i.e. white blood cells or lymphocytes, are the frontier fighters against micropathogens and thus can die easily from one of two mechanisms. One is necrosis, meaning that they can be killed in the action against micropathogens. The other is SICD [8-10]. For instance, if white blood cells are infected by, but cannot eliminate, bacteria or viruses, some programmed procedures will be triggered to eliminate the cells per se, so that they will not carry and thus spread the micropathogens to other body sites. Sometimes the micropathogens, mainly viruses, confer a form of selfishness onto the infected cells by compromising their SICD mechanism, manifested as immortalization of the cells.
Another common trait of the abovementioned curable cancers is that their normal progenitor cells have a strong mobility, i.e. can invade locally or even travel freely to and lodge in other body sites. This trait of “lodging at elsewhere” also means a strong adaptability, with white blood cells having the best adaptability as they can live happily in almost all body sites. Indeed, sperm are highly mobile in order to find an egg to fertilize. Trophoblasts are not only highly proliferative but also invasive and metastatic, as they can make an inroad into the uterine wall to establish gestation and may even encroach into the blood vessels and eventually home in on the lungs of the mother and a number of organs of the newborn [17]. Osteoclasts are generated in the bone marrow but then migrate to the bone to eat up bone tissue, which is reminiscent of the phenomenon in which cancer cells eat up the surrounding tissue. Bone-marrow protomonocytes migrate to and home in on the liver while sequentially differentiating to monoblasts, monocytes and eventually Kupffer cells during the embryonic liver development; this lineage resembles a stepwise metastasis of cancer cells that leave their hometown, migrate to a new place and make a home there with many phenotypical changes. In conclusion, invasion and metastasis are not unique properties of cancer cells but, instead, are behaviors of some normal cells as well. The phenomenon in which leucocytes migrate from bone marrow to and home in on almost everywhere in the body is the best example of “metastasis” of normal cells, which probably is a reason why in pathology textbooks all neoplastic lesions of the hematopoietic or lymphoid cell origins are classified as malignancy and none are benign. Therefore, the sequence of events in carcinogenesis in solid organs is not always from G+I-M- to G+I+M- and then G+I+M+ (here “G”, “I”, and “M” indicate growth, invasion and metastasis, respectively, whereas “+” and “-“ indicate with or without, respectively) [17]. Actually, experimental evidence has been shown that mouse mammary epithelial cells can metastasize to and colonize in the lungs before being malignantly transformed [18;19], and that dissemination of single cancer cells may occur before the primary tumor manifests invasive histology [20].
Carcinogenesis may occur by converting differentiated cells to a less differentiated status, i.e. dedifferentiation [21], such as via mutation in the PIK3CA gene [22], or by preventing differentiation of undifferentiated embryonic cells or stem cells, i.e. stop-of-differentiation. Childhood cancers may derive from a stop-of-differentiation mechanism to retain embryonic cells in their undifferentiated, i.e. cancer-resembling, morphology [11]. Sporadic cancers in adults are much more complex, as they may, somewhat similar to childhood cancers, derive from a stop of differentiation of organ- or tissue-specific stem cells or, may, probably more often, derive from dedifferentiation of differentiated cells. Many pediatric cancers are much easier to cure [23-25], such as Wilms' tumors as well as many leukemias and lymphomas [26], whereas cancers in adulthood are more often unmalleable. Our speculative explanation, which awaits verification, is that stop-of-differentiation of an embryonic cell may require fewer mutations than dedifferentiation of differentiated cells in adulthood. Regardless of how they were developed, early-stage cancers usually have a better prognosis than advanced ones, not only because earlier tumors have a better chance of being surgically removed but also largely because cancer cells at their earlier stages have fewer mutations for the establishment of contumacious phenotypes, e.g. therapy-resistance and metastasis. Axelrod et al even hypothesize that early-stage cancer cells may just have been partly transformed and need to collaborate with each other for survival and for collective manifestation of a cancer phenotype [1].
In summary, which has been done by Blagosklonny [26], some cancer types are curable, presumably because their normal progenitor cells share at least two features: First, they can die easily, and cancer cells derived from them should retain some of this “easily die” property and thus can be killed more easily by a therapy. Therefore, the “die easily” property has dual effects, i.e. on one hand allowing the cells to easily be malignantly transformed but on the other hand also rendering the transformed cells easily killed. Second, they have already equipped themselves, evolutionarily, with mobility and adaptability, thus being capable of encroachment and metastasis even before they become neoplastic. This sharply contrasts with many highly lethal solid tumors that require development and accumulation of additional mutations to be mobile and to adapt to new habitats. However, a caveat needs to be given that although the most easily curable cancers do not require additional mutations to be invasive or metastatic, some cases may still develop and accumulate such mutations, which may be a reason why some cases of these usually curable types of malignancy, such as some cases of lymphoma or leukemia, are still intractable.
Normal cells and cancer cells fight against each other for their survival
In cancer patients there exist much collaboration among different cell types, especially when a treatment is involved [27-32], as summarized by Ebos [33]. First, cancer cells recruit normal stromal cells to establish cancer niches, as exemplified by tumor angiogenesis [23]. Second, cancer cells collaborate with each other [1;2;34], which is evident not only in vivo, as in their collective invasion in patients [35] and clonal cooperation in animals [36;37], but also in vitro as demonstrated by the fact that many cancer cell lines cannot survive in a culture dish when cells are seeded in a very low density [34;38]. On the other hand, there are also many fights among different cell types. Normal cells that surround cancer cells battle the cancer, at least at early carcinogenic stages, for the fitness of the patient as a whole [7;39;40]. Even in cell culture, the media that have been used to culture normal cells have been found to contain factors such as cytokines or microRNAs that can kill or inhibit cancer cells, which somewhat resembles an antibiotic secreted by a bacterium to kill another microorganism in the nearby. For instance, normal mammary [41-43] or prostate [44] epithelial cells release such factors to the media to kill or inhibit breast or prostate cancer cells, respectively. If these observations are directly translated to clinical oncology, probably in the patients with an early breast or prostate cancer, their sera still favor the patients' health by containing cancer cell specific killing or inhibiting components released from the normal cells. These data dovetail with the so-called “neighboring suppression” notion proposed 50 years ago, which says that normal cells can cause growth inhibition of neighboring premalignant cells [45], probably by arresting them at the G2/M phase of the cell cycle [46-48]. For survival, the premalignant, probably also early malignant, cells need to find a way, such as having some additional genetic or epigenetic change, to desensitize such inhibition. Once having progressed to an advanced stage and having become dominant, cancer cells may also release factors into the blood that can eliminate normal cells in the same and other organs or tissues [7]. Cachexia, i.e. wasting syndrome, is probably one manifestation. From an ecology point of view, these complex interactions among different cell types make sense, and breaking their balances may make one cell type dominant while others become extinct, which provides us with opportunities for cancer prevention and therapy.
Are invasive and metastatic cells the initial losers in the competitions among cancer cells?
Cancer cells not only collaborate with each other as abovementioned but also compete with each other for resources [49;50], which may be due to, and is also one reason for, the intra-tumor heterogeneity [10;11;51;52]. From the viewpoint of cell-autonomy, invasive and metastatic cells are considered stronger than the others because they are more aggressive and perverse. However, from a viewpoint of biological dispersal, the winners in the cell competition are less motivated to relocate to other habitats since they adapt to the environment very well and have the privilege of the first use of nutrients and oxygen, whereas the losers may die if they do not relocate [53]. Therefore, it is possible that cells invade to the surrounding tissue or metastasize distantly because they have lost out in the competition with other cells and are forced to leave [54]. This possibility deserves exploration, not only because it may provide us with a new slant to understand cancer [54-56] but also because cells have other reasons for dispersal, including changes in metabolisms [57;58], and the winner cells may disperse as well if new habitats provide an even better living environment. From this point of view, the invasive or metastatic cells are actually weaker, although they later acquire competence in the incursive or the metastatic site where they develop colonies within which new rounds of cell-competition occur, resulting in new losers and winners. If this is the case, we may be able to kill those initial cells with invasive or metastatic potential within the primary tumor, such as by helping the winner cells to weed out the losers or by quickly depriving them from all resources before they disperse. Alternatively and conversely, slightly alleviating their hardship by providing them with basic resource may also retain them in the primary tumor without thinking of relocation to new habitats [59-61].
It is well known that the body attempts to mobilize macrophages or other predatory immune cells as scavengers to remove cancer cells [8;9;62-65]. What is unknown is whether it is the weaker (loser) or the stronger (winner) cancer cells that are scavenged first. Moreover, sometimes the scavenger engulfs a cancer cell but is unable to digest it, resulting in a hybrid (or fusion) cell that gains metastatic ability [11;62-66]. This mechanism for acquiring metastatic ability via fusion with another cell [17;67;68], especially with an immune cell that can travel freely, was already proposed one century ago [65;69] and observed 40 years ago [70] and has received ample evidence. Sometimes metastases still occur years after the primary tumor had been surgically removed at its very early stage. One possibility is that some cancer cells had already been transported to other sites by predatory immune cells. What remains unclear is whether it is the winner or the loser cancer cells that may have a better ability to resist digestion by the scavengers and further utilize the scavengers' property of evading the body's surveillance to gain metastatic ability.
Is lack of competition with normal cells a reason why most benign tumors never progress to malignancy?
Benign tumors originating from those cell types that have frequent cell turnover usually have a relatively high risk of progressing to malignancy, exemplified by colon adenomas. In contrast, the great majority of benign tumors developed from cells that have infrequent turnover do not progress to malignancy, such as most lipomas and uterine leiomyomas that originate from adipocytes and uterine smooth muscle cells, respectively. Of course, exceptions exist, such as when exogenous sex hormones are present to drive progression of hepatoma and uterine leiomyoma [71-76]. Benign cells retain most of the functions of normal cells, including restrained proliferation and nutrient uptake. These traits not only make benign cells helpful in sustaining the physiology and homeostasis of the host organ or tissue but also allow them to live in a friendly environment with little need to compete with normal cells, thus lacking a driving force for evolution to malignancy via mutations and ensuing selections. These features are also why benign cells are relatively genetically homogenous and seldom compete with each other to result in loser cells that have to disperse, i.e. invade or metastasize, and why we lack drug targets for most benign tumors. In other words, most benign tumors have two genetic features, i.e. 1) they have fewer mutations and 2) these mutations do not drive the development of more mutations. For the benign tumors with some malignant features or those cancers with a well differentiated morphology, a harsh radio- or chemo-therapy with genotoxicity may actually cause mutations, in turn prodding evolution of the cells to more virulent forms, since all such therapies promote mutation-related metastasis [33].
In what way have the curable cancers and benign tumors enlightened us?
The above sections suggest that benign tumor cells are relatively homogeneous and that some cancers are curable largely because they do not need additional mutations for invasion and metastasis, which collectively imply that “the more mutations a tumor has, the worse it is”. However, this trend of mutation-malignancy relationship probably is correct only at relatively early stages of a given tumor's progression. By continuing replication, cancer cells develop more mutations in a random and stochastic manner [10;11], leading to genetic heterogeneity among cells in the tumor. Those mutations that are good for the cell's survival are selected and passed to more progeny cells in a clonal-expansion manner and eventually are presented as more-formidable phenotypes, whereas those mutations that are deleterious will disappear along with the death of the cells. The net consequence is a higher cell turnover rate in the more-advanced tumors than in the earlier ones and in their parental normal tissue [77;78]. Many advanced tumors are even highly necrotic, although by our definition [8-10] the cells actually die of SCID, and it is a widespread misperception that SCID, commonly mistaken as apoptosis [8-10], is decreased in cancer [12]. More advanced cancers do not necessarily have a larger number of mutations than the earlier ones because they have more cellular death, although a more frequent turnover should enhance mutation formation. For this reason, it is more correct to say that “the more genetically diversified a tumor is, the worse it is”. In other words, if tumor cells in a patient develop a greater variety, but not necessarily a larger number, of mutations, the disease has a higher chance to progress to more-uncontrollable states. Moreover, one should not compare the mutation numbers between two different cases, and certainly not between two different cancer types. Cancer virulence is more closely related to its cell or tissue origin, in part because of some particular features of their normal progenitor cells as discussed in the above sections. For instance, pancreatic cancer is much worse than testicular cancer, generally speaking.
More mutations multiply the options for cancer cells to survive
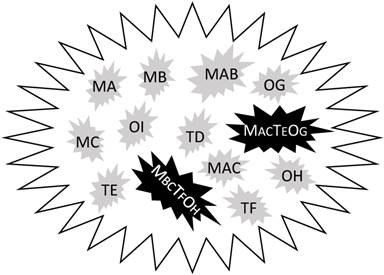
Sequencing the genomic DNA of thousands of samples from different cancer types surprisingly found only 138 genes with driver mutations, i.e. those that drive carcinogenesis [79]. Some of the mutations may already exist in normal cells [80]. In our meditation, this is because these mutated genes, coined herein as mutants, usually do not work alone. Very likely, they work not only with each other but also with many other genes that are wild type but are aberrantly expressed, in order to provide cancer cells with many more options for surviving different situations, manifested as different cancer phenotypes. We might imagine that these mutants sit at different intersections of a highly complex network of survival pathways that are familiar to us as colorful charts in many companies' catalogs or posters. These maps show clearly that another intersection or a combination of several other intersections can always establish a new bypass or detour if the one(s) currently in use are blocked. In a given tumor, different cells can use different members of these 138 mutants in different orders to establish their own survival pathways in their particular situations, which resembles different combinations (passcodes) made from these 138 mutants in 2, 3, 4 or even more digits. Very likely, these passcodes also involve many wild-type genes that are aberrantly expressed, as illustrated in figure 1, raising the complexity to a higher order. Therefore, knowing the names of the 138 genes is significant, but is not sufficient. As an analogy, a thief may have stolen many bank cards and knows that each of the cards uses a particular order of four of the ten Arabic numerals as the passcode, but this information is far from enough for him to decode the cards. A bank in the United States called “USbank” requires 8-24 letters, numbers and symbols as the passcode for the account of its customers, making it impossible to decode. Deciphering different combinations of these 138 mutants and probably also other wild type genes in different digits is even more impossible. Even if we can fortuitously decipher one combination, i.e. the order of several mutants and aberrantly-expressed wild type genes that makes a particular survival pathway, it is possible that only several of the millions of cancer cells in a given patient use this pathway, probably for just a period of time since cancer cells keep changing. This is what “intra-tumor heterogeneity” means, which may also be taken positively if one wishes: giving any chemo drug, including one of those having a particular target, to any randomly selected patient may more or less kill some cancer cells that happen to use the survival pathway sensitive to the drug. Fortunately, the 138 mutants are not used at the same frequency by different cancers to adapt to their environmental changes. In fact, some cancer types have some, often just one or two, preferred mutants in association with abnormal expression of certain wild type genes, which provides us with drug targets, as proved by some successful targeted therapies.
Illustration of hypothetic differences in mutations and aberrant gene expression between the tumor mass and individual cells. Although the tumor has been detected for mutations (M) in three genes (A, B, and C), individual cells bear only one or two, but not all three, mutations. Similarly, three (D, E, F) tumor suppressor genes (T) and three (G, H and I) oncogenes (O) are detected for decreased or increased expression, respectively, but none of the cells simultaneously shows abnormal expression of all these six genes. One therapy-resistant pathway (MACTEOG) is established in one cell by not only bearing the MA and MC mutants but also showing the decrease in TE and the increase in OG, whereas one metastatic phenotype (MBCTFOH) is established in another cell by not only bearing the MB and MC mutants but also displaying the decrease in TF and the increase in OH. One or both of these more-aggressive cells may already exist at the time of diagnosis but may also be developed during a therapy.
“Precision” is useful only when it is time-sensitive and precise to individual cells, and not just the individual patients
Cancer biomarkers resemble a person's clothes in serving as a form of identification. For example, identifying a breast cancer by its estrogen receptor α positivity, Rb negativity and p53 mutation somewhat can be analogous to identifying a lady by her “red hat, white garment and black shoes”. The phenomenon that most cancer cells in a lump express a particular marker, e.g. estrogen receptor α, resembles a situation that most enemies wear uniforms and thus can be easily identified and targeted [81]. Unfortunately, those enemies who survive our carpet bombing (e.g. with an anti-estrogen therapy) will get rid of the uniform and dress like us, making it basically impossible for us to aim with accuracy. We cannot simultaneously target 138 mutants, not even a small fraction of them. Besides, there are probably many other genes having driver mutations that occur only in a small number of cells and thus are not detected, but these mutants may become dominant to establish new complex survival pathways if due to whatever reason the 138 mutants no longer function. The “Precision Medicine Initiative” recently launched by the President Obama [82] will likely lead to the identification of more cancer biomarkers and the ensuing development of more “magic bullets”, a nice nickname for targeting drugs [83-86]. However, an oncologist still has good reason to worry whether today his patient can still be identified by the “red hat, white garment and black shoes” as shown in yesterday's lab results, since her samples were sent to the lab days ago, whereas cancer cells keep changing. Therefore, the “precision” should be dated and specific to each individual cancer cell of the patient and should be valid for only a short period of time, probably just a month or even just a week.
Another obstacle for precision medicine is that blocking one survival pathway may enhance another one, largely because evolution has rendered many, probably most, genes dually functional, i.e. one gene can appear as different forms to elicit different or even opposing functions, so that the genome can be much more efficient and thus smaller. The “different form” may be a mutant or polymorphism, an alternative splicing, a different proteolysis or phosphorylation status, or even a different subcellular location, as detailed before [12;87-89]. For instance, some p53 mutants or polymorphisms are oncogenic while its wild type usually is tumor-suppressive [12]. Cytochrome c in its location on the inner mitochondrial membrane functions to ferry electrons in the respiratory chain to power the cell, but its relocation to the cytosol makes it a killer of the cell by triggering a cascade of reactions of SICD that is commonly mistaken as apoptosis [8-10]. As an example of a functional duality of genes in cancer, activation of the well-known ERK/MAP survival pathway can also arrest cell cycle progression of some subtypes of cells in several cancer types [12;90;91], and therefore targeting this pathway for cancer therapy may have a potential side-effect.
Prevention of new mutations is probably critical for a cure
Therapy resistance can be primary. For instance, many pancreatic cancers do not show good response to any treatment. However, more often the resistance is acquired, coming from an administered therapy via one of the two mechanisms that differ mainly at when and how the resistance-responsible mutations(s) occur: One is that the therapy has killed many or even most cancer cells but allows a few cells that bear already-existing mutation(s) to populate via clonal expansion [92]. The other is that during the treatment, especially with radiation or a genotoxic agent, a few cells develop new mutation(s), in many cases due to the treatment [93], followed by their clonal expansion. Both mechanisms are likely associated with non-genetic changes, such as those described by Huang et al [94-96] and including abnormal expression of wild-type oncogenes or tumor suppressor genes. For research purpose, resistant clones of cancer cell lines are often developed intentionally through a chronic treatment with a low dose of radiation or chemotherapeutic agents [97;98]. Although the resistance of some clones so developed is reversible, the resistance of some other clones is perpetual, thus likely involving genetic alteration and supporting the existence of the second mechanism, at least in vitro. Induction of a second primary cancer by radio- or chemo-therapy, a well-known phenomenon [99-101], may be used as an in vivo evidence for therapy-induced mutation. The observation that all known resistance-responsible mutations can be detected in a tumor mass does not mean that some individual cells have already had a sufficient number of mutations for the establishment of a resistance pathway, as depicted in figure 1. Likely, individual cells still need to develop additional mutation(s) to complete a resistance pathway [11]. Hopefully, it will soon be technically possible to sequence the genome of each cell in a primary tumor to see whether there exist individual cells that bear all mutations along individual resistance pathways.
While little can probably be done for already-existing mutations, we should at least prevent new mutations from occurring since some of them may be responsible for therapy resistance or other pernicious phenotypes. Of those incurable types of cancer, many patients actually respond to the initial radio- or chemo-therapy magnificently well. However, all remedies, probably including surgery [102], change the environment and thus are also an impetus for evolution of the still-alive cells to more-aggressive phenotypes via development of mutations and selection of more-untoward clones, besides other mechanisms such as an influence on the surrounding normal cells [103]. Indeed, plentiful studies have shown that various treatments may accelerate metastases [102;104-111], as summarized by Ebos [33]. Therefore, the initial treatment is a double-edged sword, killing cancer cells while training the still-alive ones to be tougher [33]. Usually, the better the initial response a tumor shows, the more sufficient time the remaining cells have for the development of more mutations and for the ensuing expansion of the more-aggressive cells, manifested as a longer disease-free survival that is actually the latent time for the recurrence or metastases. From this viewpoint, one may say that the patient actually pays later with his life for the initial disease-free or overall survival, no matter how long it is. Indeed, for most cases of those currently incurable types of cancer, exemplified by pancreatic and liver cancers, oncologists actually know that the patients will die later, even if the efficacy of the initial treatment is marvelous. Therefore, since it takes time for cancer cells to develop mutations and then for clonal expansion of these further mutated cells, our task should not just be to kill all cancer cells but should be to kill them very quickly without leaving the not-yet-killed cells with time for these events to occur. In other words, “run against time for new mutations to emerge and be selected” should be a key principle for the development of new drugs or remedies, as we detailed elsewhere [112], since killing cancer cells massively is less difficult and has already been achieved for many cases. This hypothetical principle makes questionable the use and development of highly genotoxic drugs, although such drugs are still the major ones currently and continue to emerge.
For those advanced cases with metastases and resistance to available therapies, a second choice may be to provide a mild chemotherapy, rather than an even harsher one as our intuition may want us to do, because a tougher remedy is also a stronger goad for a quicker progression [33]. In many organisms, slowly-proliferating cells survive better than their fast-proliferating counterparts [113]. Therefore, a low dose of chemotherapy may to some extent shift the cancer cells to a slowly-proliferating or even dormant status, making low dose treatment meaningful for resistant cancers. The key point, while a difficult one, is that the mild treatment should be strong enough to let cancer cells slow down their growth but should not be so harsh as to drive the cancer cell to disperse to more body sites. Undoubtedly, it is hard for any oncologist to decide which of these two extremes should be used, i.e. a harsher treatment to kill more cells quickly or a much milder one to retrain them from proliferation and further dispersal.
Concluding remarks
Many once-existing species of organisms, like the dinosaurs, have gone extinct. These ecocides, caused by the changes of ecosystems, are precedents showing that cancers may be cured (extinguished) as well if we can make their ecological environments deleterious to the cancer cells [56;61;114], because in many aspects cancers resemble new organisms parasitizing the host [10;11;52;115]. Actually, some cancers have a good prognosis or a high chance of being cured via non-surgical approach, which is presumably attributable to the fact that their normal progenitor cells not only die easily but also have already been equipped with mobility and adaptability, thus allowing the cancers to metastasize without additional mutations. Therefore, the key for curing cancer is to kill all cancer cells before new mutations emerge, because some of the new ones may render the cells therapy-resistant or metastatic. From the viewpoint of biological dispersal, invasive and metastatic cells may be losers in competitions for resources with those cancer cells remaining in the primary tumor. If this is the case, helping the winner cells to weed out these loser ones or, conversely, slightly mitigating their hardship by such as providing some basic resource, may be an applicable approach to preventing metastasis. Similarly, to those advanced and resistant cancers, a milder chemotherapy, and not a harder blow with maximal tolerated dose as often considered, may actually be able to shift cancer cells to a slowly-proliferating or dormant status without their thinking about metastasis to more body sites, thus allowing the patients to survive for a longer time. These speculative ideas, inspired by evolution, ecology and dispersal of organisms, may provide us with new avenues leading to prevention, treatment and confinement of cancers and thus deserve further exploration.
Acknowledgements
We want to give special thanks to Dr. Fred Bogott at the Medical Center, Austin of Minnesota, and Dr. Margot Cleary at the Hormel Institute of University of Minnesota for their valuable discussions and excellent English editing of this manuscript.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Axelrod R, Axelrod DE, Pienta KJ. Evolution of cooperation among tumor cells. Proc Natl Acad Sci U S A. 2006;103(36):13474-13479
2. Axelrod R. Launching "the evolution of cooperation". J Theor Biol. 2012;299:21-24
3. Walther V, Hiley CT, Shibata D, Swanton C, Turner PE, Maley CC. Can oncology recapitulate paleontology? Lessons from species extinctions. Nat Rev Clin Oncol. 2015;12(5):273-285
4. Amend SR, Pienta KJ. Ecology meets cancer biology: the cancer swamp promotes the lethal cancer phenotype. Oncotarget. 2015;6(12):9669-9678
5. Vittecoq M, Roche B, Daoust SP, Ducasse H, Misse D, Abadie J. et al. Cancer: a missing link in ecosystem functioning? Trends Ecol Evol. 2013;28(11):628-635
6. Crespi B, Summers K. Evolutionary biology of cancer. Trends Ecol Evol. 2005;20(10):545-552
7. Aktipis CA, Nesse RM. Evolutionary foundations for cancer biology. Evol Appl. 2013;6(1):144-159
8. Liao DJ. The scavenger cell hypothesis of apoptosis: apoptosis redefined as a process by which a cell in living tissue is destroyed by phagocytosis. Med Hypotheses. 2005;65(1):23-28
9. Liu B, Xu N, Man Y, Shen H, Avital I, Stojadinovic A. et al. Apoptosis in Living Animals Is Assisted by Scavenger Cells and Thus May Not Mainly Go through the Cytochrome C-Caspase Pathway. J Cancer. 2013;4(9):716-723
10. Zhang J, Lou XM, Jin LY, Zhou RJ, Liu SQ, Xu NZ. et al. Necrosis, and then stress induced necrosis-like cell death, but not apoptosis, should be the preferred cell death mode for chemotherapy: clearance of a few misconceptions. Oncoscience. 2014;1(6):407-422
11. Zhang J, Lou XM, Zellmer L, Liu SQ, Xu NZ, Liao DJ. Just like the rest of evolution in Mother Nature, the evolution of cancers may be driven by natural selection, and not by haphazard mutations. Oncoscience. 2014;1(9):580-590
12. Lou X, Zhang J, Liu S, Xu N, Liao DJ. The other side of the coin: The tumor-suppressive aspect of oncogenes and the oncogenic aspect of tumor-suppressive genes, such as those along the CCND-CDK4/6-RB axis. Cell Cycle. 2014;13(11):1677-1693
13. Jia QW, Chen XH, Jia YP, Dou XX, Ezeogu L, Xu NZ. et al. Is type 2 diabetes one of such aging phenomena that lack an irreversible structural change? J Diabetes & Metab. 2015;6:6 543-doi:10.4172/2155-6156.1000543
14. Lovegrove BG. Cool sperm: why some placental mammals have a scrotum. J Evol Biol. 2014;27(5):801-814
15. Reyes JG, Farias JG, Henriquez-Olavarrieta S, Madrid E, Parraga M, Zepeda AB. et al. The hypoxic testicle: physiology and pathophysiology. Oxid Med Cell Longev. 2012;2012:929285-doi 10.1155/2012/929285
16. Jankovic VL, Stefanovic V. Hypoxia and spermatogenesis. Int Urol Nephrol. 2014;46(5):887-894
17. Mareel MM, Van Roy FM, De BP. The invasive phenotypes. Cancer Metastasis Rev. 1990;9(1):45-62
18. Podsypanina K, Du YC, Jechlinger M, Beverly LJ, Hambardzumyan D, Varmus H. Seeding and propagation of untransformed mouse mammary cells in the lung. Science. 2008;321(5897):1841-1844
19. Weinberg RA. Leaving home early: reexamination of the canonical models of tumor progression. Cancer Cell. 2008;14(4):283-284
20. Rhim AD, Mirek ET, Aiello NM, Maitra A, Bailey JM, McAllister F. et al. EMT and dissemination precede pancreatic tumor formation. Cell. 2012;148(1-2):349-361
21. Tenen DG. Disruption of differentiation in human cancer: AML shows the way. Nat Rev Cancer. 2003;3(2):89-101
22. Koren S, Reavie L, do Couto JP, De SD, Stadler MB, Roloff T. et al. PIK3CA induces multipotency and multi-lineage mammary tumours. Nature. 2015;525(7567):114-118
23. Kareva I. What can ecology teach us about cancer? Transl Oncol. 2011;4(5):266-270
24. Curry HL, Parkes SE, Powell JE, Mann JR. Caring for survivors of childhood cancers: the size of the problem. Eur J Cancer. 2006;42(4):501-508
25. Gatta G, Zigon G, Capocaccia R, Coebergh JW, Desandes E, Kaatsch P. et al. Survival of European children and young adults with cancer diagnosed 1995-2002. Eur J Cancer. 2009;45(6):992-1005
26. Blagosklonny MV. NCI's provocative questions on cancer: some answers to ignite discussion. Oncotarget. 2011;2(12):1352-1367
27. Marcucci F, Bellone M, Caserta CA, Corti A. Pushing tumor cells towards a malignant phenotype: stimuli from the microenvironment, intercellular communications and alternative roads. Int J Cancer. 2014;135(6):1265-1276
28. Kaminski JM, Shinohara E, Summers JB, Niermann KJ, Morimoto A, Brousal J. The controversial abscopal effect. Cancer Treat Rev. 2005;31(3):159-172
29. Marin A, Martin M, Linan O, Alvarenga F, Lopez M, Fernandez L. et al. Bystander effects and radiotherapy. Rep Pract Oncol Radiother. 2015;20(1):12-21
30. Reynders K, Illidge T, Siva S, Chang JY, De RD. The abscopal effect of local radiotherapy: using immunotherapy to make a rare event clinically relevant. Cancer Treat Rev. 2015;41(6):503-510
31. Siva S, MacManus MP, Martin RF, Martin OA. Abscopal effects of radiation therapy: a clinical review for the radiobiologist. Cancer Lett. 2015;356(1):82-90
32. McAllister SS, Weinberg RA. The tumour-induced systemic environment as a critical regulator of cancer progression and metastasis. Nat Cell Biol. 2014;16(8):717-727
33. Ebos JM. Prodding the Beast: Assessing the Impact of Treatment-Induced Metastasis. Cancer Res. 2015;75(17):3427-3435
34. Korolev KS, Xavier JB, Gore J. Turning ecology and evolution against cancer. Nat Rev Cancer. 2014;14(5):371-380
35. Friedl P, Locker J, Sahai E, Segall JE. Classifying collective cancer cell invasion. Nat Cell Biol. 2012;14(8):777-783
36. Polyak K, Marusyk A. Cancer: Clonal cooperation. Nature. 2014;508(7494):52-53
37. Cleary AS, Leonard TL, Gestl SA, Gunther EJ. Tumour cell heterogeneity maintained by cooperating subclones in Wnt-driven mammary cancers. Nature. 2014;508(7494):113-117
38. Zhu S, Wang J, Xie B, Luo Z, Lin X, Liao DJ. Culture at a Higher Temperature Mildly Inhibits Cancer Cell Growth but Enhances Chemotherapeutic Effects by Inhibiting Cell-Cell Collaboration. PLoS One. 2015;10(10):e0137042
39. Wagstaff L, Kolahgar G, Piddini E. Competitive cell interactions in cancer: a cellular tug of war. Trends Cell Biol. 2013;23(4):160-167
40. Vivarelli S, Wagstaff L, Piddini E. Cell wars: regulation of cell survival and proliferation by cell competition. Essays Biochem. 2012;53:69-82
41. Weaver VM, Petersen OW, Wang F, Larabell CA, Briand P, Damsky C. et al. Reversion of the malignant phenotype of human breast cells in three-dimensional culture and in vivo by integrin blocking antibodies. J Cell Biol. 1997;137(1):231-245
42. Furuta S, Jeng YM, Zhou L, Huang L, Kuhn I, Bissell MJ. et al. IL-25 causes apoptosis of IL-25R-expressing breast cancer cells without toxicity to nonmalignant cells. Sci Transl Med. 2011;3(78):78ra31-doi 10.1126/scitranslmed.3001374
43. Furuta S, Jiang X, Gu B, Cheng E, Chen PL, Lee WH. Depletion of BRCA1 impairs differentiation but enhances proliferation of mammary epithelial cells. Proc Natl Acad Sci U S A. 2005;102(26):9176-9181
44. Kosaka N, Iguchi H, Yoshioka Y, Hagiwara K, Takeshita F, Ochiya T. Competitive interactions of cancer cells and normal cells via secretory microRNAs. J Biol Chem. 2012;287(2):1397-1405
45. Stoker M. REGULATION OF GROWTH AND ORIENTATION IN HAMSTER CELLS TRANSFORMED BY POLYOMA VIRUS. Virology. 1964;24:165-174
46. Allard D, Stoker M, Gherardi E. A G2/M cell cycle block in transformed cells by contact with normal neighbors. Cell Cycle. 2003;2(5):484-487
47. Flaberg E, Markasz L, Petranyi G, Stuber G, Dicso F, Alchihabi N. et al. High-throughput live-cell imaging reveals differential inhibition of tumor cell proliferation by human fibroblasts. Int J Cancer. 2011;128(12):2793-2802
48. Stoker M, Gherardi E, Perryman M, Gray J. Scatter factor is a fibroblast-derived modulator of epithelial cell mobility. Nature. 1987;327(6119):239-242
49. Kareva I, Berezovkaya F, Karev G. Mixed strategies and natural selection in resource allocation. Math Biosci Eng. 2013;10(5-6):1561-1586
50. Kareva I, Morin B, Castillo-Chavez C. Resource consumption, sustainability, and cancer. Bull Math Biol. 2015;77(2):319-338
51. Knauss S, Klein A. From aneuploidy to cancer: the evolution of a new species? J Biosci. 2012;37(2):211-220
52. Vincent MD. Cancer: beyond speciation. Adv Cancer Res. 2011;112:283-350
53. Taylor TB, Johnson LJ, Jackson RW, Brockhurst MA, Dash PR. First steps in experimental cancer evolution. Evol Appl. 2013;6(3):535-548
54. Pienta KJ, Robertson BA, Coffey DS, Taichman RS. The cancer diaspora: Metastasis beyond the seed and soil hypothesis. Clin Cancer Res. 2013;19(21):5849-5855
55. Yang KR, Mooney SM, Zarif JC, Coffey DS, Taichman RS, Pienta KJ. Niche inheritance: a cooperative pathway to enhance cancer cell fitness though ecosystem engineering. J Cell Biochem. 2014;115(9):1478-1485
56. Pienta KJ, McGregor N, Axelrod R, Axelrod DE. Ecological therapy for cancer: defining tumors using an ecosystem paradigm suggests new opportunities for novel cancer treatments. Transl Oncol. 2008;1(4):158-164
57. Chen J, Sprouffske K, Huang Q, Maley CC. Solving the puzzle of metastasis: the evolution of cell migration in neoplasms. PLoS One. 2011;6(4):e17933-doi 10.1371/journal.pone.0017933
58. Aktipis CA, Maley CC, Pepper JW. Dispersal evolution in neoplasms: the role of disregulated metabolism in the evolution of cell motility. Cancer Prev Res (Phila). 2012;5(2):266-275 pii: 20140219. doi: 10.1098/rstb.2014.0219
59. Aktipis CA, Boddy AM, Jansen G, Hibner U, Hochberg ME, Maley CC. et al. Cancer across the tree of life: cooperation and cheating in multicellularity. Philos Trans R Soc Lond B Biol Sci. 2015:370 (1673)
60. Amend SR, Pienta KJ. Ecology meets cancer biology: the cancer swamp promotes the lethal cancer phenotype. Oncotarget. 2015;6(12):9669-9678
61. Walther V, Hiley CT, Shibata D, Swanton C, Turner PE, Maley CC. Can oncology recapitulate paleontology? Lessons from species extinctions. Nat Rev Clin Oncol. 2015;12(5):273-285
62. Lazova R, Chakraborty A, Pawelek JM. Leukocyte-cancer cell fusion: initiator of the warburg effect in malignancy? Adv Exp Med Biol. 2011;714:151-172
63. Pawelek JM, Chakraborty AK. The cancer cell-leukocyte fusion theory of metastasis. Adv Cancer Res. 2008;101:397-444
64. Pawelek JM, Chakraborty AK. Fusion of tumour cells with bone marrow-derived cells: a unifying explanation for metastasis. Nat Rev Cancer. 2008;8(5):377-386
65. Pawelek JM. Fusion of bone marrow-derived cells with cancer cells: metastasis as a secondary disease in cancer. Chin J Cancer. 2014;33(3):133-139
66. Carter A. Cell fusion theory: can it explain what triggers metastasis? J Natl Cancer Inst. 2008;100(18):1279-1281
67. De BP, Roos E, Brys L, Remels L, Gobert M, Dekegel D. et al. Nonmetastatic tumor cells acquire metastatic properties following somatic hybridization with normal cells. Cancer Metastasis Rev. 1984;3(1):5-24
68. Goldenberg DM. Horizontal transmission of malignancy by cell-cell fusion. Expert Opin Biol Ther. 2012;12(Suppl 1):S133-S139
69. Aichel O. About cell fusion with qualitatively abnormal chromosome distribution as cause for tumor formation. Roux W, editor. Vorträge und Aufsätze über Entvickelungsmechanik Der Organismen.Leipzig. Germany: Wilhelm Engelmann. 1911 p. 92-111
70. Goldenberg DM, Pavia RA, Tsao MC. In vivo hybridisation of human tumour and normal hamster cells. Nature. 1974;250(5468):649-651
71. Raft MB, Jorgensen EN, Vainer B. Gene mutations in hepatocellular adenomas. Histopathology. 2014 -doi: 10.1111/his.12539
72. Giannitrapani L, Soresi M, La SE, Cervello M, D'Alessandro N, Montalto G. Sex hormones and risk of liver tumor. Ann N Y Acad Sci. 2006;1089:228-236
73. Sabry M, Al-Hendy A. Medical treatment of uterine leiomyoma. Reprod Sci. 2012;19(4):339-353
74. Parker WH. Etiology, symptomatology, and diagnosis of uterine myomas. Fertil Steril. 2007;87(4):725-736
75. Liao DJ, Dickson RB. Roles of androgens in the development, growth, and carcinogenesis of the mammary gland. J Steroid Biochem Mol Biol. 2002;80(2):175-189
76. Zhang J, Sun Y, Liu Y, Sun Y, Liao DJ. Synergistic effects of androgen and estrogen on the mouse uterus and mammary gland. Oncol Rep. 2004;12(4):709-716
77. Liao DJ, Natarajan G, Deming SL, Jamerson MH, Johnson M, Chepko G. et al. Cell cycle basis for the onset and progression of c-Myc-induced, TGFalpha-enhanced mouse mammary gland carcinogenesis. Oncogene. 2000;19(10):1307-1317
78. Liao DJ, Dickson RB. Cell death in MMTV-c-myc transgenic mouse mammary tumors may not be typical apoptosis. Lab Invest. 2003;83(10):1437-1449
79. Vogelstein B, Papadopoulos N, Velculescu VE, Zhou S, Diaz LA Jr, Kinzler KW. Cancer genome landscapes. Science. 2013;339(6127):1546-1558
80. Martincorena I, Roshan A, Gerstung M, Ellis P, Van LP, McLaren S. et al. Tumor evolution. High burden and pervasive positive selection of somatic mutations in normal human skin. Science. 2015;348(6237):880-886
81. Huang S. The war on cancer: lessons from the war on terror. Front Oncol. 2014;4:293-doi 10.3389/fonc.2014.00293
82. Collins FS, Varmus H. A New Initiative on Precision Medicine. N Engl J Med. 2015;372(9):793-795
83. Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144(5):646-674
84. Hanahan D. Rethinking the war on cancer. Lancet. 2014;383(9916):558-563
85. Azmi AS. Adopting network pharmacology for cancer drug discovery. Curr Drug Discov Technol. 2013;10(2):95-105
86. Duffy MJ. The war on cancer: are we winning? Tumour Biol. 2013;34(3):1275-1284
87. Yuan C, Xu N, Liao J. Switch of FANCL, a key FA-BRCA component, between tumor suppressor and promoter by alternative splicing. Cell Cycle. 2012;11(18):3355-3356
88. Zhang J, Lou X, Shen H, Zellmer L, Sun Y, Liu S. et al. Isoforms of wild type proteins often appear as low molecular weight bands on SDS-PAGE. Biotechnol J. 2014;9(8):1044-1054
89. Jia YP, Chen LCMYK, Zhang J, Xu NZ, Liao DJ. To know how a gene works, we need to redefine it first but then, more importantly, to let the cell itself decide how to transcribe and process its RNAs. Int J Biol Sci. 2015;11(12):1413-1423
90. Deschenes-Simard X, Gaumont-Leclerc MF, Bourdeau V, Lessard F, Moiseeva O, Forest V. et al. Tumor suppressor activity of the ERK/MAPK pathway by promoting selective protein degradation. Genes Dev. 2013;27(8):900-915
91. Deschenes-Simard X, Kottakis F, Meloche S, Ferbeyre G. ERKs in cancer: friends or foes? Cancer Res. 2014;74(2):412-419
92. Foo J, Michor F. Evolution of acquired resistance to anti-cancer therapy. J Theor Biol. 2014;355:10-20
93. Heydt C, Kumm N, Fassunke J, Kunstlinger H, Ihle MA, Scheel A. et al. Massively parallel sequencing fails to detect minor resistant subclones in tissue samples prior to tyrosine kinase inhibitor therapy. BMC Cancer. 2015;15:291-doi 10.1186/s12885-015-1311-0
94. Pisco AO, Brock A, Zhou J, Moor A, Mojtahedi M, Jackson D. et al. Non-Darwinian dynamics in therapy-induced cancer drug resistance. Nat Commun. 2013;4:2467-doi 10.1038/ncomms3467
95. Brock A, Chang H, Huang S. Non-genetic heterogeneity-a mutation-independent driving force for the somatic evolution of tumours. Nat Rev Genet. 2009;10(5):336-342
96. Huang S. Tumor progression: chance and necessity in Darwinian and Lamarckian somatic (mutationless) evolution. Prog Biophys Mol Biol. 2012;110(1):69-86
97. McDermott M, Eustace AJ, Busschots S, Breen L, Crown J, Clynes M. et al. In vitro Development of Chemotherapy and Targeted Therapy Drug-Resistant Cancer Cell Lines: A Practical Guide with Case Studies. Front Oncol. 2014;4:40. doi: 10.3389/fonc.2014.00040
98. Zhang X, Yashiro M, Qiu H, Nishii T, Matsuzaki T, Hirakawa K. Establishment and characterization of multidrug-resistant gastric cancer cell lines. Anticancer Res. 2010;30(3):915-921
99. Travis LB. The epidemiology of second primary cancers. Cancer Epidemiol Biomarkers Prev. 2006;15(11):2020-2026
100. Landgren O, Mailankody S. Update on second primary malignancies in multiple myeloma: a focused review. Leukemia. 2014;28(7):1423-1426
101. Travis LB, Ng AK, Allan JM, Pui CH, Kennedy AR, Xu XG. et al. Second malignant neoplasms and cardiovascular disease following radiotherapy. Health Phys. 2014;106(2):229-246
102. Coffey JC, Wang JH, Smith MJ, Bouchier-Hayes D, Cotter TG, Redmond HP. Excisional surgery for cancer cure: therapy at a cost. Lancet Oncol. 2003;4(12):760-768
103. Sun Y, Campisi J, Higano C, Beer TM, Porter P, Coleman I. et al. Treatment-induced damage to the tumor microenvironment promotes prostate cancer therapy resistance through WNT16B. Nat Med. 2012;18(9):1359-1368
104. von Essen CF. Radiation enhancement of metastasis: a review. Clin Exp Metastasis. 1991;9(2):77-104
105. Murphy ED, Kaplan HS. Enhancement of metastasis of a mouse mammary carcinoma following roentgen irradiation. Am J Pathol. 1948;24(3):673
106. Kaplan HS, Murphy ED. The effect of local roentgen irradiation on the biological behavior of a transplantable mouse carcinoma; increased frequency of pulmonary metastasis. J Natl Cancer Inst. 1949;9(5-6):407-413
107. Brown JM, Marsa GW. Effect of dose fractionation on the enhancement by radiation or cyclophosphamide of artificial pulmonary metastases. Br J Cancer. 1978;37(6):1020-1025
108. Ruegg C, Monnier Y, Kuonen F, Imaizumi N. Radiation-induced modifications of the tumor microenvironment promote metastasis. Bull Cancer. 2011;98(6):47-57
109. Kuonen F, Secondini C, Ruegg C. Molecular pathways: emerging pathways mediating growth, invasion, and metastasis of tumors progressing in an irradiated microenvironment. Clin Cancer Res. 2012;18(19):5196-5202
110. van den Brenk HA, Crowe MC, Stone MG. Reactions of the tumour bed to lethally irradiated tumour cells, and the Revesz effect. Br J Cancer. 1977;36(1):94-104
111. Yamauchi K, Yang M, Hayashi K, Jiang P, Yamamoto N, Tsuchiya H. et al. Induction of cancer metastasis by cyclophosphamide pretreatment of host mice: an opposite effect of chemotherapy. Cancer Res. 2008;68(2):516-520
112. Liu B, Ezeogu L, Zellmer L, Yu B, Xu N, Liao DJ. Protecting the normal in order to better kill the cancer. Cancer Med. 2015;4(9):1394-1403
113. Aktipis CA, Boddy AM, Gatenby RA, Brown JS, Maley CC. Life history trade-offs in cancer evolution. Nat Rev Cancer. 2013;13(12):883-892
114. Basanta D, Anderson AR. Exploiting ecological principles to better understand cancer progression and treatment. Interface Focus. 2013;3(4):20130020-doi 10.1098/rsfs.2013.0020
115. Sprouffske K, Merlo LM, Gerrish PJ, Maley CC, Sniegowski PD. Cancer in light of experimental evolution. Curr Biol. 2012;22(17):R762-R771
Author contact
![]() Corresponding authors: Gangshi Wang, Department of Geriatric Gastroenterology, Chinese PLA General Hospital, Beijing 100853 P.R. China. Email: wanggangshicom Or Ningzhi Xu, Laboratory of Cell and Molecular Biology, Cancer Institute, Chinese Academy of Medical Science, Beijing 100021 P.R. China. Email: xuningzhiac.cn Or D. Joshua Liao, Clinical Research Center, Guizhou Medical University Hospital, Guiyang, Guizhou 550004 P.R. China. Email: djliaoedu.cn.
Corresponding authors: Gangshi Wang, Department of Geriatric Gastroenterology, Chinese PLA General Hospital, Beijing 100853 P.R. China. Email: wanggangshicom Or Ningzhi Xu, Laboratory of Cell and Molecular Biology, Cancer Institute, Chinese Academy of Medical Science, Beijing 100021 P.R. China. Email: xuningzhiac.cn Or D. Joshua Liao, Clinical Research Center, Guizhou Medical University Hospital, Guiyang, Guizhou 550004 P.R. China. Email: djliaoedu.cn.