Impact Factor ISSN: 1837-9664

 Global reach, higher impact
Global reach, higher impactJ Cancer 2016; 7(7):817-822. doi:10.7150/jca.14274 This issue Cite
Review
Warburg Effect - a Consequence or the Cause of Carcinogenesis?
Slobodan Devic ![]()
Department of Radiation Oncology, SMBD Jewish General Hospital, McGill University, Montréal, Québec, Canada.
Received 2015-10-30; Accepted 2016-3-13; Published 2016-4-26
Abstract
Ever since its discovery (1924) the Warburg effect (aerobic glycolysis) remains an unresolved puzzle: why the aggressive cancer cells “prefer” to use the energetically highly inefficient method of burning the glucose at the cellular level? While in the course of the last 90 years several hypotheses have been suggested, to this date there is no clear explanation of this rather unusual effect. Even though it is commonly assumed that Warburg effect is a consequence of carcinogenesis, yet another hypothesis could be brought up that the cellular switch to aerobic glycolysis may represent the very point in time when a normal cell becomes cancerous. Furthermore, this switch may happen at the point where the fate of pyruvic acid is determined, caused by the inadequate supply of enzymes that promote citric as opposed to lactic acid cycle. Currently, few clinical observations, like low cancer incidence in Type 1 diabetes mellitus and increased cancer incidence in people on high carbohydrate diets might be called upon to support such hypothesis.
Keywords: Warburg effect, cancer
Economy of cellular energy balance
The full oxidation of one glucose molecule (oxidative phosphorylation) within a cell in the presence of oxygen produces 38 molecules of adenosine-three-phosphate (ATP), which in turn represents the essential cellular fuel (Figure 1.a). The first step (Glycolysis, occurring in cytoplasm) of glucose cellular respiration produces only 2 ATP molecules and ends up with production of two molecules of Pyruvic acid. If a given cell has access to oxygen, the Pyruvic acid will be converted to Acetyl-coenzyme A, which enters the Krebs cycle (citric acid cycle, occurring within mitochondria) followed by the electron transport chain process (occurring on the inner mitochondrial membrane) that creates most of the ATP molecules. It is the very last step within electron transport chain that needs oxygen to collect the terminal electron from the last cytochrome (cyt a3) and become a nascent O- to pick up 2 H+ and create one of the byproducts of aerobic cellular respiration - a water molecule. On the other hand, if the cells reside under hypoxic conditions, the pyruvic acid is not converted into acetyl-coenzyme A, but into a lactic acid - the process termed anaerobic cellular respiration (lactic acid cycle). In the latter case, the net energy balance is only two ATP molecules making the anaerobic glucose metabolism energetically highly inefficient process.
Glucose metabolism at the cellular level: a) full glucose oxidative phosphorylation; b) anaerobic glycolysis (lactic acid cycle).
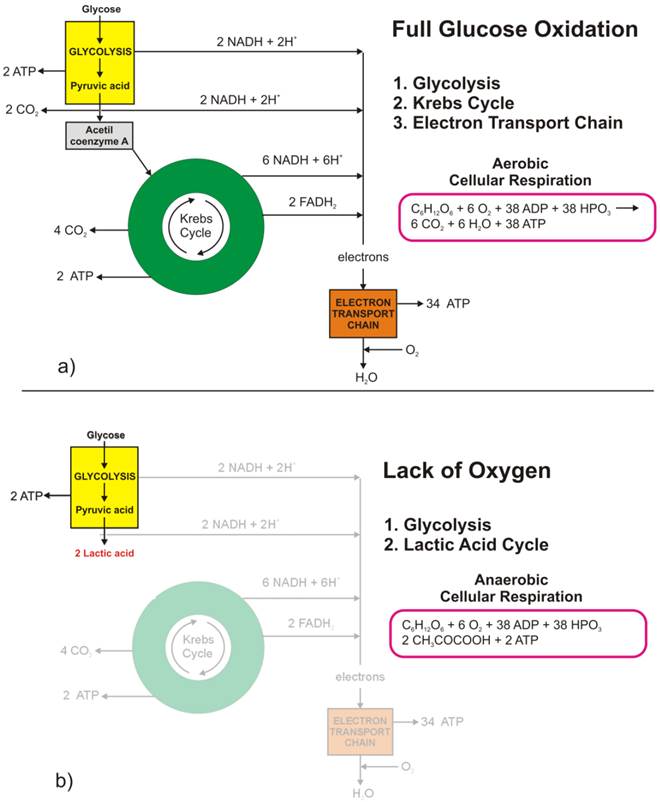
While the lack of oxygen at the cellular level can occur at times of excessive physical activity (resulting in subsequent muscle pain), it also represents a hallmark of highly invasive and fast-growing cancers [[1]]. As the cancer cells multiply, their fast multiplication rate outgrows the angiogenesis so much so that while the glucose access to fast-growing cells could be sufficient the lack of blood vessels disrupts the level of oxygen needed for the full glucose oxidation. Such a microscopic picture defines one of the fundamental properties of the cancer microenvironment that forces malignant cells to metabolize glucose through the lactic acid cycle.
Looking at chemical equations between aerobic and anaerobic glucose metabolism (Figure 1) one may conclude that cancer cells (as well as healthy ones) under hypoxic condition would need 19 times higher uptake of glucose to maintain the same metabolic level as well-oxygenated cells. This would, in turn, mean that the appropriate quantitative analysis of FDG-based PET images could help in pinpointing hypoxic segments (by abundance only [[2]]) of the tumor by solely looking into very high uptake values, which would eventually be some 20 times greater than in well-oxygenated cancer cells. However, the isolation of the hypoxic target volumes is far from being that simple.
Warburg effect, or aerobic glycolysis - hallmark of invasive cancers
Apart from the fact that acute hypoxia in tumors develop as soon as one moves few hundred microns from the blood vessels, yet another important fact prevents FDG being an ideal hypoxia marker - the Warburg effect. Recently, interest in tumor metabolism has been revived partly as a result of the widespread clinical application of PET using FDG. FDG-based PET imaging has confirmed that most primary and metastatic cancers show a significant increase in the glucose uptake when compared to normal tissues.
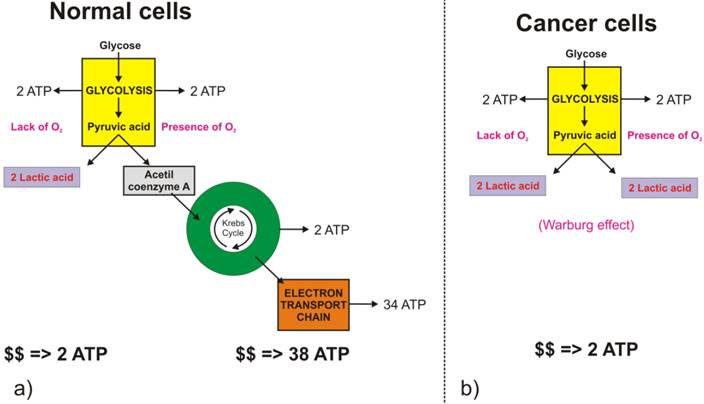
Glycolysis involves the conversion of glucose to pyruvate and then to lactic acid, the waste product. In non-cancerous cells, mitochondria oxidize pyruvate to carbon dioxide and water in the presence of oxygen (Figure 2.a), and the glycolytic reaction is inhibited (Pasteur Effect [[3]]). Conversion of glucose to lactic acid, even in the presence of oxygen is known as aerobic glycolysis (Figure 2.b) or the Warburg effect [[4], [5]]. In one of his seminal papers [[6]], Warburg suggests that carcinogenesis is a two-step process. Cancer cells originate from normal cells by firstly encountering irreversible respiration injury. The second phase of cancer formation represents a long struggle for existence by the injured cells to maintain their structure, in which a part of the cells die from lack of energy while another part succeeds in replacing the irretrievably lost respiration energy by fermentation energy (from lactic acid cycle). Warburg's initial hypothesis that cancer results from impaired mitochondrial metabolism has been shown to be incorrect, but the observation of augmented glycolysis in tumors, even in the presence of oxygen, has been continually proven [[7]]. While cancer cells do carry oxidative phosphorylation, the majority of glucose molecules taken by cancer cells (66%) are metabolized through fermentation [[8]], a process that is ten times faster than full glucose oxidation.
Difference in glucose metabolic pathways as a function of oxygen abundance in a) normal cells, and b) cancer cells.
Contemporary explanation of the Warburg effect
In addition to being energetically highly inefficient process glycolysis (either anaerobic or aerobic), with its metabolic products (such as hydrogen ions), cause constant acidification of the extracellular space, which might result in increased local toxicity [[9], [10]]. Nevertheless, despite these drawbacks, cancer cells consistently progress towards the wasteful and potentially toxic glycolytic phenotype. Gatenby and Gillies [[11]] proposed that the consistent expression of up-regulated glycolysis is not accidental but represents a solution to the environmental growth constraints during tumor development. They suggest that increased glycolysis is an essential component of the malignant phenotype and, therefore, a hallmark of invasive cancers. Transport enzymes of the Glut and hexokinase families are up-regulated in tumor cells expressing the glycolytic phenotype, and the level of Glut-1 glucose transporter expression has been shown to correlate with [18F] FDG uptake in non-small cell lung cancer, for example [[12]].
Gatenby and Gillies [11] describe the concept of carcinogenesis as a process that occurs by cellular evolution implying that common characteristics of malignant phenotypes, such as upregulation of glycolysis, are the result of active selection processes. They further argue that upregulation of glycolysis is likely to be an adjustment to hypoxia developing as pre-malignant tissue grows gradually further away from their blood supply. Also, an augmented acid production from glycolysis upregulation leads to microenvironmental acidosis and requires further adjustments through somatic evolution to phenotypes resistant to acid-induced toxicity. Finally, they conclude that cell populations that emerge from this evolutionary sequence have a compelling growth advantage, as they alter their environment through increased glycolysis in a way that is toxic to other phenotypes, but harmless to themselves.
To further support the attempt by Gatenby and Gillies [11] in explaining the cause of Warburg effect in aggressive tumors as a response to harsh environmental conditions, one might assume that cancer cells “know” a priori they will encounter severe conditions in the future. Consequently, they “decide” to switch their glucose metabolism to highly inefficient but the only possible (and highly toxic) metabolic pathway. To make their explanation more sounded, Gatenby and Gillies [11] speculate that: “… intuitively, it would seem that the Darwinian forces prevailing during the somatic evolution of invasive cancers would select against a metabolic phenotype that is more than an order of magnitude less efficient than its competitors and that is environmentally poisonous. In other words, the accepted tenet of “survival of the fittest” would seem to generally favour populations with more efficient and sophisticated substrate metabolism.” Consequently, they suggest that glycolytic phenotype in cancers is directly governed by the evolutive mechanisms, over the relatively short time frame during which tumors develop.
A new hypothesis
Figure 3 illustrates the very point within a cellular glucose metabolism where the fate of the Pyruvic acid is decided. In the presence of oxygen, the Pyruvic acid will be converted into acetyl group and attached to coenzyme A, a process mediated by the so-called Pyruvate Dehydrogenase Complex (PDC). The complex consists of three enzymes: pyruvate dehydrogenase, dihydrolipoyl transacetylase, and dihydrolipoyl dehydrogenase. On the other hand, conversion of pyruvic acid to lactic acid requires one enzyme only - lactate dehydrogenase (LDHA). From the cellular kinetics point of view, one may ask a simple question: what is the probability that something goes wrong with either of the two possible metabolic pathways depicted in Figure 3?
Enzyme-mediated cross-section between lactic and citric acid cycle.
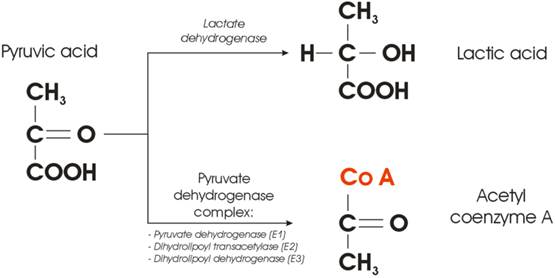
Synthesis of three enzymes, needed to provide conversion of pyruvic acid to acetyl coenzyme A, even in the presence of oxygen will be (notably three times) more prone to the errors than a transcription of only one (competing) enzyme. It was reported that many human cancers have higher LDHA levels than normal tissues [[13]], but the correlation between oncogenes and glycolysis was poorly understood. The question could be then raised whether the Warburg effect is only a consequence, or could it be at the root of the very cause of carcinogenesis? A new hypothesis could be staged that the switch from aerobic cellular respiration to aerobic glycolysis leads to carcinogenesis, and the cell begins to develop the cancerous phenotype [[14], [15]] at the point where the fate of Pyruvic acid is decided. Such a switch could be governed by the lack of complete PDC (at least one of the enzymes is missing).
While at this very moment the question whether Warburg effect is a hen, or an egg remains at the level of pure hypothesis, several clinical observations might be called upon to support such proposition indirectly. While there is no apparent relation between Type 1 diabetes mellitus patients and incidence of cancer to this date [[16]], several publications argue that there could be a lower cancer rate in patients with insulin-dependent diabetes. In 2003, Zendehdel et al. [[17]] published results on cancer incidence in patients with Type 1 (insulin-dependent) diabetes mellitus on a cohort of 29 187 patients, followed over a period of 30 years, during which they observed 355 incidences of cancer. Such a low frequency (1% over 30 years, or 0.04% per year) appears negligible when compared to 1.66 million cases of new cancer cases per year in the US (0.52% per year [[18]]). Pladys et al. [[19]] reported on the lower incidence of cancer death mortality in diabetic patients (6.7%, both Type 1 and 2) when compared to non-diabetic patients (13.4%) using a cohort of 39 811 patients with the end-stage renal disease. It might be argued that cells in diabetic patients (generally deprived of normal glucose uptake due to lacking insulin) become “trained” (to use rhetoric by Blagosklonny [[20]]) by the microenvironment and well “prepared” as soon as glucose becomes available. Once the glucose is phosphorylated by hexokinase and enters the glucose oxidation process, the cell is “prepared” not to waste the opportunity and gets the maximum out of the relatively “scars glucose supplies.” One could further argue that diabetic patient cells are making sure that the synthesis of the PDC is up and running flawlessly, to avoid wasteful pathway of cellular glucose metabolism.
On the other hand, despite a relatively small amount of data published it appears that the incidence of cancer is also correlated with the increased intake of carbohydrates [[21], [22], [23]]. One may argue that normal cells, exposed to increased supply of glucose would quickly switch towards the energetically inefficient pathway (lactic acid cycle) of burning glucose even in the presence of oxygen (Warburg effect) since the source of energy (glucose-ATP) are virtually inexhaustible. In addition, the ATP production via fermentation is much faster (as mentioned above), albeit highly ineficient, when compared to full oxydation. It is also of note that contrary to type 1 (insulin-dependent), patients with type 2 diabetes mellitus have higher probability for cancer incidence [[24]]. Yet another detail deserves attention: type 1 diabetes is commonly regarded as a juvenile-onset diabetes as it often begins in childhood while the type 2 diabetes was considered an adult-onset diabetes. However, type 2 diabetes is becoming increasingly common in children [[25]] who are more obese or overweight that could be correlated with carbohydrates rich diets. Finally, the possible triggering of carcinogenesis by aerobic glycolysis, accompanied by increased glucose uptake, can be further supported by studies demonstrating increased glucose uptake observed to coincide with the transition from premalignant lesions to invasive cancer [[26], [27]].
Conclusion
Unlike Warburg's initial hypothesis that cancer cells metabolize glucose through aerobic glycolysis due to impaired mitochondrial function a new hypothesis was presented that the normal cell becomes cancerous at the point when it switches its glucose metabolism from oxidative phosphorylation to aerobic glycolysis. The new hypothesis that Warburg effect corresponds to the very beginning of carcinogenesis might be supported by the eventual failure in synthesis of the PDC. Such failure could be mediated by one (or multiple) of the well-known carcinogenic factors in synergy with an excessive supply of glucose in carbohydrates rich diets.
The new hypothesis revolves around a point within cellular glucose oxidation at which the fate of pyruvic acid is decided. Several observations have been presented to support such hypothesis: lower incidence of cancer in insulin-dependent type 1 diabetes mellitus patients; increased cancer incidence in societies consuming high quantities of carbohydrates; and the increased probability of synthesis failure of the pyruvate dehydrogenase complex consisting of three enzymes when compared to the synthesis of a single lactate dehydrogenase enzyme.
Contemporary agreement in explaining why tumor cells opt for aerobic glycolysis (Warburg effect) that is far less efficient than oxidative phosphorylation at producing ATP, is that it represents evolutionary adaptation to harsh microenvironmental conditions by using the carbon chains (from the lactic acid) as building blocks for synthesis of biomolecules (nucleic acids, proteins, and lipids), which are essential for cell proliferation. However, even the acceptance of the aerobic glycolysis being a more adequate glucose metabolism pathway for cancer cells, the question of a hen or an egg remains: is the Warburg effect just a consequence, or could it be the very cause of carcinogenesis (Figure 4)?
Warburg effect: a hen, or an egg?

Acknowledgements
The author would like to thank Professor Shirley Lehnert, dr Remi Nout and dr Slawomir Kumala for useful discussions. S.D. is a Research Scientist supported by the Fonds de Recherche en Santé du Québec (FRSQ) 26856. The work was supported in part by the Natural Sciences and Engineering Research Council of Canada contract No. 386009.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Hsu PP, Sabatini DM. Cancer cell metabolism: Warburg and beyond. CELL. 2008:134 703-707
2. Devic S, Faria S, Lehnert S. Reply on the Letter to the Editor on “Defining Radiotherapy Target Volumes Using 18F-Fluoro-Deoxy-Glucose Positron Emission Tomography/Computed Tomography: Still a Pandora's Box?” (Devic et al. Int J Radiat Oncol Biol Phys 2010; 78: 1555-1562). Intl J Radiat Oncol Biol Phys. 2011;81:902-903
3. Racker E. History of the Pasteur effect and its pathobiology. Mol Cell Biochem. 1974;5:17-23
4. Warburg O, Posener K, Negelein E. Über den Stoffwechsel der Carcinomzelle. Biochem Zeitschr. 1924;152:309-344
5. Warburg O. Ueber den stoffwechsel der tumoren. London, UK: Constable. 1930
6. Warburg O. On the Origin of Cancer Cells. Science. 1956;123:309-314
7. Pauwels EKJ, Ribeiro MJ, Stoot JH, McCready VR, Bourguignon M, Mazière B. FDG accumulation and tumor biology. Nucl Med Biol. 1998;25:317-322
8. Koppenol WH, Bounds PL, Dang CV. Otto Warburg's contributions to current concepts of cancer metabolism. Nat. Rev. Cancer. 2011;11(5):325-337
9. Pugachev A, Ruan S, Carlin S, Larson SM, Campa J, Ling CC. et al. Dependence of FDG uptake on tumor microenvironment. Int J Radiat Oncol Biol Phys. 2005;62:545-53
10. Smallbone K, Gatenby RA, Gillies RJ, Maini PK, Gavaghan DJ. Metabolic changes during carcinogenesis: Potential impact on invasiveness. J Theor Biol. 2007;244:703-713
11. Gatenby RA, Gillies RJ. Why do cancers have high aerobic glycolysis? Nat Rev Canc. 2004;4:891-899
12. Higashi K, Ueda Y, Sakurai A, Wang XM, Xu L, Murakami M. et al. Correlation of Glut-1 glucose transporter expression with [F-18]FDG uptake in non-small cell lung cancer. Europ J Nucl Med. 2000;27:1778-1785
13. Goldman RD, Kaplan NO, Hall TC. Lactic dehydrogenase in human neoplastic tissues. Canc Res. 1964;24:389-399
14. Christofk HR, Vander Heiden MG, Harris MH, Ramanathan A, Gerszten RE, Wei R. et al. The M2 splice isoform of pyruvate kinase is important for cancer metabolism and tumour growth. Nature. 2008;452:230-233
15. Pedersen PL. Warburg, me and hexokinase 2: Multiple discoveries of key molecular events underlying one of cancers' most common phenotypes, the "Warburg Effect", i.e, elevated glycolysis in the presence of oxygen. J Bioenerg Biomembr. 2007;39:211-222
16. Shelton N, Mindell JS. Epidemiological evidence of a relationship between type-1 diabetes mellitus and cancer: a review of the existing literature. Int J Canc. 2013;132:501-508
17. Zendehdel K, Nyrén O, Ostenson CG, Adami HO, Ekbom A, Ye W. Cancer Incidence in Patients With Type 1 Diabetes Mellitus: A Population-Based Cohort Study in Sweden. J Nat Canc Inst. 2003;95:1797-1800
18. Rebecca LS, Kimberly DM, Ahmedin J. Cancer Statistics 2015. CA Canc J Clin. 2015;65:5-29
19. Pladys A, Couchoud C, LeGuillou A, Siebert M, Vigneau C, Bayat S. Type 1 and Type 2 Diabetes and Cancer Mortality in the 2002-2009 Cohort of 39 811 French Dialyzed Patients. PLoS One. 2015;10:e0125089
20. Blagosklonny MV. Cell Immortality and Hallmarks of Cancer. Cell Cycle. 2003;2:296-299
21. Ho VW, Leung K, Hsu A, Luk B, Lai J, Shen SY. A Low Carbohydrate, High Protein Diet Slows Tumor Growth and Prevents Cancer Initiation. Canc Res. 2011;71:4484-4493
22. Klement RJ, Kammerer U. Is there a role for carbohydrate restriction in the treatment and prevention of cancer? Nutr Metabol. 2011;8:75
23. Romieu I, Lajous M. The role of obesity, physical activity and dietary factors on the risk for breast cancer: Mexican experience. Salud Pública de Mexico. 2009;51:S172-S180
24. Giovannucci E, Harlan DM, Archer MC, Bergenstal RM, Gapstur SM, Habel LA. et al. Diabetes and Cancer: A Consensus Report. CA-A Canc J Clin. 2010;60:207-221
25. Sinha R, Fisch G, Teague B, Tamborlane WV, Banyas B, Allen K. et al. Prevalence of impaired glucose tolerance among children and adolescents with marked obesity. N Engl J Med. 2002;346:802-810
26. Younes M, Lechago LV, Lechago J. Overexpression of the human erythrocyte glucose transporter occurs as a late event in human colorectal carcinogenesis and is associated with an increased incidence of lymph node metastases. Clin Canc Res. 1996;2:1151-1154
27. Yasuda S, Fujii H, Nakahara T, Nishiumi N, Takahashi W, Ide M. et al. 18F-FDG PET detection of colonic adenomas. J Nucl Med. 2001;42:989-992
Author contact
![]() Corresponding author: Slobodan Devic, Medical Physics Department, McGill University, Jewish General Hospital, 3755 Cote St-Catherine, Montreal, Québec H3T 1E2, Tel: 514-340-8222/2595 Fax: 514- 340-8642.
Corresponding author: Slobodan Devic, Medical Physics Department, McGill University, Jewish General Hospital, 3755 Cote St-Catherine, Montreal, Québec H3T 1E2, Tel: 514-340-8222/2595 Fax: 514- 340-8642.