Impact Factor ISSN: 1837-9664

 Global reach, higher impact
Global reach, higher impactJ Cancer 2016; 7(8):1010-1019. doi:10.7150/jca.14425 This issue Cite
Research Paper
An Investigation on a Novel Anti-tumor Fusion Peptide of FSH33-53-IIKK
Runlin Yang1, Ping Liu2, Donghui Pan1, Pengjun zhang2, Zhicheng Bai3, Yuping Xu1, Lizhen Wang1, Junjie Yan1, Yongjun Yan4, Xingdang Liu5, ![]() , Min Yang1,2,3,
, Min Yang1,2,3, ![]()
1. Key Laboratory of Nuclear Medicine, Ministry of Health, Jiangsu Key Laboratory of Molecular Nuclear Medicine, Jiangsu Institute of Nuclear Medicine, Wuxi 214063, China.
2. School of Pharmaceutical Science, Zhengzhou University, Zhengzhou 450000, China.
3. The First School of Clinical Medicine, Nanjing Medical University, Nanjing 210029, China.
4. Department of Medical Physics, University of Wisconsin, Madison, WI 53705, USA.
5. Department of Nuclear Medicine, Hua Shan Hospital, Fudan University, Shanghai 200040, China.
Received 2015-11-15; Accepted 2016-3-15; Published 2016-5-24
Abstract
A novel fusion peptide FSH33-53-IIKK was designed and expected to combine the follicle stimulating hormone receptor (FSHR) targeting and tumor toxicity. In vitro and in vivo study showed the anti-tumor activity of FSH33-53-IIKK was enhanced compared to that of IIKK only. FSH33-53-IIKK could inhibit the growth of tumor via apoptosis and autophagy pathways. In summary, combining the tumor marker-target peptide and anti-tumor peptide together may be an efficient way to search for better anti-tumor candidates.
Keywords: FSH33-53-IIKK, Fusion peptide, FSHR, Anti-tumor, Apoptosis, Autophagy
Introduction
Tumor markers on the cell membrane of tumors are the key targets for tumor imaging and therapy. The follicle stimulating hormone receptor, which binding to the FSH and belonging to the family of G-protein coupled membrane receptor, has been considered as a potential target for prostate cancer [1].
Under physiological condition, FSHR exists mainly in the sertoli cells of testis [2] and the granulosa cells of ovary [3]. Also it can be detected in ovarian epithelial cells [4], endometrium [5], placental endothelial [6], endothelium of ovaries [7] and testis [8]. FSHR commences and maintains spermatogenesis and development of ovotid in each stage via classical cAMP-PKA signal pathway upon binding FSH. However, FSHR is also expressed in the vascular endothelium of various human solid tumors such as prostate, breast, colorectal, lung, testis, and even in that of most of metastatic tumors (79% of 209 examined patients) [9]. Moreover, FSHR has been expressed in Caov-3, OVCAR-3, PC3 cancer cells constitutively [10, 11]. In addition, FSHR demonstrates expression in endothelial cells of the tumor vasculature in 11 subtypes of soft tissue sarcomas and in tumor cells of all examined sarcoma subtypes, except for well-differentiated sarcoma [12]. Further, FSHR also increases the proliferation of cancer cells and overexpressed FSHR may enhance the potential oncogenic pathway [13-16]. Therefore, FSHR, closely related with tumor growth, invasion and metastasis, becomes an important target for clinical imaging and theranostics.
As an endogenous target ligand of FSHR, FSH is a glycoprotein and composed of α and β chains. Amino acids sequence 33-53 and 81-95 of the β chain have been identified as FSHR-binding domains, and the former has the highest binding affinity to its receptor [17, 18]. Based on that, FSH33-53 (YTRDLVYKDPARPKIQKTCTF) has been employed as a ligand targeting to FSHR recently. PET probe 18F-Al-NOTA-MAL-FSH33-53 targeting to FSHR specifically is prepared and succeeded in microPET imaging for FSHR overexpressed human prostate cancer cell, PC3 cells, in xenograft model [19], which provides the basis for the specific clinical diagnosis of prostate cancer.
For avoiding the side effects, such as toxicity from nonspecific distribution of conventional chemotherapeutic drugs [20, 21] to normal cells, FSH33-53 has been used in drug delivery systems in therapeutic research for FSHR-expressing tumors. FSH33-53-NP-PTX, FSH33-53 connected with PTX (Paclitaxel) through nanoparticles, improves the targeting ability and treatment of PTX to CAOV-3 tumor cells [10]. Fluorescein-labeled generation 5 (G5) PAMAM dendrimers that are conjugated with FSH33-53 significantly enhances the uptake of dendrimers and down-regulation of Survivin, an anti-apoptotic protein, in FSHR-expressing OVCAR-3 cells, but not in SKOV-3 cells which don't express FSHR [22]. Also, FSH33-G-NP, FSHβ33-53 peptide-conjugated gro-α siRNA-loaded PEG-PEI nanoparticles, significantly silences the expression of oncogene gro-α in ovarian cancer cell, ES-2 and consequently suppresses the proliferation, migration and invasion of the cell greatly in comparison to G-NP alone [23].
Despite the enhanced permeation retention (EPR) effect [24], the limitations of the NPs, such as limited diffusivity [25] and the instability of nucleic acid in vivo restrict the development of the delivery systems.
Small molecular peptides, existing widely in nature, attract attention of researchers for theirs low toxicity to normal tissue [26]. Cationic antimicrobial peptides are special peptides in anti-tumor peptides screening and they show toxicity to both virus [27] and cancer cells [28-30]. Lu, et al, have imitated the structural characteristics of natural antimicrobial peptides and have developed a series of short cationic peptides, G(IIKK)nI-NH2 (n=1-4) [31]. This series of peptides, with the same effects as to other natural cationic antimicrobial peptides, are capable of binding and penetrating into the negatively charged membrane of bacteria, but they do not destroy the normal mammalian cell membrane with zwitterionic structure [31-34]. Moreover, G(IIKK)nI-NH2 show strong anticancer activities and good inhibition ability for tumors in tumor bearing mice without causing specific immune response. G(IIKK)3I-NH2, named IIKK in this paper, shows the best effects on anticancer.
In this paper, we designed a novel targeting-therapeutic fusion peptide FSH33-53-IIKK and tried to use FSH33-53 subunit as a potential delivery peptide targeting to FSHR-expressing tumor cells and IIKK as an inhibiting part for tumor. By this way, the novel fusion peptide with high efficiency and low toxicity to normal tissue was expected to increase the therapeutic ability of IIKK to FSHR-expressing tumors. We also evaluated its mechanism for anti-tumor activity in vitro and in vivo.
Materials and Methods
Materials
DDP was purchased from hansoh pharmaceutical, Jiangsu. MTT, DMSO, Hoechst 33258 solution, BCA protein kit, PI, AO and ECL system (BeyoECL plus) were purchased from Beyotime Biotechnology, China. Rabbit anti-FSHR antibody was obtained from Abcam, USA. Rabbit anti-Bax, Bcl-2, Caspase-3, Caspase-8, Bad, LC3 and mouse anti-β-Actin were purchased from Beyotime Biotechnology, China. Annexin V-FITC / PI double staining apoptosis kit was from Roche, Germany and Novex NuPAGE Gel Electrophoresis Systems was from Life Technologies, USA. And male BALB/c nude mice were purchased from Cavens Lab Animal, Changzhou, China.
Preparation of peptides
Peptide IIKK (G(IIKK)3I-NH2) and FSH33-53-IIKK (YTRDLVYKDPARPKIQKTCTFGGG(IIKK)3I-NH2) were synthesized and purified by Apeptide Co., Ltd (Shanghai, China).
Methods
Cell culture
Human carcinoma cell lines PC3, SKOV3, MCF7 and HeLa, human embryo kidney cell 293, and mouse fibroblast cell L929 were purchased from the Cell Bank in Shanghai Institute of Cell Biology, China, and were cultured according to the guides of the Cell Bank.
Western Blot
Cells were cultured in 6 cm-dishes. After reaching to 80-90% confluence, cells were washed with ice-cold PBS for 3 times and lysed in RIPA lysis buffer with 1 mM PMSF on ice. Discarding the debris, and the supernatant was transferred to a new tube after centrifugation at 14,000 rpm for 5 min at 4 ℃. Protein concentrations were determined by BCA protein assay kit. Then the same amount of lysates with loading buffer were heated for 10 min at 70 ℃ according to the user guides from Invitrogen. After running on 10 % SDS-PAGE, proteins were transferred to PVDF membranes, which were subsequently blocked with 5% non-fat milk buffer for 1 h. And then membranes were incubated with each primary antibody over night at 4 ℃. Following that, the membranes were then blotted with HRP-labeled secondary antibodies for 1 h at room temperature. The blotting patterns were detected using ECL system and analyzed on LAS-3000 lumino image analyzer.
Cell cytotoxicity assay
MTT assay was carried out to measure the cytotoxicity of peptides in vitro. PC3, HeLa, SKOV3, MCF7, L929 and 293 cell lines were seeded respectively into 96-well plates with a density of 5 × 103 cells. After 24 h incubation, the cells were then treated with 0 ~ 100 μM of FSH33-53-IIKK, IIKK or DDP for 24 h. Then 20 μL MTT (5 mg/ml dissolved in PBS) was added to each well. And cells were incubated for 4 h. Subsequently, the supernatants were removed and the crystals were dissolved completely with 150 μL DMSO. Microplate reader (BioTek, uQuant, USA) was used to record the absorbance at 570 nm. The wells without cells were used as blank. IC50 was calculated. Experiments were repeated 3 times. And data were reported as means ± SD. Statistical differences between the two groups were evaluated using the paired Student t-test.
Cell staining and fluorescence microscopy
PC3 cells were seeded into 6-well plate with 5 × 105 per well and incubated for 24 h. Increasing concentration of FSH33-53-IIKK (0, 7.5, 15, 30 μM) treated the cells for another 24 h. After discarding medium, cells were washed with PBS for 2 times, followed by treated with Hoechst 33258 solution for apoptosis staining or AO for autophagy. At last, cells were washed with PBS to remove unbound dyes and observed by fluorescence microscope.
Flow cytometry
Incubated PC3 cells in 6-well plate were treated with FSH33-53-IIKK (0, 7.5, 15, 30 μM) or IIKK (0, 7.5, 15, 30 μM) for 24 h. Cells floating in the medium and adherent cells were harvested and centrifuged at 1,500 rpm for 4 min. The supernatants were discarded and the precipitations were washed with PBS. After adding 100 μL Annexin V-FITC / PI double staining reagent in binding buffer for apoptosis, cells were re-suspended and cultured for 15 min in dark. The effects were evaluated with flow cytometry. Experiments were repeated 3 times. And data were reported as means ± SD. Statistical differences between the two groups were evaluated using the paired Student t-test.
Inhibition of tumor growth in human PC3 cell xenografts
All animal experiments were approved by the local animal welfare committee and performed according to national regulations. 3-4 week-old male BALB/c nude mice were injected subcutaneously into the right front armpit with PC3 cells (1 × 107 each) in PBS. Vernier caliper was used for measurement of tumor length (L, the longest diameter) and width (W, the shortest diameter perpendicular to length). When the volume of tumors reached a mean size of 80 ~ 100 mm3, the mice were assigned to 5 groups. Each group contained 5 mice with similar average tumor volume and was administered respectively to with FSH33-53-IIKK (5 mg/kg and 10 mg/kg), IIKK (5 mg/kg), DDP (3mg/kg) as positive control and saline as negative control by tail intravenous injection 3 times a week for 3 weeks. During the therapy, the tumor volume was measured every 2 days with the same as to the body weight of each. Then the mice were sacrificed for removing, photographing, and weighing the tumors. Some formulas were used for calculating the tumors as follows.
The volume (V) of each tumor: V = L × W2 /2
Relative tumor volume (RTV): RTV = Vt / V0
V0 represents the tumor volume at the beginning of treatment and Vt does for the day t of treatment.
Tumor inhibition rate: Tumor inhibition rate=((average weight of saline control - average weight of treatment group) / average weight of saline control) × 100 %
Data were expressed as means ± SD.
Statistical analysis
For all data are represented as means ± SD. Student's t-test was used to analyze the difference between two groups. GraphPad Prism 5.0 (GraphPad Software) was used for all statistical analysis.
Results
Expression of FSHR in several cells
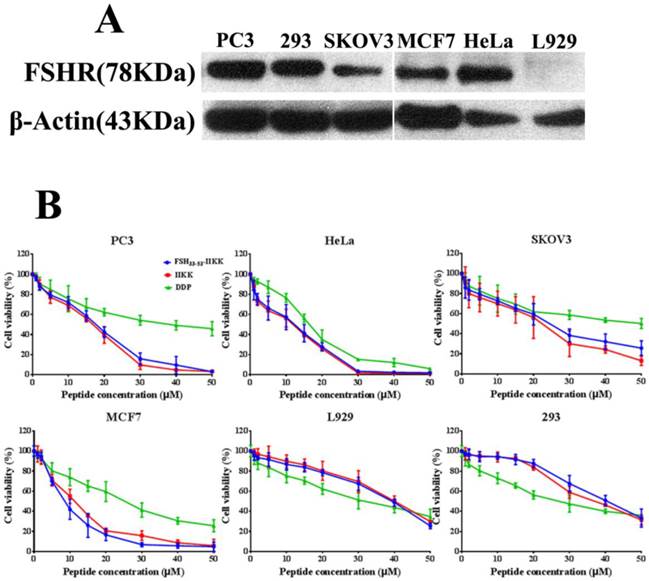
At first, Western Blot was employed to identify the level of FSHR in several cell lines. As shown in Fig.1A, human prostate cancer cell PC3, human cervical cancer cell HeLa, human breast cancer cell MCF7, and human embryo kidney cell 293 all expressed FSHR highly. But the expression in human ovarian cancer cell SKOV3 was lower than them. In mouse fibroblast cell L929, FSHR level was not identified.
The cytotoxicity of the peptides on FSHR expressed or unexpressed cells (A) Western Blot analysis of FSHR expression in some cells. FSHR has overexpressed in PC3, HeLa, MCF7, 293 cells. But low expression level in SKOV3. In L929, FSHR was not identified. (B) All cells were treated with peptides or DDP for 24 h, and the proliferation of the cells were assessed by MTT method.
Cell cytotoxicity in vitro
Then the cytotoxicity of FSH33-53-IIKK, IIKK or DDP to HeLa, PC3, SKOV3, MCF7, 293 and L929 cells were determined by MTT method. Fig.1B showed that FSH33-53-IIKK, IIKK and DDP had inhibitory effects on tumor cell proliferation significantly in a dose-dependent manner. FSH33-53-IIKK and IIKK both have shown stronger effect than DDP, but there was no significant difference between them in FSHR expressed or unexpressed cells. The data also appeared that the toxicity of FSH33-53-IIKK, IIKK on normal cell were lower than that of DDP. IC50 of FSH33-53-IIKK, IIKK and DDP on several cells were shown in Table 1.
The IC50 of peptides or DDP on various cell lines
| IC50 (mean ± SD, μM) | |||
|---|---|---|---|
| cell | FSH33-53-IIKK | IIKK | DDP |
| PC3 | 13.30 ± 1.21 | 14.20 ± 1.15 | 37.06 ± 1.59*** |
| HeLa | 7.16 ± 0.86 | 8.20 ± 0.91 | 14.08 ± 1.15* |
| SKOV3 | 21.40 ± 1.33 | 16.06 ± 1.21 | 48.85 ± 1.69** |
| MCF7 | 8.25 ± 0.92 | 10.12 ± 1.00 | 22.41 ± 1.35** |
| L929 | 43.50 ± 1.64 | 41.19 ± 1.62 | 32.03 ± 1.51* |
| 293 | 42.49 ± 1.56 | 39.68 ± 1.60 | 27.32 ± 1.44** |
(* p < 0.05, ** p < 0.01, and *** p < 0.001 vs FSH33-53-IIKK)
p-values were determined by student's t-test between the IC50 values of both cell lines
Apoptosis of PC3 cells induced by the peptides
The following experiments were completed in PC3 cells. For studying the mechanism of fusion peptide, apoptosis of PC3 cells was detected firstly. The morphological changes of the nucleus in PC3 cells treated with FSH33-53-IIKK were detected by using Hoechst 33258 fluorescence staining. Under fluorescence microscope, most of cell nucleus showed a uniform and full around or oval in control group. Very few cells showed strong staining and irregular nucleus perhaps because of its spontaneous programmed cell death. With the increase of peptide amount, cells shrinkage, chromatin collapse, fluorescence densification with obvious granular phosphor aggregation and nuclear fragmentation became apparent. And some cells decreased and followed by breaking into pieces when the concentration of peptide reached 30 μM (Fig.2A).
FSH33-53-IIKK fusion peptide inducing the apoptosis of PC3 cells (A) PC3 cells were incubated with FSH33-53-IIKK at the concentration of 0, 7.5, 15, and 30 μM for 24 h. And cells nucleus were stained by Hoechst 33258 and observed with fluorescence microscope. Arrows indicate some of apoptotic cells. (B) The expression of apoptosis related proteins, Caspase-3, Caspase-8, Bcl-2, Bax, Bad in PC3 cells treated by increasing FSH33-53-IIKK, were detected using Western Blot. And gray analysis of the bands was made by image analyzer, β-Actin as control. * P < 0.05, ** P < 0.01, and *** P < 0.001 vs 0 μM FSH33-53-IIKK. (C) Annexin V-FITC/PI double staining detected and flow cytometry analyzed the apoptosis induced by FSH33-53-IIKK or IIKK in PC3 cells. Apoptosis of PC3 cells was induced by FSH33-53-IIKK in a dose-dependent manner, and the effect was enhanced compared to IIKK at the concentration of 15 and 30 μM. a, control, b, 7.5 μM FSH33-53-IIKK, c, 15 μM FSH33-53-IIKK, d, 30 μM FSH33-53-IIKK, e, 7.5 μM IIKK, f, 15 μM IIKK, g, 30 μM IIKK.
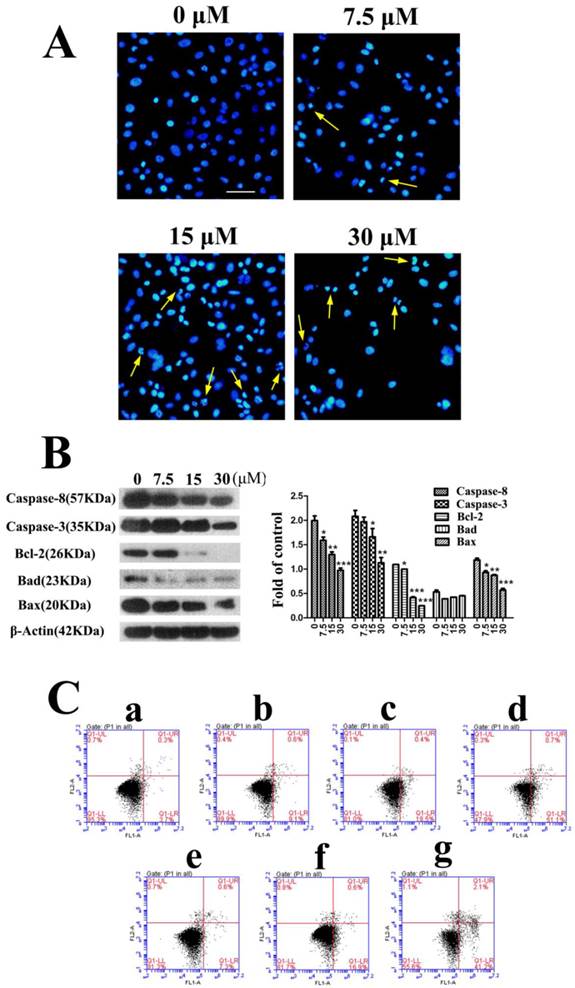
Proteins associated with apoptosis were also measured by Western Blot. As shown in Fig.2B, inactivated Caspase-8(57 KDa) and Caspase-3(35 KDa) proteins level decreased with the increase of peptide concentration after treatment for 24 h. In spite of the decrease of Bcl-2 and Bax proteins, the ratio of Bcl-2/Bax reduced in a dose-dependent manner, which means the increase of mitochondrial membrane permeability. In addition, the expression of Bad appeared no changes. The results showed the caspase cascade could be activated through mitochondrial pathway that caused cell apoptosis by FSH33-53-IIKK peptide in PC3 cells.
We then compared the apoptosis effects induced by FSH33-53-IIKK and IIKK peptides. Flow cytometry experiment shows the apoptosis respective rate is (9.1 ± 1.35) %, (18.5 ± 1.50) %, (51.1 ± 2.00) % and (7.3 ± 1.08) %, (16.9 ± 0.81) %, (41.2 ± 2.61) % at concentrations of 7.5, 15, 30 μM. Also, compared to IIKK, FSH33-53-IIKK promoted the apoptosis significantly in PC3 cells at 15 and 30 μM. (P<0.05) (Fig.2C)
Autophagy in PC3 cells
Autophagy was observed in PC3 cells that were treated with FSH33-53-IIKK. AO staining can label the autolysosome in autophagocyte. In our experiments, through AO staining, cell nucleus and cytoplasm in control, untreated with the peptide, presented bright green. A few of autolysosomes or autophagosomes with AO appeared weak orange red in some cells at 7.5 μM of FSH33-53-IIKK. With the increase amount of the peptide, the number of autophagosomes structure and fluorescence intensity both increased. Furthermore, at the concentration of 30 μM, adherent cells decreased with bright red fluorescence in the cytoplasm of them (Fig.3A).
Autophagy induced by FSH33-53-IIKK on PC3 cells (A) PC3 cells were treated with 0, 7.5, 15 and 30 μM FSH33-53-IIKK for 24 h. AO staining showed enhanced autophagy with the increasing amount of FSH33-53-IIKK. Arrows indicate some AO labeled autolysosomes or autophagosomes in PC3 cells. (B) The expression of autophagy related proteins, LC3-I and LC3-II in PC3 cells treated by increasing FSH33-53-IIKK, were detected using Western Blot. β-Actin was used as control.
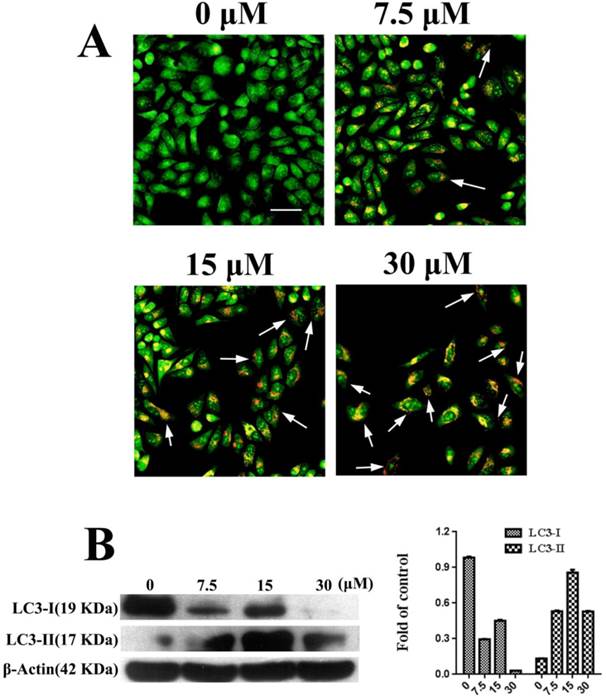
Meanwhile, we detected the expression of LC3-I and LC3-II related to autophagy under the action of FSH33-53-IIKK. With the increase of peptide concentration, the expression of LC3-I protein showed a downward trend, while LC3-II tended to increase at 0-15 μM. Opposite to the trend of LC3-II, the protein level reduced at 30 μM of peptide (Fig.3B). The results showed that the effect of FSH33-53-IIKK with low concentration can induce autophagy in PC3 cells, otherwise inhibited cell growth probably mainly by apoptosis at high concentration.
FSH33-53-IIKK anti-tumor performance in vivo
We also investigated the anti-tumor effects of fusion peptide in vivo. Male Balb/c nude mice were injected subcutaneously into the right front armpit with PC3 cells after tumors reached a mean size of 80 ~ 100 mm3, FSH33-53-IIKK (5 mg/kg, 10 mg/kg), IIKK (5 mg/kg) and DDP (3 mg/kg) were intravenously injected for anti-tumor. The tumor volume and body weights were measured every 2 days. Relative tumor volume (RTV) was chosen as the indicator for the growth of tumor. Lastly, the mice were sacrificed and the tumors were weighed. The tumor inhibition rates were calculated. Fig.4A showed the curve of tumor growth. And Table 2 showed the inhibitory effect of FSH33-53-IIKK on tumor was enhanced with the increase of concentration. And at the same concentration (5 mg/kg), the average tumor inhibition rate of FSH33-53-IIKK is better than that of IIKK. In spite of these, the DDP, 3 mg/kg as positive control, still have done the best. On the other hand, according to the curve of nude mice body weights (see Fig.4B), the average weight of DDP group mice decreased significantly in comparison with groups of FSH33-53-IIKK (P < 0.05). Also the average weight of each peptide group has no significant differences compared to saline group (P > 0.05). The above results suggested that the peptides toxicity to normal tissue in vivo were lower than DDP. At last, the tumors were weighed and photographed (Fig.4C).
The anti-tumor effects of FSH33-53-IIKK in vivo (A) Tumor growth curves of mice treated by peptides. Male nude mice were injected with PC3 cells. And when tumors were estabished. Peptides, DDP, or saline were administrated. Tumor volumes were measured and caculated. And growth curves of tumors were formed. (B) The body weights for each group during administration with drugs were measured. DDP group was decreased significantly and other groups had no obvious differences compared to saline group. (C) The tumors were removed from mice after administration and photographed.
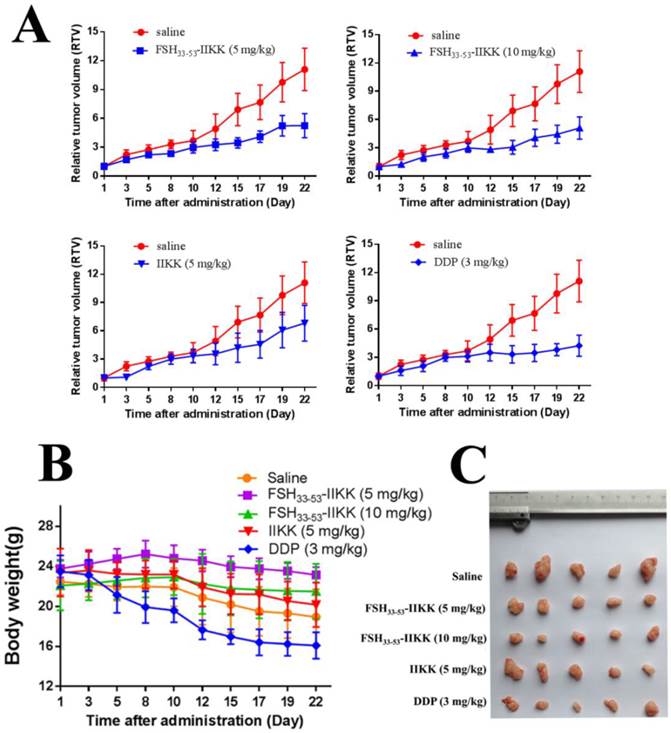
PC3 Inhibition rate of tumor triggered by drugs
| Group | Tumor inhibition rate (%) |
|---|---|
| FSH33-53-IIKK (5mg/kg) | 42.30 ± 11.10 |
| FSH33-53-IIKK (10mg/kg) | 50.75 ± 9.15 |
| IIKK(5mg/kg) | 25.15 ± 17.54 |
| DDP(3mg/kg) | 55.54±15.51 |
Discussion
As mentioned above, FSHR, just as some well-known receptors (somatostatin receptor, glucagon-like peptide-1 receptor, vascular endothelial growth factor receptor, and so on), is significantly overexpressed in many types of tumors compared to normal tissues with limited expression. We also tested and verified the high expression of FSHR in PC3, HeLa, MCF-7 cancer cells and low level in SKOV3. And it also expressed in 293 cells as reported by Rozell [35]. FSHR associated Gαs/cAMP/protein kinase A had been considered as the sole signal pathway of FSH/FSHR but the β-arrestin-dependent pathway and PI3K/mTOR pathway have been discovered recently [36]. And overexpression of FSHR increases the levels of epithelial growth factor, HER-2/neu, c-myc and ERK1/2 in ovarian surface epithelial cells [37] and in benign ovarian epithelial tumors cells [38], which associated with oncogenic pathway. Furthermore, it is proved that FSH could stimulate the proliferation of PC3 and DU145 cancer cells [39]. More importantly, EMT of epithelial ovarian cancer cells can be induced by FSH through the FSHR-PI3K/Akt-Snail signaling pathway [40]. And EMT is the early stage of metastasis in cancer. Then FSHR has been reasonably considered as a target for diagnosis, imaging and therapy of tumors.
Several classes of compounds that are considered as negative allosteric modulators of FSHR have been identified [41-46]. But these small molecules do not compete with FSH for interaction with the receptor and, especially, some were only shown functional inhibition for part of reproduction in vivo.
As a segment of FSHβ chain, FSH33-53 can bind to FSHR and was confirmed as an effective antagonist of FSH [17]. So FSH33-53 was selected as a target unit to conjugate with drugs or drug anchors for delivery in FSHR expressed cancer cells [10, 22, 23]. But for the drawbacks of chemotherapeutics and nucleic acid in vivo, we modified the approach by conjugating FSH33-53 with short peptides drugs.
Short peptide drugs have attracted the attention on theoretical and applied researches in a variety of fields [47-50], especially including tumor imaging and therapy. Just as some mature drugs which are vintage tools commandeered for new purposes in clinical, some of antimicrobial peptides (AMPs) and their synthetic analogs have been demonstrated toxicity to cancer cells [29, 30, 51]. Furthermore, these AMPs mainly use cations on surface to act on the target cancer cell membranes with a little negative charge via a non-receptor-mediated pathway. So, unlike conventional chemotherapeutic drugs, the resistance to cancer cells is not easily developed [31]. Among them, a new serious of short α-helical peptides G(IIKK)nI-NH2 (n=1-4) have been designed and demonstrated high selectivity and toxicity against cancer cells [32]. The peptides can combine tumor cell membrane which has some net negative charge by electrostatic interaction and selectivity, permeate the membrane and accumulate in tumor cells. Subsequently, the peptides induce cell apoptosis and disrupt the cell membrane.
So we combined the G(IIKK)3I-NH2 and FSHR target peptide FSH33-53 into a new fusion peptide which was expected to target and therapy FSHR-overexpressing tumor with even higher efficiency. In fact, FSH33-53-IIKK fusion peptide enhanced the anti-tumor effects compared to IIKK in PC3 xenografted mice. Meanwhile, according to the weight changes of therapeutic mice, the toxicity to mice treated by fusion peptide was not only obviously lower than that by DDP, but also inferior to that by IIKK though there was no obvious difference between them. In spite of the encouraging results in vivo, the FSH33-53-IIKK has not shown a significant advantage relative to IIKK with toxicity to tumor cells in vitro. We consider that maybe peptides in culture medium directly touched cultured cells so that they lost or greatly reduced the selectivity occurring in vivo. It may be another reason that FSH33-53-IIKK is more stable than IIKK in vivo for its longer amino acid sequence and folding structure.
Our study also revealed that FSH33-53-IIKK induced the apoptosis in PC3 cancer cells just as that by IIKK [52]. Apoptosis related proteins level, such as inactivated Caspase-3, 8, Bcl-2 and Bax, declined with the increase of FSH33-53-IIKK concentration. The ratio of Bcl-2/Bax also reduced in a dose-dependent manner. Mitochondrial pathway of apoptosis was activated and then caspase cascade appeared. So, in a way, the fusion peptide did not influence the effects of IIKK part. Furthermore, we also proved that autophagy appeared in PC3 cells treating by FSH33-53-IIKK. With the low dosage of the peptide, LC3-I/II was detected to change oppositely with the enhancement of autophagy. But the changes did not appear with high dosage, while the apoptosis was still exiting and augmenting. Then it may be explained the function of the peptides is not exclusive for the complexity of cancer cells.
From our results, combining the tumor marker-target peptide and anti-tumor peptide into fusing multi-function peptide is an efficiency model to develop anti-tumor drugs. But some deficiencies still exists. For example, the application of small molecular peptides is easy to be hampered by their degradation induced by proteolytic enzymes in vivo [53]. Also the expression of specific tumor tissue receptors has individual differences and heterogeneity which increase uncertainty of drug delivery mediated by peptides [54]. Meanwhile, the designed fusion peptides also may lose its functions. FSH33-53-Lytic fusion peptide cannot achieve the tumor-target (FSH33-53) and tumor-lytic (Lytic) bi-functions [55]. On the contrary, EGF-Lytic [56], HER2-Lytic [57] and NRP1-Lytic [58] fusion peptides achieve targeting and lysis effects. These peptides can target tumor marker EGFR, HER2 or NRP1 respectively and lytic the membrane of tumor cells. So the design of peptide drugs is still complex and all kinds of models shall be attempted.
Abbreviations
FSH, follicle stimulating hormone; PET, positron emission tomography; PTX, paclitaxel; PEG, polyethylene glycol; PEI, polyethylenimine; NPs, nanoparticles; EPR, enhanced permeability and retention; DMSO, dimethyl sulfoxide; HRP, horse radish peroxidase; DDP, casplatin; MTT, 3- (4,5-dimethylthiazol-2-yl)-2, 5-diphenyltetrazolium bromide; PI, Propidium Iodide; AO, acridine orange; EMT, Epithelial-Mesenchymal Transition.
Acknowledgements
This work was supported by National Natural Science Foundation (81171399, 51473071, 81472749, 81401450, 21401084, 81471691), Jiangsu Province Foundation (BE2012622, BL2012031, BE2014609, BM2012066), General program of Jiangsu Provincial Commission of Health and Family Planning (RC2011095, Q201406, H201529).
Author Contributions
R.Y., X.L. and M.Y. conceived the experiments. R.Y. and P.L. wrote the paper. R.Y. and P.L. performed most of the experiments. D.P., Z.B., Y.X., J.Y. and Y.Y contributed to the analysis of data and the preparation of manuscript. P.Z., L.W., D.P. and Y.X. provided technical support for animal experiments. All authors reviewed the manuscript.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Gartrell BA, Tsao CK, Galsky MD. The follicle-stimulating hormone receptor: A novel target in genitourinary malignancies. Urol Oncol. 2012;31:1403-7
2. Plant TM, Marshall GR. The functional significance of FSH in spermatogenesis and the control of its secretion in male primates. Endocr Rev. 2001;22:764-86
3. Macklon NS, Fauser BC. Follicle development during the normal menstrual cycle. Maturitas. 1998;30:181-8
4. Zheng W, Magid MS, Kramer EE, Chen YT. Follicle-stimulating hormone receptor is expressed in human ovarian surface epithelium and fallopian tube. Am J Pathol. 1996;148:47-53
5. La Marca A, Carducci Artenisio A, Stabile G, Rivasi F, Volpe A. Evidence for cycle-dependent expression of follicle-stimulating hormone receptor in human endometrium. Gynecol Endocrinol. 2005;21:303-6
6. Radu A, Pichon C, Camparo P, Antoine M, Allory Y, Couvelard A. et al. Expression of follicle-stimulating hormone receptor in tumor blood vessels. N Engl J Med. 2010;363:1621-30
7. Vannier B, Loosfelt H, Meduri G, Pichon C, Milgrom E. Anti-human FSH receptor monoclonal antibodies: immunochemical and immunocytochemical characterization of the receptor. Biochemistry. 1996;35:1358-66
8. Vu Hai Mt, Lescop P, Loosfelt H, Ghinea N. Receptor-mediated transcytosis of follicle-stimulating hormone through the rat testicular microvasculature. Biol Cell. 2004;96:133-44
9. Siraj A, Desestret V, Antoine M, Fromont G, Huerre M, Sanson M. et al. Expression of follicle-stimulating hormone receptor by the vascular endothelium in tumor metastases. BMC Cancer. 2013;13:246
10. Zhang XY, Chen J, Zheng YF, Gao XL, Kang Y, Liu JC. et al. Follicle-stimulating hormone peptide can facilitate paclitaxel nanoparticles to target ovarian carcinoma in vivo. Cancer Res. 2009;69:6506-14
11. Mariani S, Salvatori L, Basciani S, Arizzi M, Franco G, Petrangeli E. et al. Expression and cellular localization of follicle-stimulating hormone receptor in normal human prostate, benign prostatic hyperplasia and prostate cancer. J Urol. 2006;175:2072-7
12. Renner M, Goeppert B, Siraj MA, Radu A, Penzel R, Wardelmann E. et al. Follicle-stimulating hormone receptor expression in softtissue sarcomas. Histopathology. 2013;63:29-35
13. Choi JH, Wong AS, Huang HF, Leung PC. Gonadotropins and ovarian cancer. Endocr Rev. 2007;28:440-61
14. Ji Q, Liu PI, Chen PK, Aoyama C. Follicle stimulating hormone-induced growth promotion and gene expression profiles on ovarian surface epithelial cells. Int J Cancer. 2004;112:803-14
15. Li Y, Ganta S, Cheng C, Craig R, Ganta RR, Freeman LC. FSH stimulates ovarian cancer cell growth by action on growth factor variant receptor. Mol Cell Endocrinol. 2007;267:26-37
16. Syed V, Ulinski G, Mok SC, Yiu GK, Ho SM. Expression of gonadotropin receptor and growth responses to key reproductive hormones in normal and malignant human ovarian surface epithelial cells. Cancer Res. 2001;61:6768-76
17. Agris PF, Guenther RH, Sierzputowska-Gracz H, Easter L, Smith W, Hardin CC. et al. Solution structure of a synthetic peptide corresponding to a receptor binding region of FSH (hFSH-h 33-53). J Protein Chem. 1992;11:495-507
18. Santa Coloma TA, Reichert LE Jr. Identification of a follicle-stimulating hormone receptor-binding region in hFSH-beta-(81-95) using synthetic peptides. J Biol Chem. 1990;265:5037-42
19. Xu YP, Pan DH, Zhu C, Xu Q, Wang LZ, Chen F. et al. Pilot study of a novel (18)F-labeled FSHR probe for tumor imaging. Mol Imaging Biol. 2014;16:578-85
20. Schally AV, Nagy A. Chemotherapy targeted to cancers through tumoral hormone receptors. Trends Endocrinol Metab. 2004;15:300-10
21. Smith LL, Brown K, Carthew P, Lim CK, Martin EA, Styles J. et al. Chemoprevention of breast cancer by tamoxifen: risks and opportunities. Crit Rev Toxicol. 2000;30:571-94
22. Modi DA, Sunoqrot S, Bugno J, Lantvit DD, Hong S, Burdette JE. Targeting of follicle stimulating hormone peptide-conjugated dendrimers to ovarian cancer cells. Nanoscale. 2014;6:2812-20
23. Hong S, Zhang X, Chen J, Zhou J, Zheng Y, Xu C. Targeted gene silencing using a follicle-stimulating hormone peptide-conjugated nanoparticle system improves its specificity and efficacy in ovarian clear cell carcinoma in vitro. J Ovarian Res. 2013;6:80
24. Peer D, Karp JM, Hong S, Farokhzad OC, Margalit R, Langer R. Nanocarriers as an emerging platform for cancer therapy. Nat Nanotechnol. 2007;2:751-60
25. Wong C, Stylianopoulos T, Cui J, Martin J, Chauhan VP, Jiang W. et al. Multistage nanoparticle delivery system for deep penetration into tumor tissue. Proc Natl Acad Sci U S A. 2011;108:2426-31
26. Munshi A, McDonnell TJ, Meyn RE. Chemotherapeutic agents enhance TRAIL-induced apoptosis in prostate cancer cells. Cancer Chemother Pharmacol. 2002;50:46-52
27. Falco A, Ortega-Villaizan M, Chico V, Brocal I, Perez L, Coll JM. et al. Antimicrobial peptides as model molecules for the development of novel antiviral agents in aquaculture. Mini Rev Med Chem. 2009;9:1159-64
28. Wang FH, Wang Y, Zhang X, Zhang WJ, Guo SG, Jin F. Recent progress of cell-penetrating peptides as new carriers for intracellular cargo delivery. J Controlled Release. 2014;174:126-36
29. Schweizer F. Cationic amphiphilic peptides with cancer-selective toxicity. Eur J Pharmacol. 2009;625:190-4
30. Hoskin DW, Ramamoorthy A. Studies on anticancer activities of antimicrobial peptides. Biochim Biophys Acta. 2008;1778:357-75
31. Hu J, Chen C, Zhang S, Zhao X, Xu H, Zhao X. et al. Designed antimicrobial and antitumor peptides with high selectivity. Biomacromolecules. 2011;12:3839-43
32. Shai Y. Mechanism of the binding, insertion and destabilization of phospholipid bilayer membranes by alpha-helical antimicrobial and cell non-selective membrane-lytic peptides. Biochim Biophys Acta. 1999;1462:55-70
33. Matsuzaki K. Why and how are peptide-lipid interactions utilized for self-defense? Magainins and tachyplesins as archetypes. Biochim Biophys Acta. 1999;1462:1-10
34. Zachowski A. Phospholipids in animal eukaryotic membranes: transverse asymmetry and movement. Biochem J. 1993;294(Pt1):1-14
35. Rozell TG, Davis DP, Chai Y, Segaloff DL. Association of gonadotropin receptor precursors with the protein folding chaperone calnexin. Endocrinology. 1998;139:1588-93
36. Gloaguen P, Crepieux P, Heitzler D, Poupon A, Reiter E. Mapping the follicle-stimulating hormone-induced signal networks. Front Endocrinol(Lausanne). 2011;2:45
37. Choi JH, Choi KC, Auersperg N, Leung PC. Overexpression of follicle-stimulating hormone receptor activates oncogenic pathways in preneoplastic ovarian surface epithelial cells. J Clin Endocrinol Metab. 2004;89:5508-16
38. Zhang Z, Jia L, Feng Y, Zheng W. Overexpression of follicle-stimulating hormone receptor facilitates the development of ovarian epithelial cancer. Cancer Lett. 2009;278:56-64
39. Ben-josef E, Yang SY, Ji TH, Bidart JM, Garde SV, Chopra DP. et al. Hormone-refractory prostate cancer cells express functional follicle-stimulating hormone receptor (FSHR). J Urol. 1999;161:970-6
40. Yang Y, Zhang J, Zhu Y, Zhang Z, Sun H, Feng Y. Follicle-stimulating hormone induced epithelial-mesenchymal transition of epithelial ovarian cancer cells through follicle-stimulating hormone receptor PI3K/Akt-Snail signaling pathway. Int J Gynecol Cancer. 2014;24:1564-7
41. Arey BJ, Deecher DC, Shen ES, Stevis PE, Meade EH Jr, Wrobel J. et al. Identification and characterization of a selective, nonpeptide follicle-stimulating hormone receptor antagonist. Endocrinology. 2002;143:3822-9
42. van Straten NC, van Berkel TH, Demont DR, Karstens WJ, Merkx R, Oosterom J. et al. Identification of substituted 6-amino-4-phenyltetrahydroquinoline derivatives: potent antagonists for the follicle-stimulating hormone receptor. J Med Chem. 2005;48:1697-700
43. Arey BJ, Yanofsky SD, Claudia Perez M, Holmes CP, Wrobel J, Gopalsamy A. et al. Differing pharmacological activities of thiazolidinone analogs at the FSH receptor. Biochem Biophys Res Commun. 2008;368:723-8
44. Yanofsky SD, Shen ES, Holden F, Whitehorn E, Aguilar B, Tate E. et al. Allosteric activation of the follicle-stimulating hormone (FSH) receptor by selective, nonpeptide agonists. J Biol Chem. 2006;281:13226-33
45. Wrobel J, Green D, Jetter J, Kao W, Rogers J, Perez MC. et al. Synthesis of (bis)sulfonic acid, (bis)benzamides as follicle-stimulating hormone (FSH) antagonists. Bioorg Med Chem. 2002;10:639-56
46. Dias JA, Bonnet B, Weaver BA, Watts J, Kluetzman K, Thomas RM. et al. A negative allosteric modulator demonstrates biased antagonism of the follicle stimulating hormone receptor. Mol Cell Endocrinol. 2011;333:143-50
47. Lien S, Lowman HB. Therapeutic peptides. Trends Biotechnol. 2003;21:556-62
48. Chromek M, Slamova Z, Bergman P, Kovacs L, Podracka L, Ehren I. et al. The antimicrobial peptide cathelicidin protects the urinary tract against invasive bacterial infection. Nat Med. 2006;12:636-41
49. Kumar P, Wu H, McBride JL, Jung KE, Kim MH, Davidson BL. et al. Transvascular delivery of small interfering RNA to the central nervous system. Nature. 2007;448:39-43
50. Fuessel S, Meye A, Schmitz M, Zastrow S, Linne C, Richter K. et al. Vaccination of hormone-refractory prostate cancer patients with peptide cocktail-loaded dendritic cells: results of a phase I clinical trial. Prostate. 2006;66:811-21
51. Mader JS, Hoskin DW. Cationic antimicrobial peptides as novel cytotoxic agents for cancer treatment. Expert Opin Investig Drugs. 2006;15:933-46
52. Chen C, Hu J, Zeng P, Pan F, Yaseen M, Xu H. et al. Molecular mechanisms of anticancer action and cell selectivity of short α-helical peptides. Biomaterial. 2014;35:1552-61
53. Chatalic KL, Konijnenberg M, Nonnekens J, de Blois E, Hoeben S, de Ridder C. et al. In vivo stabilization of gastrin-releasing peptide receptor antagonist enhances PET imaging and radionuclide therapy of prostate cancer in preclinical studies. Theranostics. 2016;6:104-17 doi: 10.7150/thno.13580. eCollection 2016
54. Ng CK, Pemberton HN, Reis-Filho JS. Breast cancer intratumor genetic heterogeneity: causes and implications. Expert Rev Anticancer Ther. 2012;12:1021-32
55. Liu P, YANG RL, Pan DH, Xu YP, Zhu C, XU Q. et al. An investigation on the anti-tumor properties of FSH33-53-Lytic. J Radioanal Nucl Chem. DOI: 10.1007/s10967-015-4143-0.
56. Kohno M, Horibe T, Haramoto M, Yano Y, Ohara K, Nakajima O. et al. A novel hybrid peptide targeting EGFR-expressing cancers. Eur J Cancer. 2011;47:773-83
57. Kawamoto M, Horibe T, Kohno M, Kawakami K. HER2-targeted hybrid peptide that blocks HER2 tyrosine kinase disintegrates cancer cell membrane and inhibits tumor growth in vivo. Mol Cancer Ther. 2013;12:384-93
58. Ueyama H, Horibe T, Nakajima O, Ohara K, Kohno M, Kawakami K. Semaphorin 3A lytic hybrid peptide binding to neuropilin-1 as a novel anti-cancer agent in pancreatic cancer. Biochem Biophys Res Commun. 2011;414:60-6
Author contact
![]() Corresponding authors: Xingdang Liu, xingdliucom, Min Yang, yangminorg
Corresponding authors: Xingdang Liu, xingdliucom, Min Yang, yangminorg