Impact Factor ISSN: 1837-9664

 Global reach, higher impact
Global reach, higher impactJ Cancer 2016; 7(9):1142-1151. doi:10.7150/jca.10047 This issue Cite
Research Paper
Liver Label Retaining Cancer Cells Are Relatively Resistant to the Reported Anti-Cancer Stem Cell Drug Metformin
Hong-Wu Xin1, 2*, Chenwi M. Ambe1*, Tyler C. Miller1, Jin-Qiu Chen3, Gordon W. Wiegand1, Andrew J. Anderson1, Satyajit Ray1, John E. Mullinax1, Danielle M. Hari1, Tomotake Koizumi1, Jessica D. Godbout1, Paul K. Goldsmith3, Alexander Stojadinovic5, Udo Rudloff1, Snorri S. Thorgeirsson4 ![]() , Itzhak Avital1,6
, Itzhak Avital1,6 ![]()
1. Surgery Branch, Center for Cancer Research, National Cancer Institute, National Institutes of Health, Bethesda, MD 20892, USA;
2. Laboratory of Molecular Oncology, Key Laboratory of Carcinogenesis and Translational Research (Ministry of Education), Peking University Cancer Hospital and Institute, Beijing 100142, China;
3. Collaborative Protein Technology Resource, National Cancer Institute, National Institutes of Health, Bethesda, MD 20892, USA;
4. Laboratory for Experimental Carcinogenesis, Center for Cancer Research, National Cancer Institute, National Institutes of Health, Bethesda, MD 20892, USA;
5. Department of Surgery, Uniformed Services University of the Health Sciences, Bethesda, MD, USA;
6. Department of Surgery, Saint Peter's Healthcare System, Rutgers University, New Brunswick, NJ 08901, USA.
*Authors contributed equally.
Published 2016-6-6
Abstract
Background & Aims: Recently, we reported that liver Label Retaining Cancer Cells (LRCC) can initiate tumors with only 10 cells and are relatively resistant to the targeted drug Sorafenib, a standard of practice in advanced hepatocellular carcinoma (HCC). LRCC are the only cancer stem cells (CSC) isolated alive according to a stem cell fundamental function, asymmetric cell division. Metformin has been reported to preferentially target many other types of CSC of different organs, including liver. It's important to know if LRCC, a novel class of CSC, are relatively resistant to metformin, unlike other types of CSC. As metformin inhibits the Sorafenib-Target-Protein (STP) PI3K, and LRCC are newly described CSC, we undertook this study to test the effects of Metformin on Sorafenib-treated HCC and HCC-derived-LRCC.
Methods: We tested various STP levels and phosphorylation status, associated genes' expression, proliferation, viability, toxicity, and apoptosis profiles, before and after treatment with Sorafenib with/without Metformin.
Results: Metformin enhances the effects of Sorafenib on HCC, and significantly decreased viability/proliferation of HCC cells. This insulin-independent effect was associated with inhibition of multiple STPs (PKC, ERK, JNK and AKT). However, Metformin increased the relative proportion of LRCCs. Comparing LRCC vs. non-LRCC, this effect was associated with improved toxicity and apoptosis profiles, down-regulation of cell death genes and up-regulation of cell proliferation and survival genes in LRCC. Concomitantly, Metformin up-regulated pluripotency, Wnt, Notch and SHH pathways genes in LRCC vs. non-LRCC.
Conclusions: Metformin and Sorafenib have enhanced anti-cancer effects. However, in contradistinction to reports on other types of CSC, Metformin is less effective against HCC-derived-CSC LRCC. Our results suggest that combining Metformin with Sorafenib may be able to repress the bulk of tumor cells, but as with other anti-cancer drugs, may leave LRCC behind leading to cancer recurrence. Therefore, liver LRCC, unlike other types of CSC, are relatively resistant to the reported anti-cancer stem cell drug metformin. This is the first report that there is a type of CSC that is not relatively resistant to the CSC-targeting drug. Our findings suggest that a drug targeting LRCC may be critically needed to target CSC and prevent cancer recurrence. These may significantly contribute to the understanding of Metformin's anti-cancer effects and the development of novel drugs targeting the relatively resistant LRCC.
Keywords: Metformin, sorafenib, PKC/ERK/JNK/AKT phosphorylation, MAPK, stem-like label-retaining cancer cells, LRCC, HCC, cancer-stem-cells.
Introduction
Metformin is an oral hypoglycemic. It is used in type-2 diabetes, polycystic ovarian syndrome and obesity. Epidemiologic studies associated metformin with decreased incidence of cancer [1]. Bowker et al. looked into 10,309 diabetic patients and found that patients treated with metformin had a significantly lower incidence of cancer-related mortality compared with patients treated with sulfonylurea or insulin [1]. Metformin enhanced the effects of cisplatin, paclitaxel, doxorubicin and tamoxifen in lung, breast, pancreas, liver, glioblastoma, prostate and ovarian cancer cells [2-5]. Metformin's anti-cancer effects are attributed to direct inhibition of the NF-κB (nuclear-factor-kappa-light-chain-enhancer-of-activated-B-cells) or LKB1/AMPK/mTOR (liver-kinase-B1/AMP-activated-kinase/mammalian-target-of-rapamycin) pathway (Supplementary Note) and possible indirect inhibition, via reduction of blood insulin, of PI3K/AKT/mTOR (Phosphatidylinositol-3-kinases/V-akt-murine-thymoma-viral-oncogene-homolog/mTOR) and RAS/MEK/ERK (Rat-sarcoma/Mitogen-activated-protein-kinase-kinase/Extracellular-signal-regulated-kinase) [2, 3]. However, there is a paucity of evidence suggesting that metformin enhances targeted cancer drugs [5-7] or inhibits independently of blood insulin PKC/ERK/JNK/AKT (Protein-kinase-C/ERK/c-Jun-N-terminal-kinase/AKT) phosphorylation [2, 8, 9].
Recent data suggested that cancers contain cells with stem-like characteristics, or cancer stem cells (CSC) [10, 11]. It's proposed that CSC are responsible for cancer initiation, maintenance, metastasis and therapeutic failure [10-12]. However, there have been no reports of any drug targeting CSC until 2009 when Hirsch et al. showed that the anti-diabetes drug Metformin selectively targets CSC (CD44high/CD24low cells) in breast cancer cell lines [13]. It's suggested that metformin targets breast CSC by reversal of the epithelial-mesenchymal transition (EMT) status, and suppression of NF-κB or self-renewal [3, 14]. Bao et al. demonstrated that metformin selectively targets pancreatospheres of gemcitabine-resistant pancreatic CSC [15]. Recently, Saito et al. reported that metformin preferentially represses the CSC marker EpCam+ cells of hepatocellular carcinoma (HCC) [5].
HCC represents the third most common cause of cancer death worldwide [16, 17]. Sorafenib is a tyrosine kinase inhibitor and the standard of care for patients with advanced HCC [18]. Improvement in outcomes is modest; on average, sorafenib increase survival by 2.4 months [18]. Most patients will suffer disease recurrence and die. Label-retaining cancer cells (LRCC) were recently described as novel class of liver derived CSC and found relatively resistant to sorafenib [10, 12, 19]. LRCC can initiate tumors with only 10 cells and are the only CSC isolated alive according to a stem cell fundamental function, asymmetric cell division [10, 12, 19, 20]. As described above, metformin has been reported to preferentially target many other types of CSC of different organs, including liver. It's important to know if LRCC, a novel class of CSC, are relatively resistant to metformin, unlike other types of CSC. We undertook this study to test the effects of Metformin on HCC and on HCC derived LRCC.
Here we show that Metformin enhances sorafenib. It enhances the anti-proliferative effects of sorafenib possibly via inhibiting phosphorylation of several tyrosine-kinases-related-proteins (PKC, ERK, JNK and AKT). However, in contradistinction to other reports, we found that metformin suppressed the whole population of cancer cells but it increased the subpopulation of LRCC. Studying LRCC vs. non-LRCC, this phenomenon was associated with decreased cell toxicity and apoptosis, and up-regulation of cell-survival, pluripotency, stem-cells, Wnt (Wingless-type-MMTV-integration-site-family), Notch (Notch-homolog) and SHH (Sonic-hedgehog) associated genes and down-regulation of cell-death genes. These findings may contribute to better understanding of the anti-cancer effects of metformin. It further suggests that metformin has limitation. It suggests that Combining Metformin with Sorafenib may be able to repress the bulk of tumor cells, but may leave CSC LRCC behind leading. Therefore, it is critical to develop novel drugs targeting the relatively resistant LRCC.
Materials and Methods
Human liver cancer cells lines
Human HCC cells PLC/PRF/5 and SK-Hep-1 (ATCC), and HuH-7 (Japan Health Sciences Foundation) were cultured in 44% DMEM, 44% Ham's F-12, 10% FCS, 1% Glutamine, and 1% Ampicillin/streptomycin (Invitrogen).
Growth curves
Growth curves were generated with/without clinically relevant metformin concentrations: 200uM. Numbers of live cells were determined on day 0, day 1, day 2, day 5, day 6 and day 7 (no day 7 for SK-Hep-1) (n=3), using Cellometer Auto T4 (Nexcelom Bioscience). Acquired numbers were averaged and plotted as previously described (Supplemental Fig. S1)[12].
Isolation and analysis of live LRCC and non-LRCC with or without sorafenib treatment
Live LRCC and Non-LRCC were isolated from HCC cell lines as described previously (Fig. 4A-B) [10, 19].
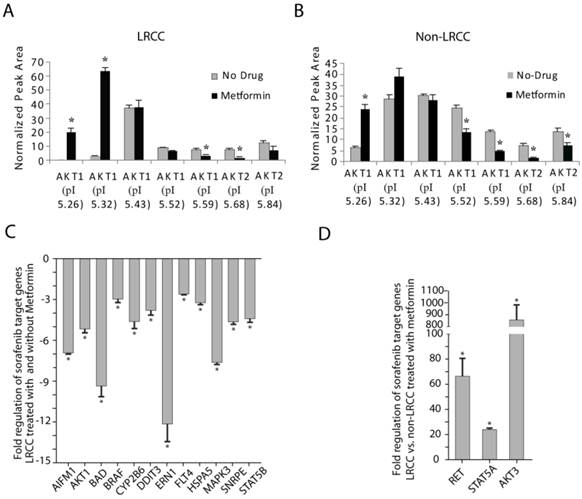
STP and STG expression in LRCC vs. non-LRCC. (A-B) Compared to non-LRCC, metformin treatment of LRCC resulted in increases of the major highly phosphorylated species of AKT1. (C) STG expression in LRCC before and after treatment with metformin. (D) Compared to non-LRCC, metformin treatment of LRCC resulted in opposite effects by up-regulation of RET, STAT5A and AKT3.
Viability, toxicity and apoptosis assays
The ApoTox-Glo assay kit (Promega) was used according to manufacturer protocol. We isolated and cultured LRCC and non-LRCC from HCC cell lines in 96-well plates with or without Meformin (200uM) during the last two cell cycles of the total eight cell cycles [12].
MEK, ERK and AKT protein kinase analysis
NanoPro 1000 is an automated capillary based isoelectric focusing (IEF) immunoassay system (ProteinSimple). Protein isolation, detection and quantification were done according to manufacturer's instructions as previously described [12].
Real-time qPCR
Pre-amplification of cDNA target templates, real-time qPCR and gene expression data analysis were done as previously described [12].
Statistics
For comparisons between two samples, we used the two-tailed Student-T Test to test the null hypothesis that the means of the two samples are equal. Two tailed p<0.05 was considered statistically significant. In all Figures, an asterisk was added when p<0.05. The error bars represent Standard Error of Mean (SEM) for Figures 1, 2, 4, 5, Figure S1 and Figure S11, and Standard Deviation (SD) for Figure 3 and Figure S5.
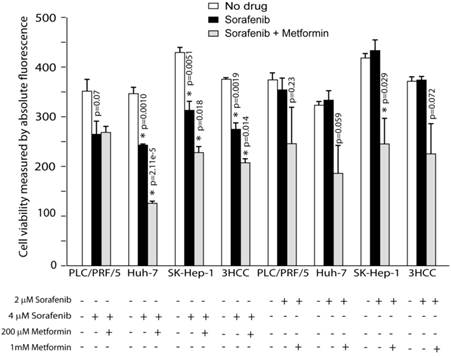
Metformin enhances cancer therapy with sorafenib. Metformin addition to sorafenib cause significant reduction of viable cells compared to sorafenib treatment alone after 48 hours of treatment.
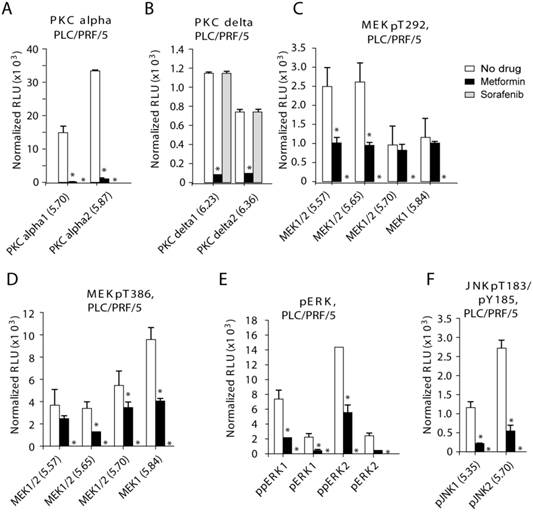
PKC-alfal/delta, MEK1/2, ERK1/2 and JNK1/2 are inhibited in PLC/PRF/5 cells after treatment with metformin. After treatment with metformin phosphorylation of multiple protein kinases were inhibited in PLC/PRF/5 cells: (A) PKC-alfal, (B) PKC-delta, (C) MEK1/2 (pT292), (D) MEK1/2 (pT386), (E) ERK1/2 and (F) JNK1/2. Metformin was used at 200 uM and sorafenib at 4 uM.
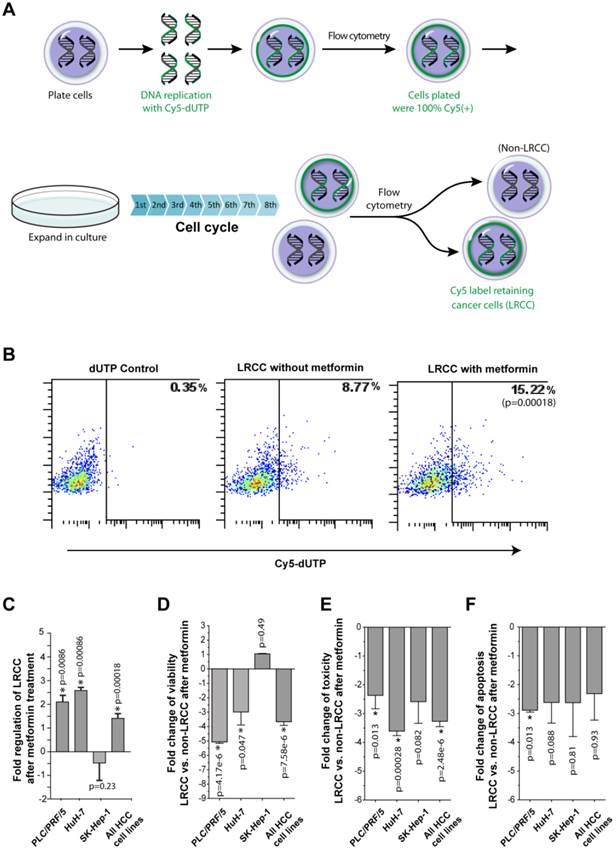
Metformin ineffectively kills LRCC. (A) Isolation of HCC derived label-retaining-cancer-cells (LRCC). The full method is described in Daniel et al. [10, 19]. Whole cell populations of HCC cells were labeled with Cy5-DNA-nucleotides (pulse phase). Subsequently, Cy5-positive-high cells were sorted and grown for 8 cell cycles (chase phase). Finally, Cy5-positive-high cells (99% pure) and Cy5-negative cells were sorted as LRCC and non-LRCC, respectively. (B)-(C) The relative proportion of LRCC is increased after metformin treatment. HuH-7 is shown in (B). Compared to non-LRCC, LRCC exhibit reduced cell viability (D), cell toxicity profile (E), and apoptosis profile (F).
Results
Metformin and sorafenib have enhanced anti-cancer effects
We tested the combination of sorafenib and metformin because metformin was reported to repress some of the sorafenib target genes and CSC. We tested the effects of metformin and sorafenib on HCC cells. First, we tested the effects of metformin (200uM) on HCC cells alone (PLC/PRF/5, HuH-7 and SK-Hep-1). We chose this relatively low concentration because there are reports of metformin-induced liver injuries [21]. Cell growth curves were plotted with or without metformin (Materials and Methods). Metformin suppressed the growths of PLC/PRF/5 (p=1.11e-2) and SK-Hep-1 (p=0.012), but not HuH-7 (p=0.32, Supplemental Fig. S1). Interestingly, HuH-7 was reported to be relatively resistant to sorafenib [12].
Next, we compared cell viability of HCC cells cultured without drugs, sorafenib alone and sorafenib plus metformin (Materials and Methods). 2uM of sorafenib alone did not affect HCC cells' viability (Fig. 1), however, 4uM of sorafenib treatment resulted in statistically significant reduction in viability of all HCC cells tested (p=0.0019, Fig. 1). The addition of metformin (200uM) to sorafenib (4uM) further reduced viability of HuH-7 (p=0.00002) and SK-Hep-1 (p=0.0082) cells compared to sorafenib alone (Fig. 1). While 2uM of sorafenib alone didn't affect viability, the addition of Metformin resulted in statistically significant reduction in viability of SK-Hep-1 (p=0.029), and a statistical trend in reduction of viability in PLC/PRF/5 and HuH-7 (p=0.23, p=0.059 respectively; Fig. 1). Overall, when all 3 HCC lines computed Metformin enhanced the effects of sorafenib in terms of HCC cells viability.
To further investigate the effects of Metformin, we tested whether Metformin affects viability by direct increased cell toxicity or induction of apoptosis. We tested apoptosis by measuring the activity of the effector caspases 3/7 and toxicity by dead cells' protease release (Materials and Methods). We tested HCC cells treated with-or-without sorafenib plus metformin (Supplemental Fig. S2). We detected statistically significant increase in the activities of caspases 3/7 only in HuH-7 cells at 4uM of sorafenib plus 200uM of metformin compared to sorafenib treatment alone (108598.3 ± 16091.5 vs, 186932.9 ± 20629.8, p=0.040). Interestingly, we detected statistically significant decrease in dead cells' protease release only in 4uM of sorafenib treatment plus 200 uM of metformin compared to sorafenib treatment alone in both HuH7 (380765.9 ± 38385.5 vs. 38065.6 ± 435.2, p=0.00067) and SK-Hep-1 (402167.8 ± 44454.5 vs. 92693.9 ± 769.1, p=0.0022). The increase of apoptosis in the combination treatment of HuH-7 cells may explain the decreased viability, while the decreases of dead cells' protease release in the combination treatment of HuH-7 and SK-Hep-1 cells may suggest that decreased cell growth may contribute to the reduced viability.
Metformin can inhibit sorafenib target proteins (STP)
MAPK (mitogen-activated-protein-kinase) and AKT associated pathways are important regulators of cell proliferation and are often over activated in liver cancer [22]. MAPK and AKT are targeted by sorafenib. Thus, we investigated the effects of metformin on key members of several important pathways: MEK, PKC, ERK, JNK and AKT. We quantitatively tested total MEK1/2, and phosphorylated-activated)-MEK (pS218/222, pT292, and pT386), total and phosphorylated ERK1/2, and phosphorylated PKC-alpha/delta, JNK1/2 and AKT1/2 in HCC cells that were treated with-or-without metformin or sorafenib (Materials and Methods).
In PLC/PRF/5 cells, sorafenib effectively inhibited all the kinases tested except PKC-delta (Fig. 2 and Supplemental Fig. S3-S4; Fig. 2 contains bar graph of the original Nano-Pro Western blot data in Figure S3-S4). Metformin effectively and statistically significantly inhibited levels of phosphorylated PKC-alpha/delta, MEK1/2, ERK1/2 and JNK1/2, but not AKT1/2 (Fig. 2 and Supplemental Fig. S3-S4). We detected dramatic inhibition of phosphorylated PKC by metformin (Fig. 2A-B): PKC-alpha-1 (p=0.018), PKC-alpha-2 (p=0.00014), PKC-delta-1 (p=0.0041), and PKC-delta-2 (p=0.0024). Metformin inhibited phosphorylated MEK1/2 (Fig. 2C-D): pMEK1/2-T292-pI5.57 (p=0.048), pMEK1/2-T292-pI5.65 (p=0.020), pMEK1/2-T382-pI5.65 (p=0.0093), pMEK1/2-T382-pI5.70 (p=0.018) and MEK1/2-T382-pI5.84 (p=0.0057). Finally, metformin inhibited phosphorylated ERK1/2 and JNK1/2 (Fig. 2E-F): ppERK1 (p=0.014), pERK1 (p=0.028), ppERK2 (p=0.025) and pERK2 (p=0.0063), and pJNK1-pI5.35 (p=0.027) and pJNK2-pI5.70 (p=0.014).
In HuH-7 cells, metformin and sorafenib effectively inhibited various species of MEK1/2, ERK1/2, JNK1 and AKT1 (Fig. S5A-D and Supplemental Fig. S6 and S8). Metformin inhibited phosphorylated MEK1/2 and ERK1/2 (Fig. S5A-B): pMEK1/2-pS218/222-pI5.70 (p=0.016) and pMEK1/2-pS218/222-pI5.91 (p=0.0060), and ppERK1 (p=0.00016), ppERK2 (p=0.013) and pERK2 (p=0.0019). JNK and AKT were effectively inhibited by Metformin (Fig. S5C-D): pJNK1-pI5.35 (p=0.0055), pJNK1-pI5.70 (p=0.00086), and AKT1-pI5.32 (p=0.029), pAKT2-pI5.68 (p=0.043), AKT1-pI5.52 (p=0.0070) and AKT1-pI5.59 (p=0.040). Metformin was more effective than sorafenib in inhibiting pMEK1/2-pS218/222-pI5.91, ppERK1, ppERK2, pERK2, pJNK1-pI5.35 and pJNK1-pI5.70 (Fig. S5A-D).
In SK-Hep-1 cells, both metformin and sorafenib effectively inhibited MEK1/2 and AKT1/2 (Fig. S5E-F and Supplemental Fig. S7 and S8). Metformin inhibited pMEK1/2-pT292-pI5.65 (p=0.022), pMEK1/2-pT292-pI5.84 (p=0.017), pAKT1-pI5.32 (p=0.0030), AKT1-pI5.43 (p=0.023), AKT2-pI5.68 (p=0.025), and pAKT2-pI5.84 (p=2.65e-5; Fig. S5E-F).
In conclusion, we show that after treatment with metformin the activated forms of several phosphorylated-protein-kinase species including some STP levels are decreased significantly. These results suggest that metformin inhibits phosphorylation of multiple protein kinases (PKC, ERK, JNK and AKT) independent of blood insulin, possibly having a more general anti-cancer mechanism. These data show that metformin can repress the sorafenib target protein kinases, which may indirectly provide a potential explanation for the enhanced repression of cell viability in the combination treatment.
Metformin treatment increases the relative proportion of LRCC
LRCC were recently described as a sorafenib-resistant subpopulation of HCC derived CSC [10, 12, 19]. Additionally, breast CSC were reported to be selectively targeted by Metformin [3, 14]. Thus, we investigated the effects of Metformin on LRCC. The procedure for the isolation of live LRCC is described in details in the Materials and Methods (Fig. 3A-B). We tested 3 HCC cell lines before and after metformin treatment for the presence of LRCC. Here we show that after treatment with metformin the relative proportion of LRCC is increased significantly, on average, by 1.4 ± 0.2 fold (p=0.00018, Fig. 3B-C); LRCC in PLC/PRF/5, HuH-7 and SK-Hep-1 were increased by 2.1 ± 0.3 (p=0.0086), 2.6 ± 0.1 (p=0.00086) and -0.5 ± 0.7 fold (p=0.23), respectively. LRCC from SK-Hep-1 cells are not increased or decreased statistically significantly (p=0.23). Our data demonstrate that metformin does not target the putative CSC, LRCC.
Metformin induces less cell death of LRCC comparing to non-LRCC
To further test the effects of Metformin on the CSC LRCC, we isolated live LRCC and non-LRCC from three liver cancer cell lines. LRCC and non-LRCC were tested for viability, toxicity and apoptosis before and after treatment with metformin (Fig. 3D-F, Materials and Methods).
We found that metformin treatment induces less cell viability, cell toxicity and apoptosis in LRCC comparing to non-LRCC (Fig. 3D-F). The fold changes of cell viability of LRCC and non-LRCC treated with metformin of three individual cell lines PLC/PRF/5, HuH-7, SK-Hep-1, and all the three cell lines together are -5.1 ± 0.1 (p=4.17-e6), -3.0 ± 0.9 (p=0.047), 1.0 ± 0.03 (p=0.49) and -3.7 ± 0.2 (p=7.58e-6), respectively (Fig. 3D). The fold changes of cell toxicity of the same are -2.4 ± 0.5 (p=0.013), -3.6 ± 0.15 (p=0.00028), -2.5 ± 0.8 (p=0.082) and -3.2 ± 0.2 (p=2.48e-6), respectively (Fig. 3E). The fold changes of cell apoptosis of the same are -2.9 ± 0.1 (p=0.013), -2.6 ± 0.7 (p=0.088), -2.6 ± 1.2 (p=0.81) and -2.3 ± 0.9 (p=0.93), respectively (Fig. 3F). There were no statistically significant inherent differences of viability, apoptosis or toxicity between LRCC and non-LRCC in the absence of metformin treatment [12].
Since the isolated LRCC have reduced cell toxicity and apoptosis compared to non-LRCC after metformin treatment (Fig. 3E-F), the reduction of cell viability are likely caused by reduced cell proliferation or slow cycling (Fig. 3D). Interestingly, the decreases of cell viability, toxicity and apoptosis of LRCC are more significant in PLC/PRF/5 and HuH-7 cells than in SK-Hep-1 cells (Fig. 3D-F), which has a good correlation with the significant increase of LRCC subpopulation after metformin treatment (Fig. 3C). Therefore our results show that metformin ineffectively kills LRCC. The reduced cell death (Fig. 3D-F) can contribute to the increase of LRCC subpopulation after metformin treatment (Fig. 3C). However, since both slow cycling and asymmetric cell division can potentially cause LRCC we investigated if the slow cycling of the LRCC treated with metformin after LRCC isolation here reflect the increase of LRCC subpopulation treated with metformin within their microenvironment [10].
Metformin can inhibit STP but promote AKT1 in LRCC of HuH-7 cells
As discussed earlier, we similarly tested if metformin could inhibit phosphorylation of the kinases of PKC, MEK, ERK, JNK and AKT differentially in LRCC and non-LRCC of PLC/PRF/5 and HuH-7 (Materials and Methods). Comparing to untreated cells, metformin effectively inhibited phosphorylation of MEK1/2 (pS218/222, pT292, and pT386) and ERK in both LRCC and non-LRCC (Supplemental Fig. 9), validating our earlier findings in the whole population cell (Fig. 2 and Fig. S5). However, we found that metformin treatment statistically significantly promoted phosphorylation of the major highly phosphorylated isoform of AKT1-pI5.32 (63.6 ± 2.3, p=1.19e-5) only in LRCC of HuH-7 cells (Fig. 4A-B and Supplemental Fig. S10). There were no statistically significant inherent differences of MEK-ERK-AKT relative protein levels between LRCC and non-LRCC in the absence of metformin treatment [12]. Promotion of phosphorylation of AKT only in LRCC may contribute to the reduced cell death of LRCC [23].
Metformin down-regulates cell death genes and up-regulates cell suvival genes in LRCC
Sorafenib has potential down-stream effects on 60 genes, herein sorafenib target genes (STG, Table S1) [12]. To gain further understanding of metformin effects on LRCC, we performed qRT-PCR SuperArray analysis (Fig. 4 and 5, Supplemental Table S1): STG (60 genes), WNT (84 genes), and stem cells associated genes (100 genes) in LRCC and non-LRCC of all the three HCC cell lines after treatment with metformin (Materials and Methods).
Wnt pathway and stem cells associated genes expression. (A) Wnt pathway genes' expression: in LRCC with/without metformin, and (B) in LRCC vs. non-LRCC after treatment with metformin. (C) Stem cells associated genes' expression in LRCC with/without metformin, and (D) in LRCC vs. non-LRCC after treatment with metformin. (E) Stem cell pluripotency genes in LRCC vs. non-LRCC after treatment with metformin.
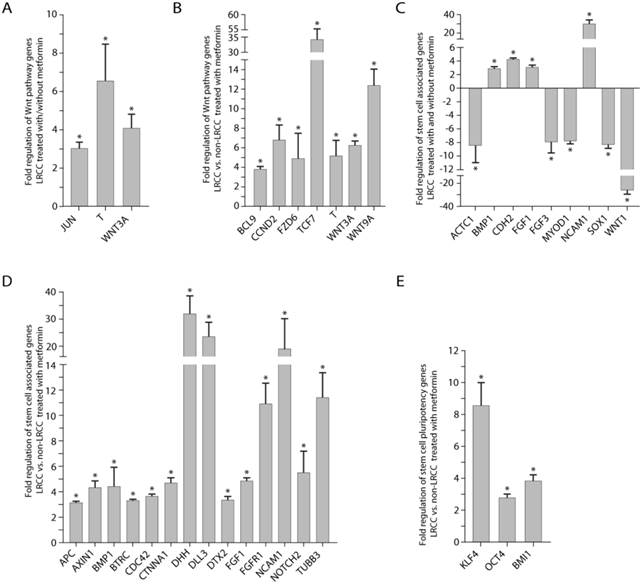
Comparing LRCC before and after treatment with metformin (Fig. 4C), the following five pro-apoptosis genes were down-regulated significantly [23]: AIFM1 (apoptosis-inducing-factor-mitochondrion-associated-1, -6.9 ± 0.1, p=0.011), BAD (-9.4 ± 0.8, p=0.045), DDIT3 (DNA-damage-inducible-transcript-3, -3.8 ± 0.3, p=0.046), ERN1 (endoplasmic-reticulum-to-nucleus-signaling-1, -12.1 ± 1.3, p=0.00081) and HSP5A (Heat-shock-70kDa-protein-5, -3.3 ± 0.1, p=0.037). We also found that seven cell proliferation genes below are down-regulated (Fig. 4C) [23]: AKT1 (-5.2 ± 0.2, p=0.027), BRAF (V-raf-murine-sarcoma-viral-oncogene-homolog-B1, -3.0 ± 0.8, p=0.000036), CYP2B6 (Cytochrome-P450-family-2-subfamily-B-polypeptide-6, -4.6 ± 0.5, p=0.0072), FLT4 (Fms-related-tyrosine-kinase-4, -2.6 ± 0.1, p=0.044), MAPK3 (-7.6 ± 0.1, p=0.019), SNRPE (Small-nuclea- ribonucleoprotein-polypeptide-E, -4.7 ± 0.2, p=0.0040), STAT5B (Signal-transducer-and-activator-of-transcription-5B, -4.4 ± 0.3, p=0.026). Our results show that comparing to untreated LRCC metformin may repress cell proliferation and cell death of LRCC. The down-regulation the AKT1 mRNA here may reflect the complex AKT1 protein level regulation (Fig. 4A-B and Supplemental Fig. S10).
Comparing LRCC vs. non-LRCC after treatment with metformin demonstrated a dramatic up-regulation of three cell proliferation and survival oncogenes (Fig. 4D) [23] of AKT3, RET (Ret proto-oncogene), and STAT5A by 860.5 ± 123.8 (p=0.048), 66.6 ± 14.2 (p=0.0028) and 24.0 ± 1.1 (p=0.0060) folds, respectively. These results suggest that LRCC may have relative proliferation and survival advantages over non-LRCC to metformin, although LRCC proliferation may be repressed after metformin treatment comparing to untreated LRCC (Fig. 4C). Since the increase of cell proliferation and survival genes here was measured from LRCC within their microenvironment in the dynamic whole population cells treated with metformin before LRCC isolation, the increase of LRCC subpopulation after metformin treatment is likely attributed to their asymmetric cell division and survival advantage [10]. Our results suggest that metformin ineffectively kills LRCC, or LRCC are relatively resistant to metformin comparing to non-LRCC.
Metformin up-regulates pluripotency, Wnt, Notch and SHH pathway genes in LRCC
It's known that Wnt, Notch, SHH, FGF (fibroblast-growth-factor), BMP (bone-morphogenic-protein), stem cells, pluripotency and EMT pathways have import role in cancer and CSC [23-25].
Comparing LRCC treated with vs. without metformin (Fig. 5A), we identified three up-regulated Wnt pathway oncogene genes or CSC markers [23, 24]: WNT3A (4.1 ± 0.7 folds, p=0.012), Brachyury homolog T (6.6 ± 1.9 folds, p=0.0077), Jun (3.0 ± 0.3 folds, p=0.011). Comparing LRCC vs. non-LRCC treated with metformin (Fig. 5B) identified six upregulated Wnt pathway oncogenes or CSC markers [23, 24]: WNT3A (6.3 ± 0.5, p=0.0097), FZD6 (frizzled-family-receptor 6, 4.9 ± 2.6, p=0.043); BCL9 (transcription-co-activators-B-cell-CLL/lymphoma9, 3.8 ± 0.3, p=0.0076), T (Brachyury-homolog, 34.3 ± 21.2, p=0.055), TCF7 (transcription-factor-7, 5.2 ± 1.6, p=0.021) and CCND2 (Cyclin D2, 6.8 ± 1.4, p=0.041), as well as the negative Wnt pathway regulator WNT9A (12.4 ± 1.6, p=0.0046).
In the stem cell qRT-PCR array, comparing LRCC treated with vs. without metformin identified (Fig. 5C) four up-regulated genes [11, 23, 25]: BMP1 (2.9 ± 0.3, p=0.039), FGF1 (3.1 ± 0.3, p=0.017), CDH2 (cadherin 2, 4.2 ± 0.2, p=0.049) and NCAM1 (neural-cell-adhesion-molecule-1, 29.9 ± 4.4, p=3e-6); and five down-regulated genes [11, 23, 24]: ACTC1 (cardiac-muscle-alpha-actin-1, -8.4 ± 2.5, p=0.026), MYOD1 (myogenic-differentiation-1, -7.7 ± 0.5, p=0.014), SOX1 (SRY-sex-determining-region-Y-box-1, -8.3 ± 0.5, p=0.039), FGF3 (-7.9 ± 1.6, p=0.017) and WNT1 (-26.1 ± 3.6, p=0.0058).
Comparing LRCC vs. non-LRCC treated with metformin identified 17 up-regulated genes (Fig. 5D-E) [11, 24, 25]: BMP1 (4.4 ± 1.5, p=0.040), DHH (desert-hedgehog, 31.9 ± 6.7, p=0.00053), DLL3 (Notch-ligand-delta-like-3, 23.5 ± 5.4, p=0.019), DTX2 (Deltex-2, 3.4 ± 0.3, p=0.010), Notch2 (5.5 ± 1.7, p=0.014), FGF1 (4.9 ± 0.3, p=0.0076), FGFR1 (fibroblast-growth-factor-receptor-1, 11.0 ± 1.6, p=0.0003), TUBB3 (tubulin-beta-3, 11.4 ± 2.0, p=0.0022), BTRC (beta-transducin-repeat-containing, 3.3 ± 0.2, p=0.026) [26], CDC42 (Rho-GTPase-cell-division-cycle-42, 3.6 ± 0.2, p=0.026), CTNNA1 (α-catenin, 4.6 ± 0.5, p=6e-6), NCAM1 (19.0 ± 11.3, p=0.014), OCT4 (POU-class-5-homeobox-1, 2.8 ± 0.2, p=0.048), KLF4 (Krüppel-like-transcription-factor-4, 8.5 ± 1.5, p=5e-6) and BMI1 (B-lymphoma-Mo-MLV-insertion-region-1-homolog, 3.9 ± 0.4, p=3.5-5), as well as the Wnt signaling negative regulators AXIN1 (auxin-1, 4.3 ± 0.6, p=3.1e-5) and APC (adenomatous-polyposis-coli, 3.1 ± 0.2, p=3.5e-5).
In summary, we find that metformin up-regulates genes of pluripotency (OCT4, KLF4, BMI1 and BMP1), Wnt (WNT3A, FZD6, T, BCL9, TCF7 and CCND2), Notch (DLL3, NOTCH2 and DTX2), SHH (DHH) and EMT (CDH2, NCAM1 and ACTC1) pathways in LRCC [11, 23-25]. These results suggest that metformin-treated LRCC may become more stem-like, less differentiated and more metastatic. To integrate these results, we generated a molecular pathway map for LRCC treated with metformin using the IPA software (Fig. S11).
Discussion
Metformin holds an increasing promise to cancer prevention and treatment [3, 13], however, there is a paucity of evidence suggesting that metformin enhances targeted cancer therapy [2, 6, 7, 27] or inhibits blood insulin-independent PKC/MEK/ERK/JNK/AKT phosphorylation [2, 8, 9, 27], and it's unknown if metformin effectively kills our recently identified CSC LRCC [10, 12, 19]. Here we find that metformin enhances HCC therapy with sorafenib likely by repressing cell proliferation via inhibiting insulin-independent phosphorylation of multiple protein kinases PKC/MEK/ERK/JNK/AKT. Unexpectedly, we show that LRCC are relatively resistant to metformin with increase of LRCC subpopulation likely via cell proliferation and survival advantage and a stem-like gene expression profile. Our findings suggest that combining metformin with sorafenib may be able to repress the bulk of tumor cells, but as with other anti-cancer drugs, may leave CSC LRCC behind leading to cancer recurrence. Therefore, a drug targeting CSC LRCC may be critically needed to prevent cancer recurrence. These findings may contribute to the understanding of metformin's anti-cancer effects, and the development of novel drugs targeting the relatively resistant LRCC.
Due to low response rate and short survival benefit of targeted cancer therapy drugs, such as sorafenib, there is an urgent need to identify drugs that can enhance them [12]. It's reported that metformin enhances the mTOR inhibitor Everolimus (Rad001) in breast cancer [6] and EGFR antibody trastuzumab [7] in melanoma. Our finding is the first report of metformin enhancement with sorafenib.
Mechanisms of metformin's anti-cancer effects are not well known. It's reported that metformin inhibits AKT in breast cancer [8] and inhibits PKC/ERK/JUN in fibrosarcoma [9], but promotes ERK in B-RAF driven melanoma cells [27]. Our finding is the first report that meformin inhibits phosphorylation of multiple protein kinases of PKC/MEK/ERK/JUK//AKT independent of insulin in HCC. Considering the frequent over-activation of these pathways in HCC, combining metformin with sorafenib may repress the bulk of the tumor cells, reduce the tumor size, and have clinical implication in the early phase of the treatment [17, 22, 23].
Evidence suggests that CSC are responsible for the major clinical challenges of cancer drug resistance and recurrence [10-12]. Recent reports show that metformin selectively target breast CD44high/CD24low and pancreatosphere CSC [3, 7, 13-15]. Recently we reported that human LRCC are a novel class of CSC that are relatively resistant to sorafenib [10, 12]. In contradiction to other reports, here we show for the first time that metformin ineffectively kills CSC (LRCC), or CSC (LRCC) are relatively resistant to metformin. The contradiction may be caused by different CSC or histologies. Since only 10 LRCC can initiate tumor, potential cancer recurrence from LRCC may be considered when metformin is used to selectively target CSC. Therefore, a drug targeting the novel class of CSC LRCC may be critically needed to prevent cancer drug resistance and recurrence.
Studying the potential mechanisms of the relative resistence of LRCC to metformin, we found that metformin increased LRCC subpopulation likely with cell proliferation and survival advantage via upregulation of AKT phosphorylation and cell survival genes, and downregulation of cell death genes. We further found that metformin upregulated genes of pluripotency, Wnt, Notch, SHH and EMT pathways in LRCC. We proposed the potential pathway map of LRCC resistance to metformin for the first time. This pathway map may pave the way for future functional analyses of the individual genes, potentially leading to development of novel cancer therapies targeting the metformin-resistant LRCC.
Supplementary Material
Supplementary tables and figures.
Acknowledgements
We thank Michelle A Herrmann for help in NanoPro data collection and Madeleine R Heldman for her help in NanoPro data analysis and figure preparation.
Financial Support
This study was supported by the intramural grant provided by the NIH/National Cancer Institute. NCI grant 1ZIABC011005.
Author Contribution
Conception and design: HWX and IA. Collection of data: HWX, CA, TCM, JQC, GWW, AJA, SR and JDG. Data analysis and interpretation: HWX, JQC, GWW, PKG, AS, UR, SST and IA. Manuscript writing: HWX, JEM, DMH, TK, AS, SST and IA. Administrative support: UR, IA and SST. Final approval of manuscript: UR, IA and SST.
Conflict of Interest
The authors who have taken part in this study declared that they do not have anything to disclose regarding funding or conflict of interest with respect to this manuscript.
References
1. Bowker SL, Majumdar SR, Veugelers P, Johnson JA. Increased cancer-related mortality for patients with type 2 diabetes who use sulfonylureas or insulin. Diabetes care. 2006;29:254-8
2. Bost F, Sahra IB, Le Marchand-Brustel Y, Tanti JF. Metformin and cancer therapy. Current opinion in oncology. 2012;24:103-8
3. Hirsch HA, Iliopoulos D, Struhl K. Metformin inhibits the inflammatory response associated with cellular transformation and cancer stem cell growth. Proceedings of the National Academy of Sciences of the United States of America. 2013;110:972-7
4. Wurth R, Barbieri F, Florio T. New molecules and old drugs as emerging approaches to selectively target human glioblastoma cancer stem cells. Biomed Res Int. 2014;2014:126586. doi:10.1155/2014/126586
5. Saito T, Chiba T, Yuki K, Zen Y, Oshima M, Koide S. et al. Metformin, a diabetes drug, eliminates tumor-initiating hepatocellular carcinoma cells. PLoS One. 2013;8:e70010. doi:10.1371/journal.pone.0070010
6. Liu H, Scholz C, Zang C, Schefe JH, Habbel P, Regierer AC. et al. Metformin and the mTOR Inhibitor Everolimus (RAD001) Sensitize Breast Cancer Cells to the Cytotoxic Effect of Chemotherapeutic Drugs In Vitro. Anticancer research. 2012;32:1627-37
7. Vazquez-Martin A, Oliveras-Ferraros C, Del Barco S, Martin-Castillo B, Menendez JA. The anti-diabetic drug metformin suppresses self-renewal and proliferation of trastuzumab-resistant tumor-initiating breast cancer stem cells. Breast cancer research and treatment. 2011;126:355-64
8. Zakikhani M, Blouin MJ, Piura E, Pollak MN. Metformin and rapamycin have distinct effects on the AKT pathway and proliferation in breast cancer cells. Breast cancer research and treatment. 2010;123:271-9
9. Hwang YP, Jeong HG. Metformin blocks migration and invasion of tumour cells by inhibition of matrix metalloproteinase-9 activation through a calcium and protein kinase Calpha-dependent pathway: phorbol-12-myristate-13-acetate-induced/extracellular signal-regulated kinase/activator protein-1. British journal of pharmacology. 2010;160:1195-211
10. Xin HW, Hari DM, Mullinax JE, Ambe CM, Koizumi T, Ray S. et al. Tumor-initiating label-retaining cancer cells in human gastrointestinal cancers undergo asymmetric cell division. Stem cells (Dayton, Ohio). 2012;30:591-8
11. Reya T, Morrison SJ, Clarke MF, Weissman IL. Stem cells, cancer, and cancer stem cells. Nature. 2001;414:105-11
12. Xin H-W, Ambe CM, Hari DM, Wiegand GW, Miller TC, Chen J-Q. et al. Label-retaining liver cancer cells are relatively resistant to sorafenib. GUT. 2013:62
13. Hirsch HA, Iliopoulos D, Tsichlis PN, Struhl K. Metformin selectively targets cancer stem cells, and acts together with chemotherapy to block tumor growth and prolong remission. Cancer research. 2009;69:7507-11
14. Vazquez-Martin A, Oliveras-Ferraros C, Cufi S, Del Barco S, Martin-Castillo B, Menendez JA. Metformin regulates breast cancer stem cell ontogeny by transcriptional regulation of the epithelial-mesenchymal transition (EMT) status. Cell Cycle. 2010;9:3807-14
15. Bao B, Wang Z, Ali S, Ahmad A, Azmi AS, Sarkar S. et al. Metformin inhibits cell proliferation, migration and invasion by attenuating CSC function mediated by deregulating miRNAs in pancreatic cancer cells. Cancer prevention research (Philadelphia, Pa. 2011
16. Jemal A, Siegel R, Ward E, Murray T, Xu J, Thun MJ. Cancer statistics, 2007. CA: a cancer journal for clinicians. 2007;57:43-66
17. Woo HG, Park ES, Lee JS, Lee YH, Ishikawa T, Kim YJ. et al. Identification of potential driver genes in human liver carcinoma by genomewide screening. Cancer research. 2009;69:4059-66
18. Llovet JM, Ricci S, Mazzaferro V, Hilgard P, Gane E, Blanc JF. et al. Sorafenib in advanced hepatocellular carcinoma. The New England journal of medicine. 2008;359:378-90
19. Hari D, Xin HW, Jaiswal K, Wiegand G, Kim BK, Ambe C. et al. Isolation of Live Label-Retaining Cells and Cells Undergoing Asymmetric Cell Division via Nonrandom Chromosomal Cosegregation from Human Cancers. Stem Cells Dev. 2011 In Press
20. Xin HW, Ambe CM, Ray S, Kim BK, Koizumi T, Wiegand GW. et al. Wnt and the cancer niche: paracrine interactions with gastrointestinal cancer cells undergoing asymmetric cell division. J Cancer. 2013;4:447-57
21. Zheng L. Metformin as a Rare Cause of Drug-Induced Liver Injury, a Case Report and Literature Review. Am J Ther. 2013 doi:10.1097/MJT.0000000000000007
22. Calvisi DF, Ladu S, Gorden A, Farina M, Lee JS, Conner EA. et al. Mechanistic and prognostic significance of aberrant methylation in the molecular pathogenesis of human hepatocellular carcinoma. The Journal of clinical investigation. 2007;117:2713-22
23. Pylayeva-Gupta Y, Grabocka E, Bar-Sagi D. RAS oncogenes: weaving a tumorigenic web. Nature reviews. 2011;11:761-74
24. Reya T, Clevers H. Wnt signalling in stem cells and cancer. Nature. 2005;434:843-50
25. Pera MF, Tam PP. Extrinsic regulation of pluripotent stem cells. Nature. 2010;465:713-20
26. Spiegelman VS, Stavropoulos P, Latres E, Pagano M, Ronai Z, Slaga TJ. et al. Induction of beta-transducin repeat-containing protein by JNK signaling and its role in the activation of NF-kappaB. The Journal of biological chemistry. 2001;276:27152-8
27. Martin MJ, Hayward R, Viros A, Marais R. Metformin Accelerates the Growth of BRAFV600E-Driven Melanoma by Upregulating VEGF-A. Cancer discovery. 2012;2:344-55
Author contact
![]() Corresponding authors: Snorri S. Thorgeirsson, M.D., Ph.D. Head, Center of Excellence in Integrative Cancer Biology and Genomics, Chief, Laboratory of Experimental Carcinogenesis, Center for Cancer Research, National Cancer Institute, NIH, 37 Convent Drive MSC 4262, Building 37, Room 4146A, Bethesda, MD 20892-4262, USA. Phone: 1+ (301) 496-1450 (O) Phone: 1+ (301)496-1935 (W) Fax: 1+ (301) 496-0734 E-mail: snorri_thorgeirssongov Or Itzhak Avital, MD. Chair and Professor, Department of Surgery, Saint Peter's Healthcare System, Rutgers University, New Brunswick, NJ 08901, USA. Phone:917-804-4362 Email:itzhak.avitalcom.
Corresponding authors: Snorri S. Thorgeirsson, M.D., Ph.D. Head, Center of Excellence in Integrative Cancer Biology and Genomics, Chief, Laboratory of Experimental Carcinogenesis, Center for Cancer Research, National Cancer Institute, NIH, 37 Convent Drive MSC 4262, Building 37, Room 4146A, Bethesda, MD 20892-4262, USA. Phone: 1+ (301) 496-1450 (O) Phone: 1+ (301)496-1935 (W) Fax: 1+ (301) 496-0734 E-mail: snorri_thorgeirssongov Or Itzhak Avital, MD. Chair and Professor, Department of Surgery, Saint Peter's Healthcare System, Rutgers University, New Brunswick, NJ 08901, USA. Phone:917-804-4362 Email:itzhak.avitalcom.