Impact Factor ISSN: 1837-9664

 Global reach, higher impact
Global reach, higher impactJ Cancer 2016; 7(10):1205-1214. doi:10.7150/jca.14604 This issue Cite
Research Paper
MET Inhibition in Clear Cell Renal Cell Carcinoma
Zuoquan Xie1,5, Young H. Lee2, Marta Boeke1, Lucia B. Jilaveanu3, Zongzhi Liu4, Donald P. Bottaro2, Harriet M. Kluger3, Brian Shuch1 ![]()
1. Department of Urology, Yale School of Medicine
2. Urologic Oncology Branch, National Cancer Institute
3. Department of Medicine, Section of Medical Oncology, Yale School of Medicine
4. Department of Pathology, Yale School of Medicine, New Haven, USA
5. Division of Antitumor Pharmacology, State Key Laboratory of Drug Research, Shanghai Institute of Materia Medica, Chinese Academy of Sciences, Shanghai, China.
Received 2015-12-4; Accepted 2016-4-26; Published 2016-6-18
Abstract
Background: Clear cell renal cell carcinoma (ccRCC) is the most lethal form of kidney cancer. Small molecule VEGFR inhibitors are widely used but are not curative and various resistance mechanisms such as activation of the MET pathway have been described. Dual MET/VEGFR2 inhibitors have recently shown clinical benefit but limited preclinical data evaluates their effects in ccRCC.
Methods: An interrogation of the Cancer Genome Atlas (TCGA) dataset was performed to evaluate oncogenic alterations in the MET/VEGFR2 pathway. We evaluated the in vitro effects of Cabozantinib, a dual MET/VEGFR2 inhibitor, using a panel of ccRCC cell lines. Drug effects of cell viability and proliferation, migration, cell scatter, anchorage independent growth, and downstream MET/VEGFR2 signaling pathways were assessed.
Results: Twelve percent of TCGA cases had possible MET/HGF oncogenic alterations with co-occurrence noted (p<0.001). MET/HGF altered cases had worse overall survival (p=0.044). Cabozantinib was a potent inhibitor of MET and VEGFR2 in vitro in our cell line panel. PI3K, MAPK and mTOR pathways were also suppressed by cabozantinib, however the effects on cell viability in vitro were modest. At nanomolar concentrations of cabozantinib, HGF-stimulated migration, invasion, cellular scattering and soft agar colony formation were inhibited.
Conclusions: We provide further preclinical rationale for dual MET/VEGFR2 inhibition in ccRCC. While the MET pathway is implicated in VEGFR resistance, dual inhibitors may have direct anti-tumor effects in a patient subset with evidence of MET pathway involvement. Cabozantinib is a potent dual MET/VEGFR2 inhibitor, significantly inhibits cell migration and invasion in vitro and likely has anti-angiogenic effects similar to other VEGFR tyrosine kinase inhibitors. Future work involving in vivo models will be useful to better define mechanisms of potential anti-tumor activity.
Keywords: HGF, MET, clear cell Carcinoma, VEGFR2, XL184, cabozantinib.
Introduction
In the past several years VEGF Receptor (VEGFR) tyrosine kinase inhibitors have become a standard of care in metastatic renal cell carcinoma (RCC) [1]. These agents are believed to act through targeting the tumor endothelium and pericytes rather than directly on the cancer cell itself [2, 3]. While many of these agents cause tumor regression, responses are rarely durable due to upregulation of resistance pathways. MET signaling is one of the pathways implicated in resistance to VEGFR therapy in clear cell RCC [4]. The MET pathway is also important in papillary RCC where activating germline and somatic MET mutations have been described [5, 6].
The tyrosine kinase receptor MET is a cell surface molecule present in most epithelial cells. Dysregulated HGF/MET signaling is found in various carcinomas and contributes to invasiveness and metastasis [7]. Binding of its ligand, hepatocyte growth factor (HGF), activates multiple pathways involved in cell proliferation, differentiation, survival, and cytoskeletal rearrangement [7]. The HGF/MET pathway also plays an important role in the regulation of angiogenesis [8].
Various agents have been developed to target the MET pathway including tyrosine kinase inhibitors and monoclonal antibodies to either the HGF or the MET receptor. Dual MET/VEGFR2 inhibitors also are in development and are in Phase III trials in a variety of solid tumors. One such agent, cabozantinib (XL184), is a highly potent inhibitor of MET/VEGFR2 that is now FDA approved for medullary thyroid cancer. Cabozantinib has been studied in a small phase I trial in renal cell carcinoma (RCC). Although these patients were heavily pre-treated, substantial anti-tumor activity was observed (28% response rate) and progression-free survival (PFS) was longer than expected [9]. Initial results from a large phase III trial of cabozantinib showed improvement in response rate and PFS compared to everolimus in the second line setting, however, overall survival follow-up in ongoing [10].
To date there has been little investigation into the downstream molecular effects of dual MET/VEGFR2 inhibition in clear cell RCC. We set out to assess the influence of dual MET/VEGFR2 inhibition on tumor proliferation, migration, anchorage independence growth, colony formation, and signaling pathways in pre-clinical models.
Materials and Methods
Oncogenic alterations in the cancer genome atlas
The Cancer Genome Atlas (TCGA) provisional dataset contains published clinical information and cross-platform genomic data on a total of 413 Stage I-IV, central-pathology reviewed clear cell renal tumors [11, 12]. All cases contain extensive clinical annotation that can be linked to previously performed analyses of somatic copy number and exome and RNA-sequencing. Detailed information regarding the cohort and the available testing platforms has been previously described [12]. This dataset was interrogated for potential alterations (mutations or mRNA overexpression in MET, HGF, VEGFR2 (KDR), and VEGFA on cBioPortal [11, 12]. Co-occurrence in oncogenic alterations was evaluated by Fisher's exact test. Overall survival for cases with and without alterations was assessed by the Kaplan-Meier method and compared by log-rank test. Differences in mRNA expression from RNA-seq data were compared by independent T testing. Statistical significance was considered for p<0.05.
Reagents
Cabozantinib was purchased from Selleck Chemicals (Houston, TX, USA). Antibodies against pMET, total ERK, pERK, total AKT, and pAKT were obtained from Cell Signaling Technology (Danvers, MA, USA). A total MET antibody was obtained from Santa Cruz Biotechnology (Santa Cruz, CA, USA). Purified recombinant human HGF was obtained from EMD Millipore Corporation (Temecula, CA, USA). Crystal violet solution was purchased from (Sigma-Aldrich, St. Louis, MO).
Cell culture
We chose clear cell lines deficient in the VHL gene as a model of the majority of ccRCC. Clear cell kidney cancer cell lines 786-0 and A498 and kidney epithelial cell lines HK2 were previously purchased from American Type Culture Collection (ATCC) and RCC4 was purchased from the European Collection of Cell Cultures. Cell line controls for the MET and VEGFR2 pathway included mammary gland cell line (184B5) (ATCC) and 293/KDR, HEK-293 cells previously transfected with KDR (293/KDR) [13]. 786-0 cells were maintained in RMPI-1640 with 10% fetal bovine serum (FBS), A498 cells were maintained in EMEM with 10% FBS and HK2 and RCC4 cells were maintained in high glucose DEME in a standard 37˚ humidified 5% CO2 incubator.
MET and VEGFR2 protein/phospho-protein quantification
Human KDR Base Kit, Phospho-VEGFR-2 Whole Cell Lysate Kit, and Phospho/Total Met Whole Cell Lysate Kit (Meso Scale Discovery, Rockville, MD) were used for total and phospho-protein quantification. Whole cell lysates were prepared using Triton X-100 containing buffer with protease and phosphatase inhibitors (Pierce, Springfield, Il) [14]. The ratio of phosphorylated to total MET was calculated by the signal intensity from total and phospho-MET levels and total and phospho-KDR as previously described [14]. The 293/KDR cell line was used to assess the effects of cabozantinib on VEGFR2 phosphorylation due to the lack of expression in the kidney cancer cell lines.
Western blot
Serum starved cells were treated with cabozantinib for one hour and then stimulated with HGF for 20 minutes to assess the downstream mTOR, AKT and ERK pathways. A total of 10 μg of proteins were resolved using sodium dodecyl sulfate-polyacrylamide electrophoresis. Proteins were transferred to a nitrocellulose membrane and then blocked with 5% nonfat milk in TBST (10 mM Tris-HCl, 100 mM NaCl, 0.1% Tween 20). Membranes were then incubated with primary antibodies at 4˚C overnight and incubated with horseradish-peroxidase linked secondary antibody. Detection was performed as described previously [15].
Cell viability and proliferation assays
Cell viability assays were determined by a novel tetrazolium compound (3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H tetrazolium, inner salt; MTS) and an electron coupling reagent (phenazine methosulfate; PMS) using CellTiter 96 Aqueous Non-Radioactive Cell Proliferation Assay Kit (Promega, Madison, WI, USA). Briefly, cells were plated in 96-well plate overnight at a concentration of 4000 cells/well and exposed to different concentrations of cabozantinib. After 72 hours cells were treated with 20 µl of MTS for 2 hours, and OD490 was measured by multiwell plate reader (SpetraMAX250; Molecular Devices, Sunnyvale, CA). Experiments were carried with four replicates in three independent experiments. The control group was considered 100%, and other groups were calculated as the percentage of the control.
For cell proliferation, 2.5 x 105 cells/well were seeded and left overnight and then on day zero, treatment was initiated. At this time, culture media was replaced with 2% FBS that with and without 50 ng/ml of HGF stimulation. Two cabozantinib concentrations were used in the presence of HGF, 100 nM and 3 µM of cabozantinib added. On days 1, 3 and 5, cells were washed with PBS, trypsinized, resuspended in media, and counted with a TC10TM Automated Cell Counter (Bio-Rad, Hercules, CA). Both sets of experiments were carried out with three replicates.
Migration and invasion assays
Cell migration and invasion assays were performed using 8 μm pore size 24-well transwell plate (Corning Inc., Corning, NY). For the cell invasion assay, Matrigel (Corning Inc., Corning, NY) was coated onto the top chamber at a concentration of 1 mg/ml overnight. For migration and invasion assays, cells suspended in serum-free medium at a density of 1.5×105 and 5×104 cells/well, respectively. For both experiments, cells were added to the top chamber in the absence or presence of 10 nM or 100 nM of cabozantinib. Serum-free medium (0.7 ml) was added to the lower chamber with or without 50 ng/ml of HGF stimulation. After 24 hours (migration) and 40 hours (invasion), cells were fixed by 90% ethanol for 1 hour and stained by 0.1% crystal violet for 15 minutes, followed by removal of non-motile cells at the top of the filter with a cotton swab, and images were captured with light microscopy (160X magnification). The crystals stained on the lower side of the well were dissolved by 100 µl of 10% acetic acid, and the absorbance of the resulting solution was measured at 595 nm using a multi-well spectrophotometer (SpetraMAX250; Molecular Devices, Sunnyvale, CA). The HGF-stimulated group was considered 100%, and other groups were calculated as percentage of control.
Cell scatter assay
A498 cells (500 cells per well) were plated into 96-well plates and grown for 2 days. Fifty ng/ml HGF was used to stimulate cell scattering in the presence or absence of 10 nM or 100 nM of cabozantinib under 100 µl of DMEM with 10% FBS (A498) or serum-free RPMI-1640 (786-O). After 24 h, cells were fixed with 90% ethanol for 1 hour and stained with 0.1% crystal violet for 15 minutes, and washed with PBS, then photographed under microscope (160X total magnification). The experiments were carried out in four replicates in two independent experiments.
Colony formation assay
Individual cell lines were plated in six well plates at a concentration of 500 cells/well in growth media. Cells were treated with Cabozantinib at various concentrations (10, 100, and 1000 nM) and along with untreated control under 2% FBS. After 14 days, colonies were fixed with 90% ethanol for 1 hour. Fixed cells were stained with 0.1% crystal violet solution and counted. All experiments were carried out in triplicate.
Anchorage-independent growth assay
A 96 well plate assay was used to quantitatively assess anchorage independent growth. Phenol-red free DMEM was used for these experiments. A base layer of 0.5% Noble agarose (Difco, Franklin Lakes, NJ) was added to each well. 1×104 cells were suspended in 0.3% agarose were added to the base layer and allowed to set. Cells were treated with fresh DMEM, HGF, and cabozantinib every 3 days. After 2 weeks, MTT was added to the plate to quantify viable colonies by absorbance using a Victor2 plate reader (PerkinElmer, Hopkinton, MA).
Statistical analysis
SPSS 19 (IBM, Chicago, IL) was used for statistical analyses of cell viability, migration, invasion, colony formation, and soft agar assays. Independent t-test and One-Way ANOVA analyses were used for comparison of continuous variables between two or more groups, respectively. Statistical significance was considered when p<0.05.
Results
Evidence of MET and VEGR2 signaling in the TCGA
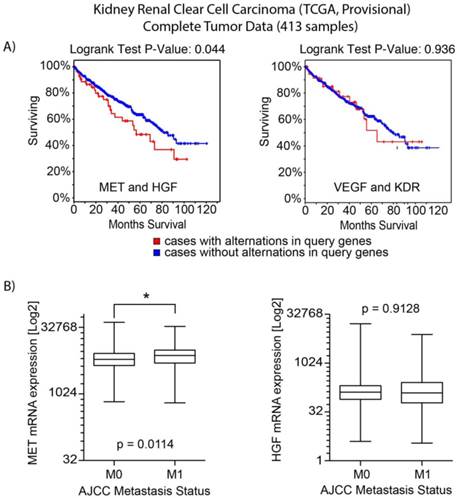
An interrogation of the provisional ccRCC dataset in The Cancer Genome Atlas (TCGA) was performed to evaluate potentially oncogenic alterations in both MET and VEGFR2 pathways. MET and HGF had alterations (amplification, mutation and/or overexpression) in 12% of cases (n=413) [12, 16]. There was no evidence of significantly reduced expression or gene deletion for either HGF or MET, as well as evidence of significant co-occurrence of HGF/MET alterations (Odds Ratio of 2.566, p<0.001). Cases with HGF or MET alterations had a significantly decreased median overall survival (54.6 vs. 80.6 months, p=0.044) (Figure 1A). Analysis by metastatic stage (M0 vs M1) demonstrated cases with metastatic disease had greater MET mRNA expression (p =0.0114) (Figure 1B). Similarly, 8% of cases had oncogenic alterations in VEGFA or KDR. Evidence of co-overexpression of mRNA transcripts encoding VEGFA and KDR was not found (p>0.4). There was no difference in overall survival between those with and without VEGFA/KDR alterations (p=0.936). While there was a trend of co-occurrence for the MET and VEGFR pathways (receptors and ligands), it was not statistically significant (p>0.16).
Analysis of TCGA data for clear cell kidney cancer. a) Overall survival analysis for MET/HGF and VEGFA/KDR (VEGFR2) oncogenic alterations. For MET/HGF altered tumors, survival was significantly decreased (p=0.044). b) Analysis of mRNA expression by M stage (M0 vs M1) demonstrated MET was increased in M1 tumors (p=0.0114). No significant difference was observed for HGF expression.
Cabozantinib mediated inhibition of MET and VEGFR2 activity and their downstream effectors
To determine the level of MET and VEGR2 and their activity in ccRCC cell lines, a two-site electroluminescent two-site immunoassay was performed. For all cell lines, treatment with HGF (1 nM) induced significant MET kinase activation within 20 min, which was completely suppressed by cabozantinib at 10 and 100 nM (Figure 2A). VEGFR2 levels in the clear cell lines lysate were undetectable despite excellent assay sensitivity (lower limit of detection <0.0007 pg/µl). In order to determine whether cabozatinib could inhibit VEGFR2 in our hands, HEK293 cells overexpressing KDR were used. This HEK293 transfected cell line (HEK/KDR) had KDR levels of 30 pg/ul. Brief VEGF stimulation demonstrated a rapid increase in pKDR levels. Treatment with 10 nM of cabozantinib inhibited VEGF stimulation by approximately 50%. At 100 nM of cabozantinib, VEGF stimulation of its receptor was abolished (Figure 2B). With all experiments total MET and total KDR did not change with HGF stimulation or cabozantinib inhibition.
Assessment of cabozantinib's inhibitory effects on a) the MET receptor on HGF stimulated clear cell RCC lines b) VEGFR2 in 293/KDR cells and c) downstream effects on control and HGF stimulated 786-0 and A498 cell lines.*P<0.05, HGF/VEGF/XL184 treated group vs control group, #P<0.05, XL184 treated group (under HGF/VEGF) vs HGF/VEGF stimulated group. Comparisons were made by One-Way ANOVA analysis.
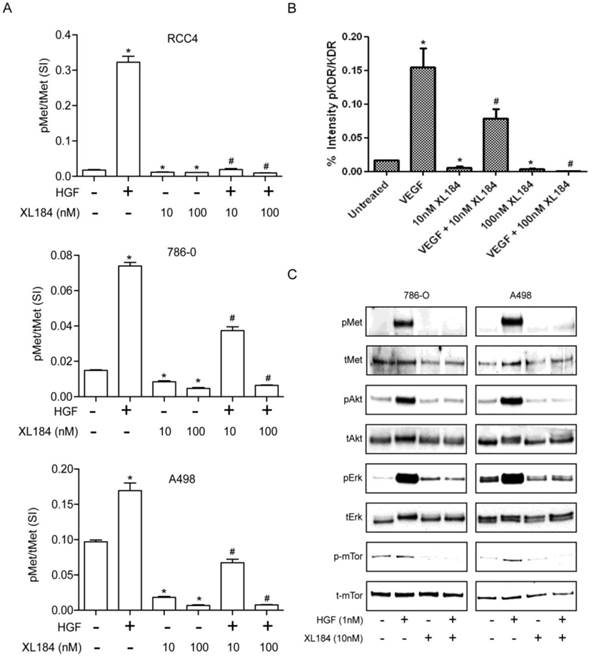
Activation of the phosphotidylinositol 3-kinase (PI3K), MAPK and mTOR pathways downstream of MET mediates HGF-driven cell survival, migration, invasion and viability. We therefore studied HGF stimulated MET, AKT, ERK and mTOR phosphorylation in 786-0 and A498 cell lines. Treatment with 10 nM cabozantinib suppressed HGF-activated pMET, pAKT, pERK and p-mTOR. (Figure 2C).
Cabozantinib effects on cell viability and proliferation
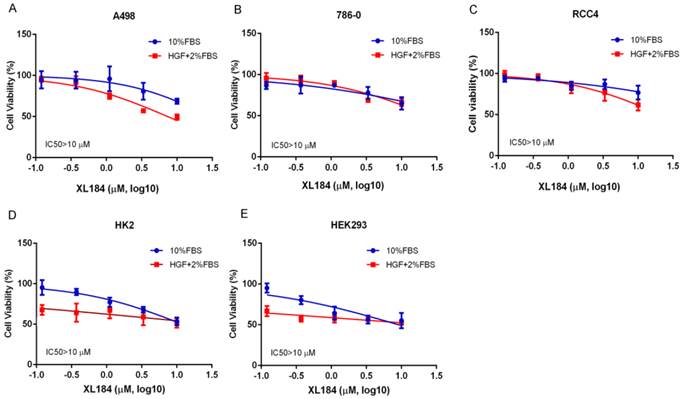
We assessed the effects of cabozantinib on cell viability at 72 hours. Treatment with cabozantinib in clear cell RCC lines had minimal effects on cell viability until concentrations were >1 µM (Figure 3A-C). For all cancer cell lines, 50% inhibition was not reached despite using concentrations as high as 10 µM. As inhibition of the MET pathway was observed at concentrations several logs less than this, blocking this pathway in vitro does not appear to potently affect viability, at least in the absence of added HGF. To assess cabozantinib effects with HGF stimulation, we lowered the concentration of FBS to 2% and added 50 ng/ml of HGF. Cell viability with cabozantinib with HGF was similar to that observed with 10% FBS except for A498, which appeared to be more sensitive at concentrations of 3.33 and 10 µM when compared with HGF-free group. For non-malignant, kidney epithelial cell lines HK2 and HEK293 (Figure 3D, 3E), 50% inhibition was also not reached at 10 µM in 10% FBS. While these non-cancer cell lines were more sensitive to a reduction of FBS with HGF stimulation, no dramatic cytotoxicity was observed with increasing concentrations of XL184.
Cell viability assays at 72 hours with cabozantinib treatment. a-c demonstrates the effects in normal growth media (10% FBS) or performed with 50 ng/ml of HGF stimulation with culture medium containing 2% FBS.
Cell proliferation was assessed in 786-0 and A498 at 72 and 120 hours with and without HGF stimulation. HGF increased cell proliferation in both cell lines. For both cell lines, at 100 nM, there was no difference in cell proliferation compared to the HGF-stimulated group, indicating HGF-stimulated cell proliferation was not inhibited by cabozantinib. At higher concentrations (3 µM) both cell lines had significantly reduced cell proliferation (Supplementary Figure 1).
Effects of cabozantinib on migration, invasion, and cell scatter
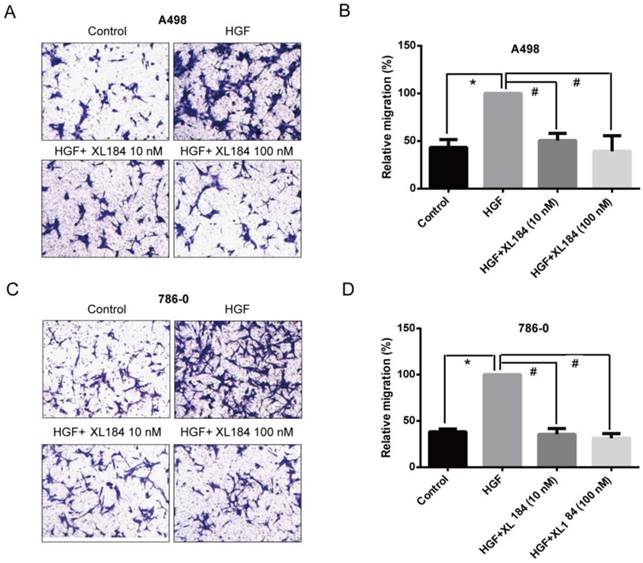
The effect of cabozantinib on HGF-stimulated RCC cell migration and invasion were determined using Transwell chamber assays. As shown in Figure 4, HGF significantly enhanced migration by A498 and 786-0 cells. After 24h, HGF stimulation resulted in a two-fold increase compared to the unstimulated conditions for A498 and 786-0 respectively. Cabozantinib treatment, however, fully suppressed HGF-mediated A498 and 786-0 migration to the level of untreated controls (Fig 4A-D). In parallel, a Matrigel invasion assay showed that HGF stimulation significantly increased A498 cell invasion, however, cabozantinib treatment suppressed HGF-induced RCC cell invasion across matrigel-coated Boyden chambers (Figures 5A-B).
Assessment of cell migration through Boyden chambers using 10 and 100 nM cabozantinib with and without HGF stimulation for A498 (a and b) and 786-0 (c and d). 160X magnification. *P<0.05 as compared to control group, #P<0.05 as compared to HGF stimulated group, comparison were made by One-Way ANOVA analysis.
Assessment of cell invasion through Matrigel using 10 and 100 nM cabozantinib with and without HGF stimulation for A498 (a and b). The effects of HGF on cellular scatter with and without cabozantinib are demonstrated for A498 (c) and 786-0 (d). 160X magnification. *P<0.05 as compared to control group, #P<0.05 as compared to HGF stimulated group, comparison were made by One-Way ANOVA analysis.
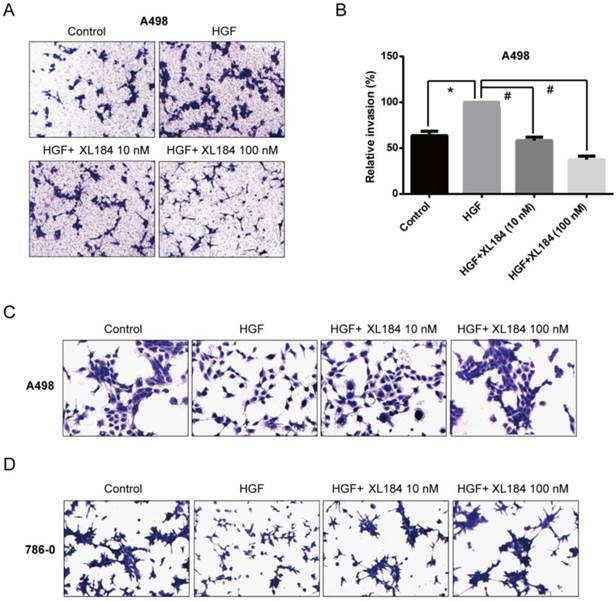
As the HGF/MET pathway is important in the process of individual cell migration from colonies, we performed an HGF-induced cellular scattering assay. Treatment with HGF stimulated cellular scattering in both A498 and 786-0. Pre-treatment with 10 or 100 nM cabozantinib blocked these effects (Figure 5C and D).
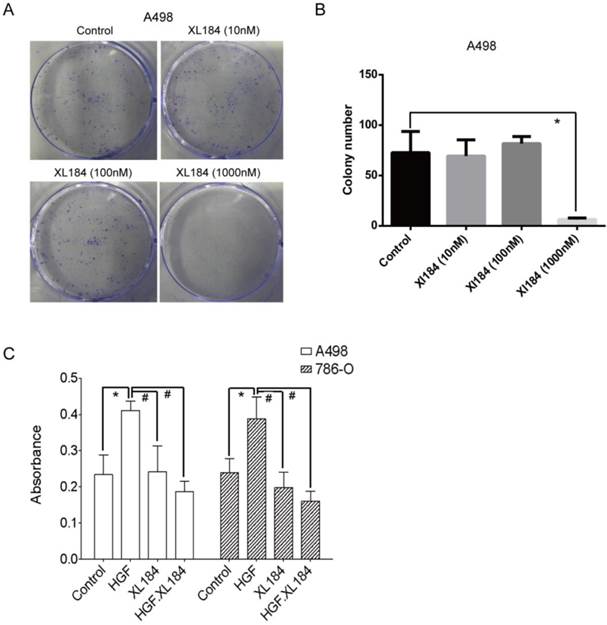
Cabozantinib inhibition of ccRCC colony formation
Colony formation assays were carried out with A498 as they readily form colonies in culture in serum-containing media in the absence of HGF. At low concentrations of cabozantinib (10 and 100 nM) there was no effect on colony formation (Figure 6A and B). At 1000 nM of treatment, colony formation was completely abolished, although at this concentration it is unclear which cabozantinib target(s) may be affected. Soft agar growth was also quantitated using a 96-well plate-based assay method. HGF stimulation significantly increased the number of colonies for both 786-0 and A498 and this increase was inhibited by cabozantinib at a concentration of 10 nM (Figure 6C).
(a and b) Colony formation assays with various concentrations of cabozantinib. Anchorage independent colony formation on soft agar in the presence of cabozantinib with and without HGF stimulation(c). *P<0.05 as compared to control group, #P<0.05 as compared to HGF stimulated group, comparison were made by One-Way ANOVA analysis.
Discussion
Novel dual MET/VEGFR2 inhibitors are particularly exciting for treatment of clear cell RCC, as these tumors have limited sensitivity to VEGFR2-inhibition alone. Clear cell is the most common subtype of renal cell carcinoma (RCC) and is characterized by VHL alterations in over 80% of tumors [17, 18]. Loss of VHL leads to upregulation of VEGF as well aberrant MET pathway activation [19-21]. In vitro studies demonstrate that in ccRCC cell lines, loss of VHL activates the MET pathway and drives a more invasive phenotype [20]. As indicated by TCGA results, a subset of ccRCC (12%) may also have oncogenic alterations in the MET/HGF pathway, including co-occurrence potentially indicative of autocrine signaling. Patients with MET/HGF altered tumors have worse survival than those without alterations, and those tumors with greater MET mRNA expression are more likely to be metastatic.
Various potent inhibitors of the MET pathways are in development [7]. Currently two randomized trials evaluate cabozantinib in clear cell RCC. The METEOR study investigates cabozantinib vs. everolimus for previously treated patients with metastatic kidney cancer (NCT01865747). With an improvement in PFS and a trend towards improvement in survival prior to cross-over [10], cabozantinib is anticipated to be available in the clinic in the second-line setting. In the front line setting, CABOSUN evaluates cabozantinib vs. sunitinib (NCT01835158) and the trial is currently ongoing. However, despite these agents moving forward clinically, there has been limited preclinical data demonstrating their effects in clear cell RCC.
We analyzed cabozantinib, a potent inhibitor of both VEGFR2 and MET kinase activity and found that at concentrations as low as 10 nM, cabozantinib can nearly abolish phosphorylation of both receptors. Prior to testing the in vitro effects on our VHL deficient clear cell lines, we measured baseline levels of MET and VEGFR2 expression. In all clear cell lines, VEGFR2 was undetectable despite using a highly sensitive assay, although expression and activity might be induced in tumors in vivo. Therefore, assessment of the in vitro effects of VEGFR tyrosine kinase inhibitors may result from their off target effects. Huang and colleagues reported similar findings and found that stimulation with VEGF, VEGFR knockdown, or administration of inhibitory concentrations of VEGFR small molecule inhibitors did not affect cell proliferation [22]. In vitro analyses evaluating the direct effects of VEGFR2 inhibition likely requires a model system utilizing human endothelial cells. Based on these findings, we expected that our in vitro effects of MET/VEGFR2 inhibition with cabozantinib was due to direct inhibition of the MET pathway in clear cell RCC.
We interrogated the effects of dual MET/VEGFR2 inhibition in our clear cell lines in the presence of HGF stimulation. With HGF stimulation, treatment with a low dose of cabozantinib (10 nM) was able to inhibit MET phosphorylation in all cell lines. Well-known mediators of the MET pathway, mTOR, AKT and ERK, were investigated, as these signaling pathways are important mediators of tumorigenesis. Treatment was sufficient to block the stimulatory effects of HGF in each of these downstream signaling pathways. Knowing that 10 nM or greater was sufficient to inhibit MET signaling, we used similar concentrations to investigate the in vitro effects on growth, invasion, migration, colony formation, and agar growth. Treatment with cabozantinib did not appear to affect cell viability until >1µM concentrations were used. Other VEGFR inhibitors have demonstrated in vitro effects on cell proliferation at µM concentrations, well above the concentrations necessary to block VEGFR2 signaling [15, 22]. These findings may also suggest other targets are responsible for growth inhibitory effect in vitro.
HGF stimulation increases cellular migration, invasion, and scattering in clear cell RCC. Blockage with cabozantinib abolished many of these effects likely due to direct inhibition of the MET pathway. As the MET signaling plays a critical role in cancer progression [7, 23], the direct tumor effects of this class of agent may serve an important therapeutic role. Despite limited cytotoxic effects, the ability to inhibit cellular motility and possibly the development of metastases makes MET inhibition an interesting target in clear cell RCC. It has been suggested that these properties make MET inhibitors an interesting choice in the adjuvant setting [24], an unmet need in kidney cancer where no agent has yet proven efficacious [25, 26].
Limited preclinical studies have been conducted on MET and MET/VEGFR2 inhibition in ccRCC. Gibney and colleagues have investigated MET inhibition using ARQ197 and SU11274 on a panel of clear cell RCC cell lines [15]. Both agents demonstrated MET inhibition, but SU11274 exhibited inhibition in the nM range and had downstream effects in pAKT pathway. Similar to our findings with cabozantinib, SU11274 also did not affect cell viability in concentrations under 5 µM. ARQ197, which had less potent MET inhibition, had a better influence on cell viability, with an IC50 of around 500nM for all cell lines [15]. This is likely due to off-target effects, as ARQ197 also has direct cytotoxic effects through tubulin rather than through the MET pathway [27, 28].
In summary, although dual MET/VEGFR2 inhibition has limited effect on viability in ccRCC in vitro, the effects on migration, cell scatter, anchorage independent growth, and signaling pathways downstream of MET/VEGFR2 are seen in low nano-molar concentrations of cabozantinib. Agents targeting both pathways therefore hold promise in kidney cancer for the effects on cell migration and invasion in addition to the known in vivo effects on angiogenesis. Limitations of our study include the lack of VEGFR2 expression in our clear cell RCC lines, likely hindering efforts to study direct cellular effects. While prior immunohistochemical studies demonstrate cytoplasmic and membranous expression of VEGFR2 within the tumor [29], perhaps in vivo expression is influenced by the tumor microenvironment. Ideally it would be useful to have human clinical specimens before and after initiation of MET-targeted systemic therapy but no such cohort exists at this time. Neoadjuvant protocols prior to nephrectomy have been proposed with this class of agent, but none has been initiated to date. Future work involving endothelial cell models or murine models will be useful to better define the mechanisms of anti-tumor activity.
Supplementary Material
Supplementary figure 1.
Abbreviations
ccRCC: clear cell renal cell carcinoma
MET: Hepatocyte Growth Factor Receptor
HGF: Hepatocyte growth factor
VEGFR/KDR: Vascular endothelial growth factor receptor
VHL: Von Hippel-Lindau
TCGA: The Cancer Genome Atlas
VEGF: Vascular endothelial growth factor
PI3K: Phosphotidylinositol 3-kinase
MAPK: Mitogen-activated protein kinase
mTOR: Mammalian target of rapamycin
AKT: RAC-alpha serine/threonine-protein kinase
ERK: Extracellular Signal-Regulated Kinase
IC50: Half Maximal Inhibitory Concentration
MTS: 3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H tetrazolium, inner salt.
FDA: Food and Drug Administration.
Acknowledgements
This publication was made possible by CTSA Grant Number KL2 TR000140 from the National Center for Advancing Translational Science (NCATS), a component of the National Institutes of Health (NIH). This work was also supported in part by the Intramural Research Program of the NIH, National Cancer Institute, Center for Cancer Research. Its contents are solely the responsibility of the authors and do not necessarily represent the official view of NIH. We acknowledge the funding of China Scholarship Council for Zuoquan Xie (NO.201404910330).
Author Contributions
BS, YL, DB, ZX, and HK assisted in the conception and design of the study. ZX, BS, YL, and MB performed acquisition of data. LJ, HK, MB, and DB also provided materials and technical support. All authors (BS, YL, MB, LJ, DB, ZX, and HK) provided critical input into the drafting of the manuscript and the final approval.
Competing interests
All authors report no competing interests.
References
1. (NCCN) NCCN. Kidney Cancer. NCCN Clinical Practice Guidelines in Oncology. 2012
2. Bergers G, Song S, Meyer-Morse N, Bergsland E, Hanahan D. Benefits of targeting both pericytes and endothelial cells in the tumor vasculature with kinase inhibitors. The Journal of clinical investigation. 2003;111:1287-95
3. Rini BI. Vascular endothelial growth factor-targeted therapy in renal cell carcinoma: current status and future directions. Clinical cancer research: an official journal of the American Association for Cancer Research. 2007;13:1098-106
4. Shojaei F, Lee JH, Simmons BH, Wong A, Esparza CO, Plumlee PA. et al. HGF/c-Met acts as an alternative angiogenic pathway in sunitinib-resistant tumors. Cancer Res. 2010;70:10090-100
5. Schmidt L, Duh FM, Chen F, Kishida T, Glenn G, Choyke P. et al. Germline and somatic mutations in the tyrosine kinase domain of the MET proto-oncogene in papillary renal carcinomas. Nature genetics. 1997;16:68-73
6. Schmidt L, Junker K, Nakaigawa N, Kinjerski T, Weirich G, Miller M. et al. Novel mutations of the MET proto-oncogene in papillary renal carcinomas. Oncogene. 1999;18:2343-50
7. Cecchi F, Rabe DC, Bottaro DP. Targeting the HGF/Met signalling pathway in cancer. European journal of cancer. 2010;46:1260-70
8. Zhang YW, Su Y, Volpert OV, Vande Woude GF. Hepatocyte growth factor/scatter factor mediates angiogenesis through positive VEGF and negative thrombospondin 1 regulation. Proc Natl Acad Sci U S A. 2003;100:12718-23
9. Choueiri TK, Kumar Pal S, McDermott DF, Morrissey S, Ferguson KC, Holland J. et al. A phase I study of cabozantinib (XL184) in patients with renal cell cancer. Ann Oncol. 2014
10. Choueiri TK, Escudier B, Powles T, Mainwaring PN, Rini BI, Donskov F. et al. Cabozantinib versus Everolimus in Advanced Renal-Cell Carcinoma. N Engl J Med. 2015;373:1814-23
11. Cerami E, Gao J, Dogrusoz U, Gross BE, Sumer SO, Aksoy BA. et al. The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer discovery. 2012;2:401-4
12. Cancer Genome Atlas Research N. Comprehensive molecular characterization of clear cell renal cell carcinoma. Nature. 2013;499:43-9
13. Cecchi F, Pajalunga D, Fowler CA, Uren A, Rabe DC, Peruzzi B. et al. Targeted disruption of heparan sulfate interaction with hepatocyte and vascular endothelial growth factors blocks normal and oncogenic signaling. Cancer Cell. 2012;22:250-62
14. Lee YH, Apolo AB, Agarwal PK, Bottaro DP. Characterization of HGF/Met Signaling in Cell Lines Derived From Urothelial Carcinoma of the Bladder. Cancers. 2014;6:2313-29
15. Gibney GT, Aziz SA, Camp RL, Conrad P, Schwartz BE, Chen CR. et al. c-Met is a prognostic marker and potential therapeutic target in clear cell renal cell carcinoma. Ann Oncol. 2013;24:343-9
16. Gao J, Aksoy BA, Dogrusoz U, Dresdner G, Gross B, Sumer SO. et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Science signaling. 2013;6:pl1
17. Nickerson ML, Jaeger E, Shi Y, Durocher JA, Mahurkar S, Zaridze D. et al. Improved identification of von Hippel-Lindau gene alterations in clear cell renal tumors. Clinical cancer research: an official journal of the American Association for Cancer Research. 2008;14:4726-34
18. Sato Y, Yoshizato T, Shiraishi Y, Maekawa S, Okuno Y, Kamura T. et al. Integrated molecular analysis of clear-cell renal cell carcinoma. Nat Genet. 2013;45:860-7
19. Nakaigawa N, Yao M, Baba M, Kato S, Kishida T, Hattori K. et al. Inactivation of von Hippel-Lindau gene induces constitutive phosphorylation of MET protein in clear cell renal carcinoma. Cancer research. 2006;66:3699-705
20. Peruzzi B, Athauda G, Bottaro DP. The von Hippel-Lindau tumor suppressor gene product represses oncogenic beta-catenin signaling in renal carcinoma cells. Proceedings of the National Academy of Sciences of the United States of America. 2006;103:14531-6
21. Maxwell PH, Wiesener MS, Chang GW, Clifford SC, Vaux EC, Cockman ME. et al. The tumour suppressor protein VHL targets hypoxia-inducible factors for oxygen-dependent proteolysis. Nature. 1999;399:271-5
22. Huang D, Ding Y, Li Y, Luo WM, Zhang ZF, Snider J. et al. Sunitinib acts primarily on tumor endothelium rather than tumor cells to inhibit the growth of renal cell carcinoma. Cancer Res. 2010;70:1053-62
23. Benvenuti S, Comoglio PM. The MET receptor tyrosine kinase in invasion and metastasis. Journal of cellular physiology. 2007;213:316-25
24. Surriga O, Rajasekhar VK, Ambrosini G, Dogan Y, Huang R, Schwartz GK. Crizotinib, a c-Met inhibitor, prevents metastasis in a metastatic uveal melanoma model. Mol Cancer Ther. 2013;12:2817-26
25. Haas N, Manola J, Uzzo R, Atkins M, Wilding G, Pins M. et al. Initial results from ASSURE (E2805): Adjuvant sorafenib or sunitinib for unfavorable renal carcinoma, an ECOG-ACRIN-led, NCTN phase III trial. J Clin Oncol. 2015:33
26. Messing EM, Manola J, Wilding G, Propert K, Fleischmann J, Crawford ED. et al. Phase III study of interferon alfa-NL as adjuvant treatment for resectable renal cell carcinoma: an Eastern Cooperative Oncology Group/Intergroup trial. J Clin Oncol. 2003;21:1214-22
27. Basilico C, Pennacchietti S, Vigna E, Chiriaco C, Arena S, Bardelli A. et al. Tivantinib (ARQ197) displays cytotoxic activity that is independent of its ability to bind MET. Clinical cancer research: an official journal of the American Association for Cancer Research. 2013;19:2381-92
28. Aoyama A, Katayama R, Oh-Hara T, Sato S, Okuno Y, Fujita N. Tivantinib (ARQ 197) exhibits antitumor activity by directly interacting with tubulin and overcomes ABC transporter-mediated drug resistance. Molecular cancer therapeutics. 2014;13:2978-90
29. Kluger HM, Siddiqui SF, Angeletti C, Sznol M, Kelly WK, Molinaro AM. et al. Classification of renal cell carcinoma based on expression of VEGF and VEGF receptors in both tumor cells and endothelial cells. Lab Invest. 2008;88:962-72
Author contact
![]() Corresponding author: Brian Shuch, MD. Department of Urology, Yale School of Medicine, PO Box 208058 New Haven, CT 06520-8058, Brian.shuchedu (T) 203-785-2815 (F) 203-785-6475.
Corresponding author: Brian Shuch, MD. Department of Urology, Yale School of Medicine, PO Box 208058 New Haven, CT 06520-8058, Brian.shuchedu (T) 203-785-2815 (F) 203-785-6475.