Impact Factor ISSN: 1837-9664

 Global reach, higher impact
Global reach, higher impactJ Cancer 2016; 7(10):1360-1373. doi:10.7150/jca.15155 This issue Cite
Research Paper
Transduction of Recombinant M3-p53-R12 Protein Enhances Human Leukemia Cell Apoptosis
Tsung Chi Lu1, 2, Guan- Hao Zhao1, Yao Yun Chen1, 3, Chia-Ying Chien1, Chi-Hung Huang2, Kwang Hui Lin4, 5, Shen Liang Chen1 ![]()
1. Department of Life Sciences, National Central University, Jhongli 32001, Taiwan;
2. Taiwan Advance Bio-Pharma Inc., New Taipei City 22180, Taiwan.
3. The Affiliated Jhongli Senior High School of National Central University, Jhongli 32047, Taiwan;
4. Department of Biochemistry, Chang Gung University, Taoyuan 333, Taiwan;
5. Liver Research Center, Chang Gung Memorial Hospital, Linko, Taoyuan 333, Taiwan.
Received 2016-1-31; Accepted 2016-5-22; Published 2016-6-28
Abstract
Tumor suppressor protein p53 plays important roles in initiating cell cycle arrest and promoting tumor cell apoptosis. Previous studies have shown that p53 is either mutated or defective in approximately 50% of human cancers; therefore restoring normal p53 activity in cancer cells might be an effective anticancer therapeutic approach. Herein, we designed a chimeric p53 protein flanked with the MyoD N-terminal transcriptional activation domain (amino acids 1-62, called M3) and a poly-arginine (R12) cell penetrating signal in its N-and C-termini respectively. This chimeric protein, M3-p53-R12, can be expressed in E. coli and purified using immobilized metal ion chromatography followed by serial refolding dialysis. The purified M3-p53-R12 protein retains DNA-binding activity and gains of cell penetrating ability. Using MTT assay, we demonstrated that M3-p53-R12 inhibited the growth of K562, Jurkat as well as HL-60 leukemia cells carrying mutant p53 genes. Results from FACS analysis also demonstrated that transduction of M3-p53-R12 protein induced cell cycle arrest of these leukemia cells. Of special note, M3-p53-R12 has no apoptotic effect on normal mesenchymal stem cells (MSC) and leukocytes, highlighting its differential effects on normal and tumor cells. To sum up, our results reveal that purified recombinant M3-p53-R12 protein has functions of suppressing the leukemia cell lines' proliferation and launching cell apoptosis, suggesting the feasibility of using M3-p53-R12 protein as an anticancer drug. In the future we will test whether this chimeric protein can preferentially trigger the death of malignant cancer cells without affecting normal cells in animals carrying endogenous or xenographic tumors.
Keywords: apoptosis, cancer, leukemia, MyoD, p53, and protein drug.
Introduction
The human tumor suppressor gene p53 is located on the short arm of chromosome 17 and encodes a DNA-binding transcription factor [1], which is considered as “the guardian of the genome” and plays critical roles in recognizing and repairing DNA damage by DNA damage response (DDR) pathway. p53 also induces programmed cell death when the genetic damages of cells are beyond repair. The functions of p53 protein are modulated by various post-modifications, including phosphorylation, ubiquitination, ubiquitin-like modifications (sumoylation, and neddylation), acetylation and methylation, which can affect p53's stability, DNA binding and transcriptional activation, and even promoter selectivity. For instance, phosphorylated p53 is translocated to nucleus, where it functions as a transcription factor to launch cell autophagy and apoptosis [2-4]. Zinc ion is crucial for the folding and stabilization of p53 and it also contribute to the DNA-binding activity of p53 [5, 6]. Former studies have found that chelating zinc ions from p53 protein by metallothionein, a recombinant chelator protein, reduces the transcriptional activities [7]. Meanwhile, zinc ion can also restore misfolded and mutant p53 conformation to trigger cancer cells death through apoptotic cascade [8].
p53 initiates apoptosis by down-regulating Bcl-2 [9] and up-regulating Bax, Bid, PUMA, and NOXA [10-12]. The accumulated PUMA and NOXA bind to the Bcl-2 family and Mcl-1, respectively, and block their anti-apoptotic activities to trigger the apoptotic cascade in mitochondria [13]. In addition to initiating apoptosis transcriptionally, p53 can also trigger apoptosis by direct binding to Bcl-2 to shut down anti-apoptosis signaling or interacting with Bax to result in mitochondrial outer membrane permeabilization (MOMP) [14]. Accumulated evidences show that transcription-independent functions of p53 co-operate with its transcription-dependent functions in the induction of apoptosis. Therefore, p53 plays a decisive role in crossing the cytoplasmic and mitochondrial apoptotic threshold as well as triggering the program of cell death.
In normal cells, the level of p53 protein is too low to be detected due to MDM2 mediated ubiquitination and subsequent degradation in proteasome; therefore, MDM2 serves as a p53 negative regulator [15]. Mono-ubiquitination of p53 by basal level of MDM2 results in nuclear export and accumulation in cytosol, which leads to mitochondrial p53 translocation as well. On the other hand, higher levels of MDM2 promote p53's poly-ubiquitination and degradation in nucleus [16, 17]. Besides, MDM2 is also upregulated by p53 transcriptionally; therefore the appropriate 'digital quanta' of p53 in p53-MDM2 dynamics feedback loop determines the cell's fate of survival (damage repaired) or death (apoptosis) [18]. RITA and Nutlins are two small molecules that induce cancer cell apoptosis by reactivating p53 transcriptional conformation and abrogating p53-MDM2 complex, respectively [19]. Collectively, no matter p53 located in nucleus, cytoplasm or mitochondrion, it supports both transcription-dependent and -independent apoptosis in cancer cells.
While cells suffer genotoxic stress during early carcinogenesis, p53 participates in the DNA damage response (DDR) pathway to safe guard the integrity of genome. In case of DNA damage is too serious to be repaired, p53 instead induces apoptosis or senescence. Hence, p53 functions as an important anti-carcinogenesis barrier to prevent the survival and proliferation of malignant cells. As a transcription factor, the expression of p53 is induced by various stresses, resulting in cell cycle arrest, apoptosis, autophagy, and senescence. Both ATR and ATM kinase are positive regulators of p53 that phosphorylate and stabilize p53 to deter the malignant conversion while cells are suffering the genotoxic stress (DNA damage) in vivo [20]. The observation that exogenous expression of wild-type p53 inhibits the Ras and E1A mediated transformation strongly supports its tumor-suppressor properties [21, 22]. However, mutations or depletion of p53 gene generates p53 isoforms with loss-of-function or other defects contributing to tumor progression. Some aberrant p53 proteins dominant-negatively inhibit the transcriptional activity of wildtype p53 and thus unlock tumor cells from the safe guard of the DDR system [23]. Therefore, restoring the normal p53 activities might conquer the adverse effect of accumulated p53 mutation to reactivate the programed cell death of malignant cells.
Gendicine™ is an adenovirus-mediated p53 gene therapy whose clinical trial has been approved by the State Food and Drug Administration of China (SFDA) since 2003 [24]. Unfortunately, current clinical studies of p53 gene therapies, including Gendicine™, do not provide enough evidence of safety to convince FDA for their clinical application yet. In addition to gene therapy and small molecule inhibitors, restoring the p53 function in cancer cells by use of recombinant protein is another choice. Hence, developing a cell penetrable and gene regulation competent p53 protein becomes an alternative strategy to fulfill this purpose [25, 26]. Protein transductions with cell-penetrating peptides (CPP), such as poly-arginine, HIV TAT, and Antp peptides, are currently well established approaches for delivering proteins and other macromolecular biomaterials into target cells [27-29].
According to clinical statistics, p53 is frequently mutated or defective in human cancers with approximately 50%, and mostly often observed in ovary, colon, esophagus, head & neck and lung cancer [30]. Even though the prevalence of p53 mutation in hematological cancer is lower than solid tumors [31], the survival time in chronic lymphocytic leukemia (CLL) patients with wild-type p53 are double longer than patients with mutated p53 [20]. It implies that even p53 is irrelevant in the carcinogenesis of leukemia, it might at least suppress leukemia progression via transcriptional-dependent and -independent mechanisms. Thus, p53 should be an attractive tool in cancer therapy, even for treating leukemia.
In this study, we would like to create a chimeric p53 protein with enhanced activity and cell-penetrating capability so it can be applied as a recombinant protein. A poly-arginine domain (R12) is used as cell penetrating signal and the M3 activation domain derived from MyoD is used as an effective transcriptional activation domain [32]; therefore, coupling these two domains to p53 might create an dominant active p53 protein that can enter target cells automatically to selectively kill cancer cells. Here, we successfully cloned, expressed, and purified the chimeric protein, M3-p53-R12. Transduction of M3-p53-R12 into tumor cells preferentially repressed cell growth and initiated cell apoptosis in p53 mutated or null leukemia cell lines. Thus, recombinant M3-p53-R12 protein might have great potential for cancer treatment in the future.
Materials and Methods
Plasmid construction
The pET32a-LpRH plasmid was derived from pET32a vector by inserting a pfu amplified PCR product (encoding a linker, 12 arginines, and 7 histidines) into the blunted NdeI site, which inactivated the downstream NdeI site but kept the upstream NdeI site intact. The M3 domain was amplified by PCR and ligated into the upstream NdeI site of pET32a-LpRH to generate pET32a-M3-LpRH plasmid. The human p53 gene was amplified from pCDNA3-p53 by primers (5'-CGCGGATCCATGGAGGAGCCGCAGTCAGAT-3' and 5'-CGCGGATCCGTCTGAGTCAGGCCCTTCTGT-3') and inserted into the BamHI site of pET32a-M3-LpRH plasmid to generate pET32a-M3-p53-LpRH plasmid. The protein expressed from this plasmid is called as M3-p53-R12 in short.
Cell Lines and cell culture
The E. coli strain BL21(DE3)-CodonPlus-RP used as protein expression host was purchased form Agilent and cultured in LB broth. Three cell lines, HL-60, Jurkat and K562 cells used in this article were purchased from BCRC. These cell lines were maintained in RPMI medium supplemented with 10% fetal bovine serum (FBS), 100 units/ml penicillin, and 100 μg/ml streptomycin in a CO2 incubator with 5% CO2 at 37oC
Protein expression and purification
The BL21(DE3)-Codon Plus-RP strain (Agilent) of Escherichia coli cells were used as expression hosts. Protein was induced by 0.5 mM IPTG and 0.1 mM ZnSO4 for 3 hrs at 37 oC after OD600 reached 0.6. Cells were harvested by centrifugation and cell pellet were re-suspended in PBS and sonicated, then inclusion body was collected by centrifugation. Inclusion body was re-suspended in buffer A [100mM Tris-HCl, 100 mM NaCl, 8M urea, pH7.8] and DTT was added to final concentration of 50 mM, followed by stirring for 2hrs and titrated to pH 2.0 by 1M HCl. Then, the solution was neutralized to pH7.8 by 1M NaOH titration before centrifuged to collect the soluble fraction. The soluble fraction was applied into Ni charged Chelating Sepharose (GE healthcare) and impurities were removed by several washes with wash buffer (50 mM and 100 mM imidazole in buffer A). The M3-p53-R12 protein was eluted with 250 and 500 mM imidazole in buffer A and was concentrated before changed to refolding buffer [100mM Tris-HCl, 1M L-arginine, 10% glycerol, pH7.8] by Amicon ultra (Millipore) to refold the M3-p53-R12 protein. The refolded protein was further dialyzed against storage buffer [50 mM Tris-HCl, 100 mM NaCl, 3% glycerol, pH7.8] for long-term storage.
Protein oligomerization assay
Because the cross-link ability of glutaraldehyde was inactive in Tris buffer, hence, refolded protein was further dialyzed against D-PBS (Dulbecco's PBS) for cross-link assay. Purified protein in volume of 50 μl was treated with 2.5 μl of 0.46% glutaraldehyde and incubated on ice for 20 min. The crosslink reaction was stop by adding 10 μl of 1M Tris buffer [pH8.0], and then equal volume of 2X Laemmli sample buffer was added before analyzed in 8% SDS-PAGE to view the oligomerized proteins.
Electrophoretic mobility shift assay (EMSA)
The specific oligonucleotides containing the 41-mer consensus p53 DNA-binding sequence was generated by annealing 2 primers (5'-AgACATgTCCAgACATgTCCgAACATgTCCCAACATgTTgT-3' and 5'-ACAACATgTTgggACATgTTCggACATgTCTggACATgTCT-3') at concentration of 100 μM following the anneal program: 95 oC 3 min, 55 oC 1 min, and 25 oC 3 min. About 2 μM of DNA and 0.5 mg/ml (7.7 μM) of M3-p53-R12 protein were used in each reaction, and mixture of DNA and various amounts of M3-p53-R12 protein was incubated at 37 oC for 10 min. The reaction was stop by adding loading dye and the mixture was resolved in 1% agarose gel (1x TBE) followed by ethidium bromide staining to view the oligonucleotides.
M3-p53-R12 transduction ability and translocation analysis
Three leukemia cell lines, HL-60, Jurkat, and K562 were seeded (105 cells/well) into 6-well plate dishes one day before M3-p53-R12 protein was add to medium. After transduction for 2 hrs, cells were harvested for analyzing the protein penetrative ability. Nuclear and cytosolic fractions were prepared using the Nuclear Extraction Kit by following supplier's protocol (Nuclear Extraction Kit; Biovision). The presence of M3-p53-R12 protein in various cell fractions was analyzed by Western Blot.
Cell proliferation assay (MTT assay)
For single dose experiments, leukemia cells were seeded (104 cells/well) onto 96-well plates, and incubated with different concentrations of M3-p53-R12 protein for 24, 48, and 72 hrs. For multiple dose assays, daily renewal of protein was achieved by removing half of the medium after the plates were spun at 300xg for 5 min before medium with 2-fold concentration of M3-p53-R12 protein was added to reach the specified concentration in each well. At the end of incubation, 50 μl MTT (2 mg/ml) was added into the wells and incubated for additional 1 hr before the plates were spun at 300xg for 5 min before the medium was removed. The 100 μl DMSO was added to each well and incubated for additional 10 min and the absorbance was read at 570 nm.
FACS analysis
Cells were seeded at 105 cells/ml onto 6-well plates and incubated with 10 μg/ml M3-p53-R12 protein for additional 8 and 54 hrs. Cells treated with Doxorubicin (2 μg/ml; Sigma) for 8 hr served as a positive control. Cells were collected and washed twice with cold PBS and then re-suspended, and apoptotic cells were determined with FITC Annexin V Apoptosis Detection Kit (BD Biosciences, USA) according to the manufacture's instruction. The cell suspension was analyzed by BD FACSCanto II flow cytometer.
Soft agar assay
Leukemia cells were seeded (104 cells/well) onto 96-well plates, and transduced with 10 μg/ml M3-p53-R12 protein for overnight. A total of 200 initial cells/ well were seeded in the growth medium containing 0.5% soft agar (top layer) on top of the 0.5% soft agar (base layer) in 6-well plates and cultured for additional 10~14 days. The appeared colonies were observed under microscope and colony number was counted after stained with 0.05% (w/v) crystal violet (in 5% methanol) and washed with 20% ethanol.
Mesenchymal stem cell (MSC) isolation and culture
MSCs used in this research were derived from the Wharton's Jelly of the human umbilical cord. In brief, the cords were cut into 3~5 cm long fragments and with the blood inside the cords removed. These fragments were further cut into small pieces by scissors and followed by digestion with collagenase type II (300 unit/ml, with 3 mM CaCl2) for 30 min at 37 oC. Further digestion with 0.5% trypsin at 37 oC was lasted for 30 min before neutralized with FBS. Then, 5X volumes of PBS was added and the slurry was filtered through a nylon mesh to remove un-digested clumps. MSCs in filtrate were harvested by centrifugation and cultured in DMEM medium supplemented with 10% fetal bovine serum (FBS), 100 units/ml penicillin, 100 μg/ml streptomycin and 10 ng/ml bFGF. Use of the MSCs from human subjects has been approved by Institutional Review Board (CGHIRB number: CT100036).
Results
Expression and purification of recombinant M3-p53-R12 chimeric protein
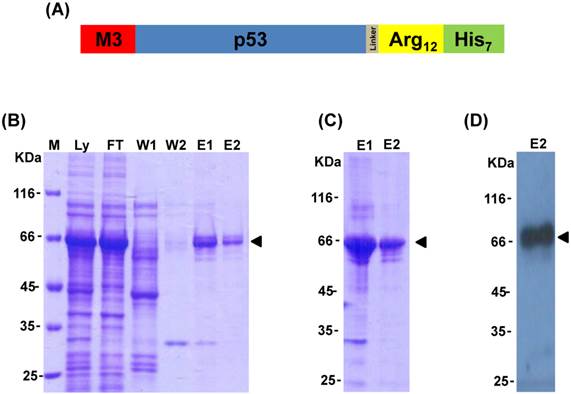
MyoD has been recognized as one of the most powerful lineage determining transcription factors, and the fusion of its N-terminal activation domain (amino acids 1-60, named as M3) to other transcription factors can highly increase their transactivational activity [32]. To enhance p53 transactivational activity, the M3 domain was fused to the N-terminus of human tumor suppressor protein p53 coding sequence that was followed by a poly-arginine domain (R12) as well as a His-tag in its C-terminus to create the M3-p53-R12 chimeric factor (Fig. 1A). A linker sequence was inserted between p53 and the R12 domains to allow their free folding. After induction with IPTG in BL21(DE3)-CodonPlus-RP cells, M3-p53-R12 protein was abundantly expressed and could be purified using immobilized metal ion chromatography followed by serial refolding dialysis procedure (Fig. 1B and C). The identity of the purified protein was confirmed by Western blot analysis using antibody against p53 (Fig. 1D). During purification procedure, we also found that zinc ion was important for stabilizing p53 core structure (data not shown), as suggested by previous studies [6, 8].
Expression and purification of M3-p53-R12 protein. (A) Schematics representing the chimeric M3-p53-R12 protein used in this study. (B) The expression of M3-p53-R12 in E.coli was induced by IPTG and later purified by Ni-NTA column. The induction and purification process was monitored with coomassie blue stained SDS-PAGE gels. Ly: E. coli cell lysate; FT: flow-through fraction; W1: 50 mM imidazole buffer wash; W2: 100 mM imidazole buffer wash; E1: 250 mM imidazole buffer eluent; E2: 500 mM imidazole buffer eluent. (C) The quality of the M3-p53-R12 protein after refolding with dialysis of the different eluted fractions. (D) Western blot analysis of refolded M3-p53-R12 protein by using anti-His tag antibody.
Characterization of refolded M3-p53 protein
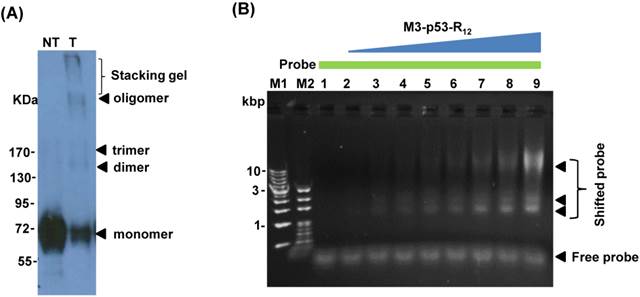
p53 binds to target cis-elements in the form of tetramer and the formation of tetramer is important to its transactivational activity on target genes. Several reports showed that purified p53 protein has quaternary structure and p53 with tetramerization activity can form oligomers in vitro [26, 33], which is a good indicator of its DNA-binding activity. To verify the oligomerization potency of purified M3-p53-R12, we performed the oligomerization test by chemical crosslinking reagent glutaraldehyde (0.02%) and we found that purified M3-p53-R12 protein form higher structures spontaneously and with structural polymorphism (Fig.2A), demonstrating its oligomerization potency. To further characterizing its DNA binding activities, the electrophoretic mobility shift assay (EMSA) was employed. We found that the purified M3-p53-R12 could bind to the consensus p53-targetting sequence (protein/DNA molar ratio from 0 to 35) and thus confirmed that this chimeric p53 recombinant protein maintained its DNA binding activity (Fig. 2B).
Characterization of the M3-p53-R12 protein. (A) Western blot analysis of purified M3-p53-R12 protein un-treated (NT) or treated (T) with glutaraldehyde at final concentration of 0.02%. Anti-p53 (DO-1) antibody was used as primary antibody. (B) EMSA analysis of M3-p53-R12 protein. About 2 μM of DNA (consensus p53-binding site sequence) and 0.5 mg/ml (7.7 μM) of M3-p53-R12 protein were used in each reaction. The reaction mixture was resolved in 1% agarose gel and DNA was viewed by ethidium bromide staining. M1 and M2 are DNA makers. Numbers on the left side represent DNA makers in kilobase. The presence of M3-p53-R12 protein in total lysate (C) as well as in nuclear and cytoplasm fractions (D) of leukemia cells after protein transduction for 2 hrs was examined with Western blot. (E, F) The stability of M3-p53-R12 protein in their culture medium (E) or in Jurkat cells (F) was analyzed by Western blot. Treatment in (F) was stop in 30 min by several washes with fresh medium and cells were harvested at the indicated time points. Anti-p53 (DO-1) antibody was used as primary antibody to detect the transduced recombinant p53 protein. The signals of GAPDH, β-Actin, BSA, and Lamin B1 serve as internal controls.
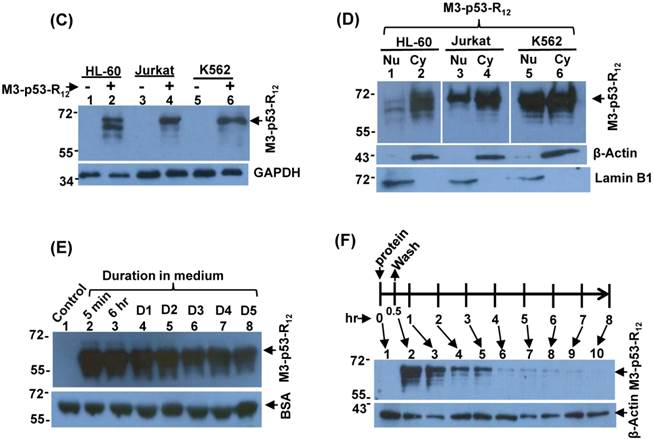
To induce cancer cells apoptosis, the recombinant M3-p53-R12 protein should has cell penetrative ability no matter it is going to trigger apoptosis via transcription-dependent or -independent pathways. We collected total cell lysates after they had been transduced with M3-p53-R12 protein for 2 hours, and we found that M3-p53-R12 can be detected in all cells tested, indicating that R12 transmembrane domain does assist M3-p53-R12 protein to enter all three test leukemia cell lines (Fig. 2C). It was of interest to know whether M3-p53-R12 can also enter nucleus to perform its transcription factor functions. So we isolated nuclear and cytoplasmic fractions of the transduced cells to examine the presence of M3-p53-R12 in the nucleus. We found that although large fraction of transduced protein accumulated in the cytoplasm, sufficient amount of this protein had translocated into nucleus within 2 hours after transduction (Fig. 2D). These results suggest that the purified M3-p53-R12 should have normal nuclear localization activity and can recognize target cis-elements in situ. The stability of M3-p53-R12 protein was also characterized and we found stable presence of this protein in culture medium after 5 days (Fig. 2E); however, once it entered the cells, this protein was gradually degraded in 2-3 days (Fig. 2F).
M3-p53-R12 protein inhibits the proliferation of leukemia cells
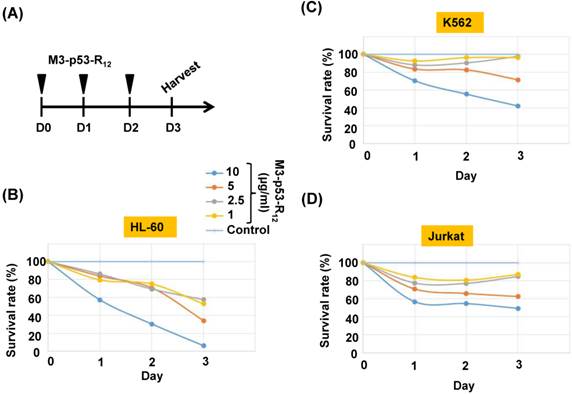
It was of interest to test whether M3-p53-R12 protein retained the tumor suppressor function of wildtype p53 so it could be used as an anti-cancer drug, and a potential target for this protein were hematopoietic cancer cells because they can contact directly with M3-p53-R12 in the blood without the worry of tissue/vascular barrier. Leukemia cells K562, Jurkat, and HL-60 carrying mutant p53 genes are treated with various does of M3-p53-R12 for 3 days and the effects on cell survival were determined by MTT assay. Daily refreshment of M3-p53-R12 protein was performed for first 48 hours and cells were harvested at 72 hours. We found that M3-p53-R12 protein did launch apoptosis in a dose-dependent manner (Fig.3A~D). The lowest concentration (1 μg/ml or 15 nM) of M3-p53-R12 used here was sufficient to trigger the programed cell death in all three cell lines in vitro, although HL-60 cells showed the strongest response. We also found that M3-p53-R12 became un-effective after 48 hours in culture medium and M3-GFP had no effects on the survival of these leukemia cells (Fig.4A~D); therefore, daily or every other day refreshment of the M3-p53-R12 in the medium might be necessary if long-term suppression of leukemia cell proliferation is intended. In addition to leukemia, the survival of cancer cells of other tissue origins was also suppressed by M3-p53-R12 treatment (Supplementary Fig. S1), implying its general application for cancer treatment.
Cell viability assay of M3-p53-R12 transduced leukemia cells. (A) Leukemia cells were respectively treated with various levels (1~10 μg/ml) of M3-p53-R12 protein daily, and then harvested on day 1~ 3. The relative survival ratio of HL-60 (B), Jurkat (C) and K562 cells (D) treated with different concentrations of M3-p53-R12 protein on each day was determined with MTT assay.
The sustained apoptotic effect of M3-p53-R12 protein on leukemia cells. (A) Leukemia cells were treated with M3-p53-R12 or M3-EGFP protein (10 μg/ml= 0.15 μM) on day 0 and harvested daily on day 1~ 3. The relative survival ratio of treated HL-60 (B), Jurkat (C), and K562 cells (D) on each day was determined with MTT assay.
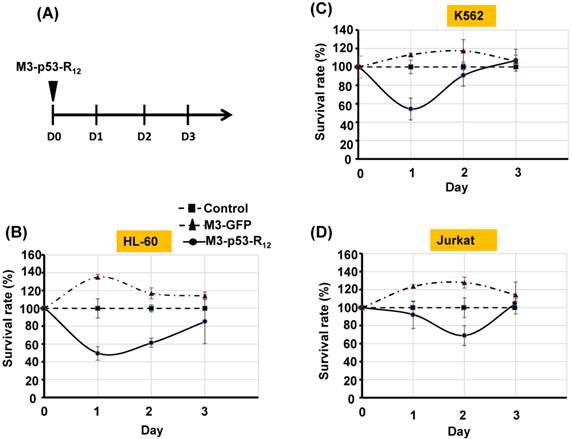
Transduction of M3-p53-R12 induces leukemia cell lines apoptosis
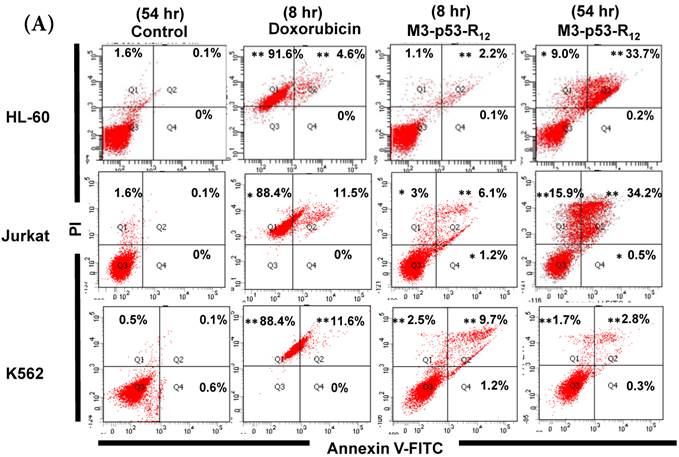
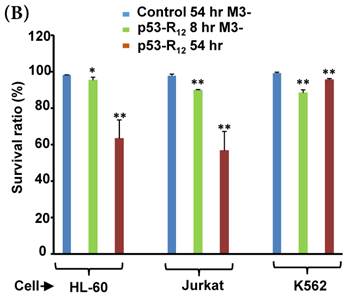
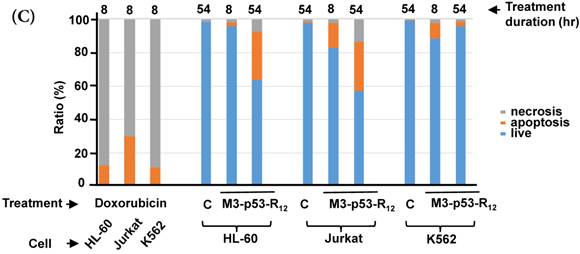
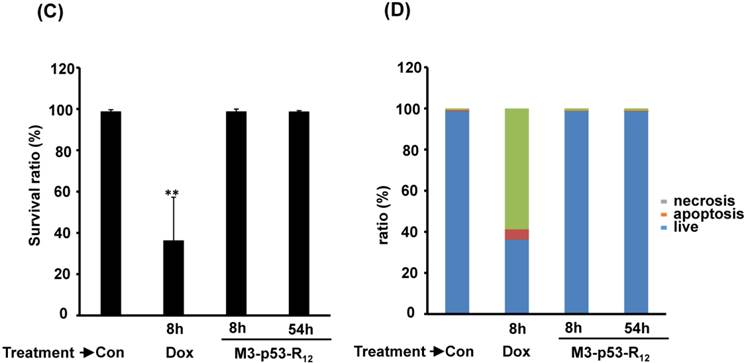
Although M3-p53-R12 did induce leukemia cell lines death, it was unknown whether it achieved this through necrosis or programmed cell death. This query was answered with flow cytometry using the Annexin V /PI double staining of leukemia cells after they had been transduced with single dose of M3-p53-R12 protein for 8 and 54 hours. After treated for 8 hr, all cell lines showed considerable amount of cell death, in which most of dead cells were stained simultaneously by both Annexin V /PI, suggesting that M3-p53-R12 protein induces cell apoptosis but not necrosis within 8 hours (Fig.5A). Among these 3 cell lines, K562 showed the strongest response to M3-p53-R12 after this short treatment. In HL-60 and Jurkart cells treated for 54 hours, the apoptotic cells were significantly increased, especially those at early apoptotic stage, and surpass that of K562 cells (Fig. 5A ~ C), suggesting accumulative killing effect of M3-p53-R12 on both cells. On the other hand, apoptotic K562 cells were dramatically reduced after treated for 54 hours, suggesting that K562 cells survived the first assault quickly recover from the apoptotic effect as observed in Fig. 4.
FACS analysis of Annexin V /PI stained cells for assessing M3-p53-R12 induced apoptosis. (A) Assessment of apoptosis of HL-60 (top panels), Jurkat (middle panels), and K562 (bottom panels) cells stained by Annexin V and propidium iodide (PI) after treated with M3-p53-R12 protein (10 μg/ml) for 8 and 54 hr. Cells treated with doxorubicin serves as positive control. Section Q1, Q2, Q3 and Q4 indicate the cells under necrosis, late apoptosis, live, and early apoptosis stages, respectively. The percentages of cells at different stages are indicated. The survival ratio of cells after treatment is shown in (B). Accumulated histograms in (C) indicate ratios cells at the stages of the necrosis, apoptosis and live after treatments with Doxorubicin or M3-p53-R12 protein. * and **: p< 0.05 and p<0.01, respectively, vs. control.
M3-p53-R12 protein represses leukemia cell's proliferation
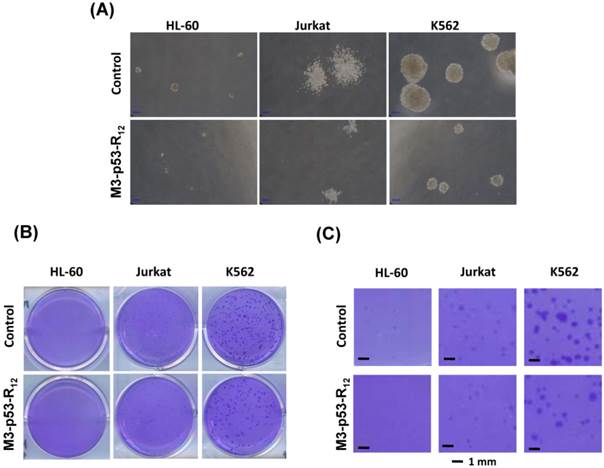
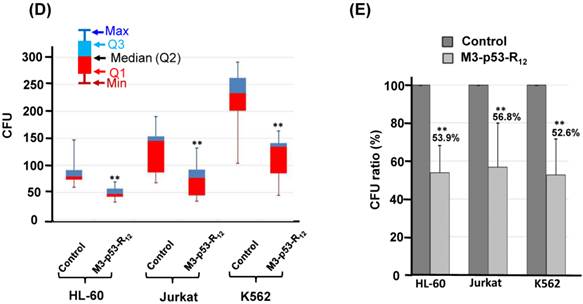
The effect of M3-p53-R12 on the proliferation of leukemia cells was examined by colony-forming unit analysis with cells grown on soft agar. Interestingly we found that each cell line had particular colony shape: Jurkat cells form loose colony, K562 cells form big compact colonies, and HL-60 cells form small but compact colonies (Fig.6A). According to the microscopic observation, the average size of colonies formed by M3-p53-R12 treated groups are dramatically smaller than untreated groups (Fig.6B & C), indicating that cells were suffering from M3-p53-R12 protein induced cell cycle arrest. Furthermore, the number of colony after M3-p53-R12 protein transduction was also dramatically reduced (Fig. 6D & E), implying that, upon the single dose challenge of M3-p53-R12 only about half of the cells escaped from the M3-p53-R12 induced apoptosis and cell cycle arrest and continued to form colonies.
The colony formation ability of leukemia cell lines is repressed by M3-p53-R12 protein. The effect of M3-p53-R12 protein (10 μg/ml, overnight) on leukemia cell colony formation ability was investigated by soft agar assay after grown on soft agar for 14 days. The morphology of the colonies formed by these 3 leukemia cell lines is shown in (A), and the crystal violet stained colonies are shown in (B, C). Images in (C) are shown at higher magnification. Numbers of colonies formed by control and treated cells is shown in (D) and their relative colony formation ability is shown in (E). The colony numbers in (C) are shown as quartiles. **: p<0.01 vs. control.
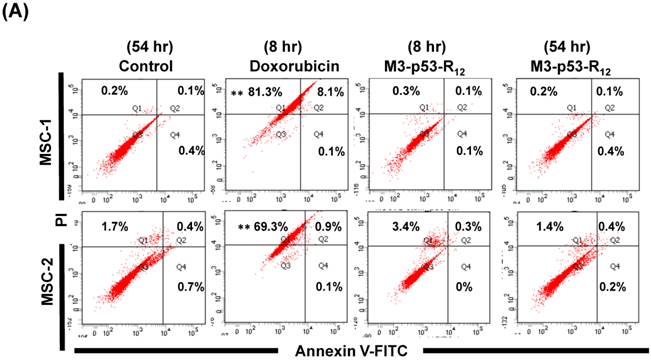
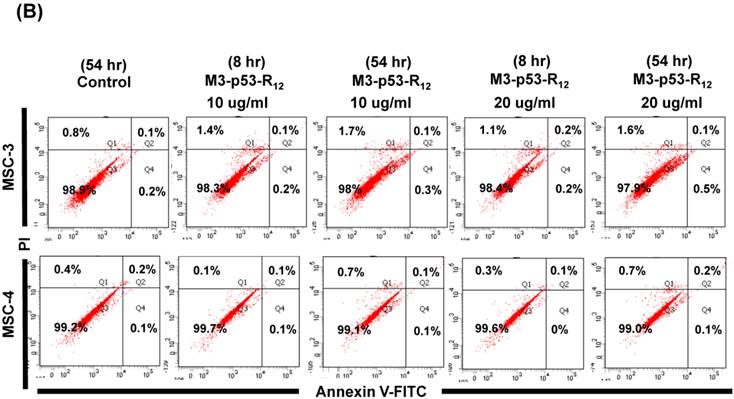
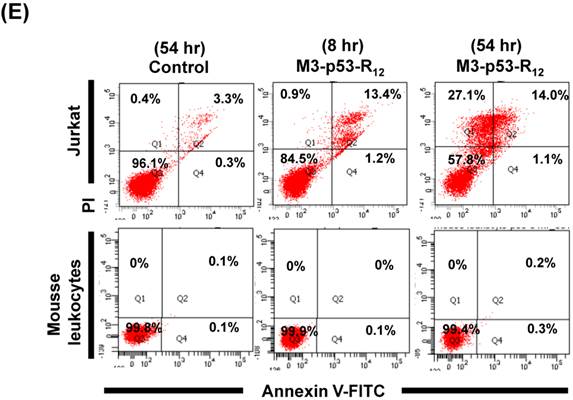
The failure of current cancer therapy is attributed very much to the indiscriminative killing effects of chemotherapy drugs that cause systemic destruction of the already weakened body and later failure of multiple organ systems. To preserve the patients' health and remove tumor cells from the patients' body, it is important to develop drugs that can discriminate normal and tumor cells. The effects of M3-p53-R12 on normal cells were tested on mesenchymal stem cells (MSC) isolated from umbilical cords of human new born babies, and we found very weak, if any, apoptotic effects at 8 and 54 hr after the treatment (Fig. 7A~D), and similar effect on mouse normal leukocytes was also observed (Fig. 7E). This discovery is very surprising but encouraging as it demonstrates the discriminating ability of M3-p53-R12 protein to specifically target tumor cells for apoptosis. In the future, we will further test its apoptotic effects on stem cells of other sources, such as hematopoietic stem cells from bone marrow, so the safety of treatments using M3-p53-R12 can be further verified.
M3-p53-R12 protein has no apoptotic effect on normal mesenchymal stem cells. (A, B) Assessment of apoptosis of normal mesenchymal stem cells (MSC) stained by Annexin V and propidium iodide (PI) after treated with M3-p53-R12 protein (10 or 20 μg/ml) for 8 and 54 hr. Cells treated with doxorubicin (Dox) served as positive control. Sections Q1, Q2, Q3 and Q4 indicate the cells under necrosis, late apoptosis, live, and early apoptosis stages, respectively. The percentages of cells at different stages are indicated. MSC-1~-4 represent MSC cells isolated form 4 different sources. The survival ratio of cells after treatment is shown in (C). Accumulated histograms in (D) indicate ratios of cells at the stages of the necrosis, apoptosis and live after treatments with Doxorubicin or M3-p53-R12 protein. **: p<0.01 vs. control (Con). (E) FACS analysis of Jurkat cells and normal mouse leukocytes after treated with M3-p53-R12 protein (10 μg/ml) for 8 and 54 hr before they were analyzed as described in (A, B). Leukocytes from mice of B6 strain were analyzed independently.
Discussion
Due to its importance in the prevention of tumorigenesis/carcinogenesis, many approaches have been attempted to deliver p53 into patients for treating cancer. To date, the most successful approach is using adenoviral vector for expressing p53 gene (called Adp53 in short). Gendicine™ was hailed as the first gene therapy drug approved by the China Food and Drug Administration in 2004. This drug is usually performed by orthotopic (intra-tumor) injection into tumors and its initially claimed success rate in combination with radiotherapy for treating head and neck squamous cell carcinoma (HNSCC) was about 96% (64% complete regression and 32% partial regression). However, after more than 10 years on the market, its efficacy and benefit on patients is still controversial [34, 35], as it does not significantly prolong survival or improve the disease-free survival rate (reviewed in [24]).
Unfortunately, similar story has been seen when Gendicine is combined with chemotherapy to treat non-small cell lung cancer (NSCLC), and the cause of its failure might be attributed to its low infection efficiency. The combination of Gendicine with transcatheter arterial chemoembolization (TACE) or fractionated stereotactic radiotherapy (fSRT) seems to improve the survival rate of hepatocellular carcinoma (HCC) patients, but the long-term effect on disease-free survival rate is waiting to be seen (reviewed in [24] ). Advexin and SCH-58500 are two other similar Adp53-expressing gene therapy drugs that have been at advanced clinical research stages. Both drugs have finished Phase I/II trials and some benefits can be seen in the response rate; however, no significant difference between patients receiving treatment and control group in the survival rate has been observed [36, 37]. The clinical trial study of SCH-58500 has been terminated in United State. Coincidentally, Advexin was refused by both FDA and EMA because of lacking evidence supporting its stability and the absence of p53-carrying replication competent adenovirus. The failure of these adenovirus based p53 gene therapy approaches demonstrates that alternative route is required to deliver p53 gene into tumor cells for combating cancers.
Cell type-specific transcription factors (TF) regulate the expression of a subset of genes that guides progenitor cells to develop/differentiate along a preset route to become functional cells, and similar mechanism is operating to keep cells staying as a stem or progenitor cell. Recent studies showed that the same work can also been achieved by ecotopic recombinant proteins. For instance, purified HOXB4 protein specifically enhances the proliferation of hematopoietic stem cells and sustain them at naïve stage [38]; myogenic cells can be induced from fibroblasts by recombinant MyoD protein [39]; somatic cells could be reprogrammed to pluripotent stem cell by recombinant Yamanaka factors transduction [40]; even use of recombinant SOX2 protein could trans-differentiate somatic cells to neurons [41]. In spite of the fact that no transcription factor protein has been launched for drug usage to date, abundant studies have disclosed its medical potency, which has made the application of transcription factor protein to drug usage a virgin territory in novel drug development. Before they can be used as clinical drugs, some hurdles, such as cell targeting and penetrating, signal amplification, transcription efficiency, and post-translational modification, will need to be crossed.
Compared to the restriction of current Adp53 gene therapy, a protein candidate drug is much suitable for intra-vein injection to treat hematologic malignancies and circulating (metastasizing) tumor cells. Recombinant protein also has advantage of avoiding tumorigenic risk caused by virus-mediated gene therapy. The cell penetrating ability also lowers immunogenicity in the circulation system. In this study, the cell entry ability of chimeric p53 protein is endowed by the fusion of a poly (arginine) peptide and its nuclear translocation ability is directed by its own NLS. By the observation of leukemia cell apoptosis, the transcriptional efficacy and bioactivity of chimeric M3-p53-R12 protein is also confirmed. Its anti-cancer growth potency against three different types of leukemia cell line, HL-60 (acute myeloid leukemia, AML), Jurkat (acute lymphoid leukemia, ALL) and K562 (chronic myeloid leukemia, CML) in vitro is an unexpected result as some of them expressing dominant-negative mutant p53. The overcome of the dominant-negative effect of endogenous mutant p53 suggests that either the addition of the M3 domain reversed the recessive role of wildtype p53 or the high dose of penetrated M3-p53-R12 overcame the dominance of the mutant allele. In the future, more endeavor on the heterodimerization between mutant p53 and M3-p53-R12 in vitro and on the regulatory cis-elements of target genes should reveal how much is contributed by either mechanism. The effect of the M3 domain on the activation of p53 target gene should also be clarified.
The suppression of cell growth and induction of apoptosis by M3-p53-R12 does not mean the same story can happen in vivo. However, the in vivo experiment is hindered by the migrating/circulating nature of leukemia cells, which avoids the measuring and characterizing the tumor growth during the treatment period. In the future, a traceable leukemia murine model should be developed to evaluate M3-p53-R12 protein therapy efficacy, in which remaining cancer cells can be measured by FACS scanning.
Apart from Adp53-based gene therapy, several reports also tried to use recombinant p53 protein for cancer therapy [42, 43]. Most of those reports only use cell-penetrating peptides (CPP) to assist protein to entry the cells without enhancing its transactivating activity. In this study, a MyoD transcriptional activation domain was fused to the N-terminus of p53 and our results revealed that M3-p53-R12 has competitive advantage at low concentration (approx. 0.014μM) to induce cancer cell death as compared to other studies (range in 0.01 and 10 μM). In the future, we also like to know whether this protein preferentially target cancer cells at this low dose. It has been shown that poly (arginine) and other CPP are recognized by cell surface heparan sulfate proteoglycans (HSPG), which serves an independent internalization receptor for uptaking extracellular macromolecules via endocytosis [44]. By using a HSPG specific monoclonal antibody, it has been found that HSPG is expressed in human myeloid leukemia cells but not in normal blood cells [45], which infers that HSPG may serve as a cancer-specific receptor for M3-p53-R12 to kill leukemia cells. Besides, previous study also demonstrated that the composition of tumor HSPG is not only different from normal tissue but also affect its growth [46], suggesting that binding of M3-p53-R12 to HSPG will certainly interfering the growth of cancer cells. Hopefully, intravenous administration of recombinant protein could cure the hematopoietic malignance and metastasis cancer cells in vivo.
Acknowledgements
The authors are grateful to our colleagues Han Fang Teng and Moo Rung Loo at NCU for their help with the construction of the pET32a-LPRH vector. The authors also like to thank the support from Taiwan Advance Bio-Pharma Inc. and the assistance by Dr. Yu-Che Cheng at CGH.
This work was supported by National Science Council of Taiwan, Contract grant number NSC-102-2311-B-008-003, MOST-103-2314-B-008-001, MOST-104-2311-008-001.
Supplementary Material
Supplementary figure S1.
Competing Interests
The authors have declared that no competing interest exists.
References
1. McBride O, Merry D, Givol D. The gene for human p53 cellular tumor antigen is located on chromosome 17 short arm (17p13). Proc Natl Acad Sci U S A. 1986;83:130-4
2. Abida WM, Gu W. p53-Dependent and p53-independent activation of autophagy by ARF. Cancer Res. 2008;68:352-7
3. Scherz-Shouval R, Weidberg H, Gonen C, Wilder S, Elazar Z, Oren M. p53-dependent regulation of autophagy protein LC3 supports cancer cell survival under prolonged starvation. Proc Natl Acad Sci U S A. 2010;107:18511-6
4. Yonish-Rouach E, Deguin V, Zaitchouk T, Breugnot C, Mishal Z, Jenkins JR. et al. Transcriptional activation plays a role in the induction of apoptosis by transiently transfected wild-type p53. Oncogene. 1995;11:2197-205
5. Cho Y, Gorina S, Jeffrey PD, Pavletich NP. Crystal structure of a p53 tumor suppressor-DNA complex: understanding tumorigenic mutations. Science. 1994;265:346-55
6. Duan J, Nilsson L. Effect of Zn2+ on DNA recognition and stability of the p53 DNA-binding domain. Biochemistry. 2006;45:7483-92
7. Meplan C, Richard MJ, Hainaut P. Metalloregulation of the tumor suppressor protein p53: zinc mediates the renaturation of p53 after exposure to metal chelators in vitro and in intact cells. Oncogene. 2000;19:5227-36
8. Puca R, Nardinocchi L, Porru M, Simon AJ, Rechavi G, Leonetti C. et al. Restoring p53 active conformation by zinc increases the response of mutant p53 tumor cells to anticancer drugs. Cell Cycle. 2014;10:1679-89
9. Wu Y, Mehew JW, Heckman CA, Arcinas M, Boxer LM. Negative regulation of bcl-2 expression by p53 in hematopoietic cells. Oncogene. 2001;20:240-51
10. Miyashita T, Reed JC. Tumor suppressor p53 is a direct transcriptional activator of the human bax gene. Cell. 1995;80:293-9
11. Nakano K, Vousden KH. PUMA, a novel proapoptotic gene, is induced by p53. Mol Cell. 2001;7:683-94
12. Oda E, Ohki R, Murasawa H, Nemoto J, Shibue T, Yamashita T. et al. Noxa, a BH3-only member of the Bcl-2 family and candidate mediator of p53-induced apoptosis. Science. 2000;288:1053-8
13. Chen L, Willis SN, Wei A, Smith BJ, Fletcher JI, Hinds MG. et al. Differential targeting of prosurvival Bcl-2 proteins by their BH3-only ligands allows complementary apoptotic function. Mol Cell. 2005;17:393-403
14. Speidel D. Transcription-independent p53 apoptosis: an alternative route to death. Trends Cell Biol. 2010;20:14-24
15. Fuchs SY, Adler V, Buschmann T, Wu X, Ronai Z. Mdm2 association with p53 targets its ubiquitination. Oncogene. 1998;17:2543-7
16. Li M, Brooks CL, Wu-Baer F, Chen D, Baer R, Gu W. Mono- versus polyubiquitination: differential control of p53 fate by Mdm2. Science. 2003;302:1972-5
17. Marchenko ND, Wolff S, Erster S, Becker K, Moll UM. Monoubiquitylation promotes mitochondrial p53 translocation. Embo j. 2007;26:923-34
18. Lahav G, Rosenfeld N, Sigal A, Geva-Zatorsky N, Levine AJ, Elowitz MB. et al. Dynamics of the p53-Mdm2 feedback loop in individual cells. Nat Genet. 2004;36:147-50
19. Chipuk JE, Green DR. Dissecting p53-dependent apoptosis. Cell Death Differ. 2006;13:994-1002
20. Zenz T, Krober A, Scherer K, Habe S, Buhler A, Benner A. et al. Monoallelic TP53 inactivation is associated with poor prognosis in chronic lymphocytic leukemia: results from a detailed genetic characterization with long-term follow-up. Blood. 2008;112:3322-9
21. Eliyahu D, Michalovitz D, Eliyahu S, Pinhasi-Kimhi O, Oren M. Wild-type p53 can inhibit oncogene-mediated focus formation. Proc Natl Acad Sci U S A. 1989;86:8763-7
22. Finlay CA, Hinds PW, Levine AJ. The p53 proto-oncogene can act as a suppressor of transformation. Cell. 1989;57:1083-93
23. Willis A, Jung EJ, Wakefield T, Chen X. Mutant p53 exerts a dominant negative effect by preventing wild-type p53 from binding to the promoter of its target genes. Oncogene. 2004;23:2330-8
24. Chen GX, Zhang S, He XH, Liu SY, Ma C, Zou XP. Clinical utility of recombinant adenoviral human p53 gene therapy: current perspectives. OncoTargets and therapy. 2014;7:1901-9
25. Inoue M, Tomizawa K, Matsushita M, Lu YF, Yokoyama T, Yanai H. et al. p53 protein transduction therapy: successful targeting and inhibition of the growth of the bladder cancer cells. Eur Urol. 2006;49:161-8
26. Lafevre-Bernt M, Wu S, Lin X. Recombinant, refolded tetrameric p53 and gonadotropin-releasing hormone-p53 slow proliferation and induce apoptosis in p53-deficient cancer cells. Mol Cancer Ther. 2008;7:1420-9
27. Cheung CH, Sun X, Kanwar JR, Bai JZ, Cheng L, Krissansen GW. A cell-permeable dominant-negative survivin protein induces apoptosis and sensitizes prostate cancer cells to TNF-alpha therapy. Cancer cell international. 2010;10:36
28. Guelen L, Paterson H, Gaken J, Meyers M, Farzaneh F, Tavassoli M. TAT-apoptin is efficiently delivered and induces apoptosis in cancer cells. Oncogene. 2004;23:1153-65
29. Han K, Jeon MJ, Kim KA, Park J, Choi SY. Efficient intracellular delivery of GFP by homeodomains of Drosophila Fushi-tarazu and Engrailed proteins. Molecules and cells. 2000;10:728-32
30. Olivier M, Hollstein M, Hainaut P. TP53 mutations in human cancers: origins, consequences, and clinical use. Cold Spring Harb Perspect Biol. 2010;2:a001008
31. Greenblatt MS, Bennett WP, Hollstein M, Harris CC. Mutations in the p53 tumor suppressor gene: clues to cancer etiology and molecular pathogenesis. Cancer Res. 1994;54:4855-78
32. Hirai H, Tani T, Katoku-Kikyo N, Kellner S, Karian P, Firpo M. et al. Radical acceleration of nuclear reprogramming by chromatin remodeling with the transactivation domain of MyoD. Stem Cells. 2011;29:1349-61
33. Bell S, Hansen S, Buchner J. Refolding and structural characterization of the human p53 tumor suppressor protein. Biophysical chemistry. 2002;96:243-57
34. Sheridan C. Gene therapy finds its niche. Nat Biotechnol. 2011;29:121-8
35. Shi J, Zheng D. An update on gene therapy in China. Current opinion in molecular therapeutics. 2009;11:547-53
36. Schuler M, Herrmann R, De Greve JL, Stewart AK, Gatzemeier U, Stewart DJ. et al. Adenovirus-mediated wild-type p53 gene transfer in patients receiving chemotherapy for advanced non-small-cell lung cancer: results of a multicenter phase II study. Journal of clinical oncology: official journal of the American Society of Clinical Oncology. 2001;19:1750-8
37. Watanabe N, Yano K, Tsuyuki K, Okano T, Yamato M. Re-examination of regulatory opinions in Europe: possible contribution for the approval of the first gene therapy product Glybera. Molecular therapy Methods & clinical development. 2015;2:14066
38. Huang CH, Chen PM, Lu TC, Kung WM, Chiou TJ, Yang MH. et al. Purified recombinant TAT-homeobox B4 expands CD34(+) umbilical cord blood and peripheral blood progenitor cells ex vivo. Tissue Eng Part C Methods. 2010;16:487-96
39. Sung MS, Mun JY, Kwon O, Kwon KS, Oh DB. Efficient myogenic differentiation of human adipose-derived stem cells by the transduction of engineered MyoD protein. Biochem Biophys Res Commun. 2013;437:156-61
40. Zhou H, Wu S, Joo JY, Zhu S, Han DW, Lin T. et al. Generation of induced pluripotent stem cells using recombinant proteins. Cell Stem Cell. 2009;4:381-4
41. Hu Q, Chen R, Teesalu T, Ruoslahti E, Clegg DO. Reprogramming human retinal pigmented epithelial cells to neurons using recombinant proteins. Stem Cells Transl Med. 2014;3:1526-34
42. Kousparou CA, Yiacoumi E, Deonarain MP, Epenetos AA. Generation of a selectively cytotoxic fusion protein against p53 mutated cancers. BMC cancer. 2012;12:338
43. Michiue H, Tomizawa K, Wei FY, Matsushita M, Lu YF, Ichikawa T. et al. The NH2 terminus of influenza virus hemagglutinin-2 subunit peptides enhances the antitumor potency of polyarginine-mediated p53 protein transduction. The Journal of biological chemistry. 2005;280:8285-9
44. Christianson HC, Belting M. Heparan sulfate proteoglycan as a cell-surface endocytosis receptor. Matrix Biol. 2014;35:51-5
45. Vongchan P, Linhardt RJ. Expression of human liver HSPGs on acute myeloid leukemia. Clinical immunology (Orlando, Fla). 2007;122:194-206
46. Kleeff J, Ishiwata T, Kumbasar A, Friess H, Buchler MW, Lander AD. et al. The cell-surface heparan sulfate proteoglycan glypican-1 regulates growth factor action in pancreatic carcinoma cells and is overexpressed in human pancreatic cancer. J Clin Invest. 1998;102:1662-73
Author contact
![]() Corresponding author: Shen Liang Chen, Department of Life Sciences, National Central University, 300 Jhongda Rd, Jhongli 32054, Taiwan (R. O. C.). Tel: 886-3-4227151 ext 65069; Fax: 886-3-4228482; E-mail: slchenncu.edu.tw.
Corresponding author: Shen Liang Chen, Department of Life Sciences, National Central University, 300 Jhongda Rd, Jhongli 32054, Taiwan (R. O. C.). Tel: 886-3-4227151 ext 65069; Fax: 886-3-4228482; E-mail: slchenncu.edu.tw.