Impact Factor ISSN: 1837-9664

 Global reach, higher impact
Global reach, higher impactJ Cancer 2016; 7(11):1374-1382. doi:10.7150/jca.14669 This issue Cite
Research Paper
The Expression Levels of XLF and Mutant P53 Are Inversely Correlated in Head and Neck Cancer Cells
Sizhe Feng1, 2, #, Ramin Rabii1, #, Guobiao Liang2, Chenxi Song1, Wei Chen1, Mian Guo1, Xuezhong Wei2, Diana Messadi1, Shen Hu1, ![]()
1. School of Dentistry and Jonsson Comprehensive Cancer Center, University of California, Los Angeles, CA 90095, USA
2. North Hospital, Shenyang, China
# These authors contribute to the work equally.
Received 2015-12-13; Accepted 2016-3-15; Published 2016-6-30
Abstract
XRCC4-like factor (XLF), also known as Cernunnos, is a protein encoded by the human NHEJ1 gene and an important repair factor for DNA double-strand breaks. In this study, we have found that XLF is over-expressed in HPV(+) versus HPV(-) head and neck squamous cell carcinoma (HNSCC) and significantly down-regulated in the HNSCC cell lines expressing high level of mutant p53 protein versus those cell lines harboring wild-type TP53 gene with low p53 protein expression. We have also demonstrated that Werner syndrome protein (WRN), a member of the NHEJ repair pathway, binds to both mutant p53 protein and NHEJ1 gene promoter, and siRNA knockdown of WRN leads to the inhibition of XLF expression in the HNSCC cells. Collectively, these findings suggest that WRN and p53 are involved in the regulation of XLF expression and the activity of WRN might be affected by mutant p53 protein in the HNSCC cells with aberrant TP53 gene mutations, due to the interaction of mutant p53 with WRN. As a result, the expression of XLF in these cancer cells is significantly suppressed. Our study also suggests that XLF is over-expressed in HPV(+) HNSCC with low expression of wild type p53, and might serve as a potential biomarker for HPV(+) HNSCC. Further studies are warranted to investigate the mechanisms underlying the interactive role of WRN and XLF in NHEJ repair pathway.
Keywords: XLF, mutant p53, biomarker, HPV, HNSCC
Introduction
Non-homologous end joining (NHEJ) is a well-known mechanism for the repair of double-strand breaks (DSBs), a lethal type of DNA damage in mammalian cells. It basically utilizes a group of enzymes to take hold of the ends of a broken DNA molecule, create a bridge between them, and subsequently re-ligate the broken DNA molecule [1]. Ku heterodimer (KU70/KU80), DNA-dependent protein kinase catalytic subunit (DNA PKcs), DNA repair protein XRCC4 (X-ray repair cross-complementing protein 4) and DNA Ligase IV constitute the core elements of the NHEJ apparatus. The repair process is initiated by binding of the ring-shaped Ku70/Ku80 heterodimer (Ku) to both ends of a DSB, followed by recruitment of DNA PKcs to the DNA-Ku complex. The DNA Ligase IV, a specialized DNA ligase that forms a complex with the cofactor XRCC4, then directly joins the two ends of a DSB. NHEJ remains active throughout the cell cycle, but is the major DSB repair mechanism during the G1 phase of the cell cycle. It is also the primary repair pathway for DSBs induced by radiation damage.
XRCC4-like factor (XLF), also known as Cernunnos, is a protein encoded by the human NHEJ1 gene and originally discovered as a molecule mutated in patients with growth retardation, microcephaly, and immunodeficiency [2]. The interaction of XLF with XRCC4 is required for the NHEJ repair of DSBs [3-5]. Studies have shown that XLF can bind to DNA and accumulated at DNA damage sites via constitutive interaction of XRCC4 in the early DSB. XLF may also stimulate ligation of complementary and non-complementary DNA ends by combining with XRCC4-DNA Ligase IV complex, enhancing the stability of XRCC4-DNA ligase IV, modulating the efficiency and/or specificity of DNA Ligase IV and promoting the Ligase IV activity in the later of DSB. In XLF-deficiency murine embryonic stem (ES) cells, there are serious defects as observed in XLF/Cer patients, including radiation sensitivity, intrinsic DSB repair defect, and increased genomic instability with elevated levels of chromosomal breaks and translocations in comparison to wild type ES cells. These results indicated that XLF/Cer plays an important role in the NHEJ process for DNA repair [6, 7]. Nevertheless, little is known about the cellular machinery that regulates the expression of XLF in mammalian cells, particularly at transcriptional regulation level.
Werner syndrome protein (WRN) is a member of the human RecQ family DNA helicases implicated in the maintenance of genome stability. Mutations of the WRN gene give rise to the Werner syndrome, a genetic disease characterized by premature aging and cancer predisposition [8]. Patients carrying WRN gene mutations also exhibit increased genomic instability and an elevated rate of cancer [9, 10]. WRN may play an important role in NHEJ because it strongly binds to Ku and is recruited to DNA ends by Ku [11-13]. In fact, Ku stimulates/alters the properties of WRN exonuclease activity on a variety of DNA substrates [14], and DNA-PKcs phosphorylates WRN and regulates its catalytic activity as well [15, 16]. Moreover, the WRN protein can form a specific interaction with tumor suppressor p53. This interaction involves the carboxyl-terminal part of WRN and the extreme carboxyl terminus of p53, a region that plays an important role in the functional state of p53. Over-expression of WRN leads to augmented p53-dependent transcriptional activity and induction of p21 protein expression. These findings imply that the cross talk between WRN and p53 may be important for maintaining genomic integrity and for preventing the accumulation of aberrations that can give rise to premature senescence and cancer [17].
In this study, we found that XLF and mutant p53 expression levels are inversely correlated in head and neck cancer cells. When compared to HPV(-) HNSCCs, XLF is over-expressed in HPV(+) HNSCCs, which normally harbor wild-type TP53 and express low levels of p53 protein. We also found that WRN binds to both mutant p53 and the NHEJ1 gene promoter, and siRNA knockdown of WRN leads to the inhibition of XLF expression in the cancer cells. Collectively, these findings suggest that WRN is potentially involved in the regulation of XLF expression and the activity of WRN may be affected by mutant p53 protein in the head and neck cancer cells with aberrant TP53 gene mutations, due to the interaction of mutant p53 with WRN. As a result, the expression of XLF in these cancer cells was significantly inhibited.
Materials and Methods
Cell culture
The head and neck cancer cell lines, UM1, UM2, UMSCC-5, UMSCC-6, UMSCC-10B, and UMSCC-17B cell lines were obtained from Dr. Yong Kim (University of California, Los Angeles, School of Dentistry). All the cancer cell lines were cultured in DMEM with 10% fetal bovine serum and 1% penicillin/streptomycin (Invitrogen, Carlsbad, CA, USA). Normal human oral keratinocytes (NHOK) were cultured in EpiLife media supplemented with the human keratinocyte growth supplement (Invitrogen, Carlsbad, CA, USA). Cell cultures were maintained in a humidified atmosphere of 5% CO2 at 37 °C.
2-Dgel electrophoresis (2-DGE) analysis
HNSCC tissues were used for the present study following the provision of the UCLA Institutional Review Board approval. Prior to 2-DGE analysis, snap-frozen cancer tissues were lysed with 2-D rehydration buffer containing 2M thiourea, 7M urea, 2% CHAPS and 50mM DTT, and the total protein concentration was measured with the 2D Quant kit (GE Healthcare). Due to small amount of tissue materials available, 2-DGE analysis was performed on two pooled cancer tissue samples (HPV(+) or HPV(-) HNSCCs, total amount of 75μg each), which were prepared by equally pooling of tissue lysates from each individual subjects (equal amount of total proteins). The pooled samples were initially processed with 2-D Clean-up Kit (GE Healthcare) to remove interfering small molecules present in the samples. Afterwards, both pooled samples (60 μg from each sample) were focused withimmobilized pH gradient strips (11-cm length, pI 3-10 NL) on a Protean IEF cell and then separated with 8-16 % Criterion SDS-PAGE gels (Bio-Rad). Finally, the PAGE gels were stained with fluorescent Sypro-Ruby stain (Invitrogen) and gel images were acquired with the GS-800 gel imager (BioRad).
Analysis of gel images was performed using the Progenesis Samespot software (Nonlinear dynamics). A matchset was created initially, and protein spots were automatically matched and further manually verified. Afterwards, normalization was performed based on the total density of the gel image and the proteins levels were quantified. Each protein sample was analyzed in triplicate and Student T-test was used for statistical analysis.
Liquid chromatography with tandem mass spectrometry (LC-MS/MS)
Protein spots of interest were excised using a spot-excision robot (Progenesis) and deposited into 96-well plates. Tryptic digestion of proteins in each gel spots were performed by first reducing protein with 10 mM DTT for 30 min, followed by alkylation with 50 mMiodoacetamide for 60 min in the dark, and then digested with 10-ng trypsin at 37°C overnight. The resulting peptides were extracted and analysed by LC-MS/MS using a nano-LC system (Eksigent Technology) and LTQ ion trap mass spectrometer (Thermo Finnigan). LC separation of peptides was performed with C18 PicoFrit capillary columns (New Objectives) at a flow rate of 400nL/min. Database search was performed against the SwissProt database using the Proteome Discoverer/SEQUEST (Thermo Finnigan).
Western blotting
Cell lysates (30μg total proteins of each sample) were separated with a 12% NuPAGE gel (Invitrogen) at 120V for approximately 60 min and then transferred to nitrocellulose membrane using the TRANS-BLOT semi-dry transfer cell (Bio-Rad). Membranes were blocked with 5% milk in TBST buffer (1×) for 2 hours and then incubated with a primary antibody overnight at 4°C. The following primary antibodies, anti-XLF (Sigma, St. Louis, MO), anti-WRN (Santa Cruz Biotech, Dallas, TX), and anti-p53 (Genetex, Irvine, CA), were used for Western blot analysis. Following incubation with secondary antibody horseradish peroxidase-conjugated mouse or rabbit antibody (GE Healthcare, NJ, USA), protein bands were detected using the ECL Kit (GE Healthcare).
Chromatin immunoprecipitation (ChIP)
ChIP assays were performed with the ChIP assay kit from Millipore (Cat # 17-295, Billerica, MA, USA). After washing with PBS, the cancer cells were cross-linked with 1% formaldehyde for 15 min at room temperature and then stopped by adding 125 mM glycine. Cells were then washed twice with cold PBS, harvested in RIPA buffer (150 mM NaCl, 1% Igepal CA-630, 0.5% deoxycholate, 0.1% SDS, 50 mM Tris-HCl at pH 8), and sonicated to generate DNA fragments between 200 and 1000 base pairs. For immunoprecipitation, 1 mg of protein extract was pre-cleared with 30 μL of Protein A Agarose/Salmon Sperm DNA (50% Slurry) for 30min. Once the agarose centrifuged and removed, the supernatant fraction was incubated with 5μg of WRN antibody (Santa Cruz Biotechnology) overnight at 4°C and then incubated with 30μL Protein A Agarose/Salmon Sperm DNA (50% Slurry) for one hour at 4°C. Following the washes with low salt, high salt, LiCl immune complex wash buffers as well as the TE buffer, the immunocomplexes were harvested, eluted with 1%SDS/0.1M NaHCO3 for 10 min at 65°C, and then treated with NaCl (200mM) at 65°C for 4 hours to reverse histone-DNA crosslinks. Finally, the DNA fragments were purified and used as templates for qPCR reactions using the following primers: GTGGGAAAGGCTTTATGCAA (forward) and TCCCATAGTCCGACCTCATC (reverse).
Co-immunoprecipitation (Co-IP)
MyOne Dynabeads (Invitrogen) were first coated with the WRN antibody (Santa Cruz Biotech) according to the manufacturer's suggested protocol. Afterwards, the coated beads were washed twice in PBS/0.1% BSA (pH 7.4) for 5 minutes at 4° C, and then incubated in 0.2M Tris/0.2% BSA (pH 8.5) for 24 hours at 4° C. Cancer cells were collected using a Cell Stripper (Cellgro, Mediatech, Manassas, VA) and washed extensively with PBS for three times, followed by re-suspension in RIPA buffer. The supernatant (2mg total proteins) was transferred and incubated with the antibody-coated Dynabeads at 4 °C for overnight, followed by extensive washes with PBS. The beads were then harvested and citric acid (100 mM, pH 2.9) was added to elute the proteins from the beads. Afterwards, the eluted samples were tested for p53 with Western blotting.
siRNA knockdown of WRN
To perform siRNA knockdown of WRN, UM1, UM2, UM-SCC5 and UM-SCC6 cells were transfected with targeted siRNA to WRN (Santa Cruz Biotech) using the Hilymax transfection regent (Dojindo Molecular Technologies, Rockville, MD) according to the company's suggested manual. Validated double-stranded siRNAs of WRN or non-target scramble control siRNAs were mixed with the Hilymax transfection reagent and then added to the cell cultures. After 24-hour incubation, the media containing siRNAs were removed and the cells were further cultured in fresh complete media for 48 hours prior to cell harvesting for Western blot analysis.
Sequencing of TP53 gene in UM1 and UM2 cell lines
DNA was extracted from UM1 and UM2 cells using the QIAamp DNA Mini kits (Qiagen, Hilden, Germany). TP53 exons was polymerase chain reaction (PCR) amplified from each sample using the following set of primers: primer set 1 - forward TCTCATGCTGGATCCCCACTT; reverse ACGGCCAGGCATTGAAGTCTCAT; primer set 2 - forward CTCTTCCTACAGTACTCCCCTGC; reverse GGCCACTGACAACCACCCTTAACC; primer set 3 - forward ACCTGATTTCCTTACTGCCTCTTGC; reverse CCACTTGATAAGAGGTCCCAAG. PCR products were purified with the DNA Clean and Concentrator Kit (Zymo Research, Irvine, CA) and sequenced on an ABI PRISM 310 Genetic Analyzer (Applied Biosystems, Foster City, CA). DNA sequences were first viewed as chromatograms using the Finch TV. Using the program, the sequences amplified by reverse primers were converted to reverse complements, and then exported as FASTA files. The exported sequences were aligned to their respective p53 sequences obtained from NCBI gene (Accession number NC_000017.11, Gene ID 7157) using the EMBOSS Water Nucleotide Alignment Tool. The ExPASy Translate Tool was used to determine amino acid sequences based on the mutant sequence, and the wild type and mutated amino acid sequences were aligned using the EMBOSS Water Protein Alignment Tool.
Results
Identification of differentially expressed proteins between HPV(+) and HPV(-) HNSCCs
Figure 1 shows 2-D gel analysis of proteins expressed in HPV(+) and HPV(-) HNSCC tissues. Following gel staining with Sypro Ruby, proteins showing differential expression levels were excised from the gels and identified with LC-MS/MS. Table 1 presents a list of identified proteins that are differentially expressed between HPV(+) and HPV(-) HNSCCs. XLF, a DNA repair protein, was found to be over-expressed in HPV(+) HNSCCs when compared to HPV(-) HNSCCs.
2-DGE of proteins in HPV(+) and HPV(-) HNSCC tissues. Equal amount of tissue proteins from 5 HPV-positive or 5 HPV-negative HNSCC patients were pooled for the comparative analysis. Differentially expressed proteins were identified and listed in Table 1. Protein identity: Spot #1: Heat shock protein beta 1; Spot #2: PARK7 protein; Spot #3: XRCC4-like factor (XLF); Spot #4: Superoxide dismutase (SOD).
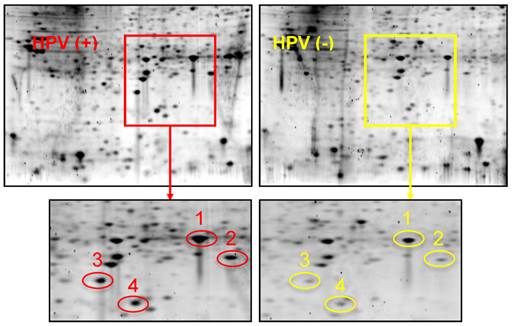
Differentially expressed proteins between HPV(+) and HPV(-) HNSCCs.
| Protein name | PI | MW (kDa) | Up- or down-regulation | Ratio HPV(+):HPV(-) |
|---|---|---|---|---|
| ADP-ribosylation factor 4 | 7.02 | 20.5 | Up-regulation | 4.1 |
| Ferritin-like protein | 5.27 | 20.0 | Up-regulation | 2.7 |
| Galectin-1 | 5.15 | 14.1 | Up-regulation | 2.7 |
| Heat shock protein beta 1 | 5.97 | 22.8 | Up-regulation | 3.0 |
| Isoform 1 of gelsolin | 5.86 | 49.2 | Up-regulation | 2.4 |
| Peroxiredoxin-6 | 5.97 | 25.0 | Up-regulation | 3.0 |
| Prohibitin | 5.46 | 29.8 | Up-regulation | 1.3 |
| PARK7 protein | 8.36 | 21.8 | Up-regulation | 5.2 |
| Superoxide dismutase | 5.95 | 25.1 | Up-regulation | 4.5 |
| XRCC4-like factor (XLF) | 6.25 | 19.6 | Up-regulation | 5.3 |
| Annexin A1 | 6.64 | 38.7 | Down-regulation | 0.6 |
| Cystatin A | 5.26 | 11.0 | Down-regulation | 0.1 |
| Fatty acid binding protein | 6.79 | 15.2 | Down-regulation | 0.3 |
| Fibrinogen beta chain | 8.23 | 55.9 | Down-regulation | 0.1 |
| Isoform 1 of 14-3-3 protein sigma | 4.53 | 27.8 | Down-regulation | 0.2 |
| Serpin B4 | 5.82 | 44.9 | Down-regulation | 0.4 |
Expression of mutant p53 and XLF in head and neck cancer cells
Using Western blotting, we measured the expression of p53, XLF and WRN in NHOKs, two wild type TP53 HNSCC lines, UM-SCC6 and UM-SCC17B, as well as two mutant TP53 HNSCC lines, UM-SCC5 and UM-SCC10B. As expected, mutant TP53 HNSCC lines displayed a dramatic over-expression of p53 versus wild type TP53 HNSCC lines which had very low p53 expression (Figure 2A). On the other hand, XLF expression was found to be much lower in mutant TP53 HNSCC lines versus wild type TP53 HNSCC lines.
(A) Western blot analysis of p53, WRN and XLF in wild-type and mutant TP53 head and neck squamous cell carcinoma (HNSCC) cells. (B) Western blot analysis of p53, WRN and XLF between UM1 and UM2 oral cancer cells.
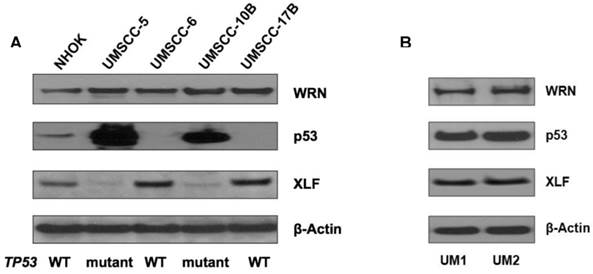
We also compared the expression of p53, WRN and XLF betweenUM1 and UM2 oral cancer cell lines which were initially established from a same patient's tumor. UM1 cells are highly invasive whereas UM2 cells are low invasive. Our invasion assay results indicate that UM1 cells have more than 8-fold invasive potential than UM2 cells (data not shown). As shown in Figure 2B, both p53 and XLF expression levels are not significantly different between UM1 and UM 2 cells.
Sequencing of TP53 gene in UM1 and UM2 cells
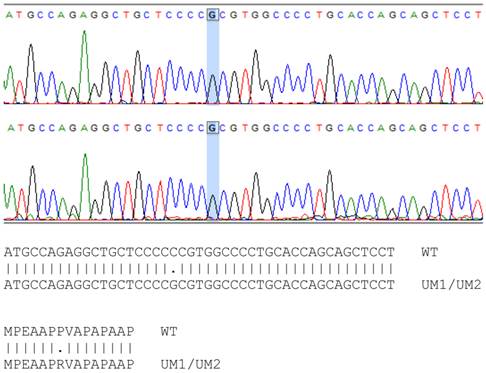
The mutation sites of TP53 in UMSCC-5 and UMSCC-10B cells have been identified previously [18]. However, whether the TP53 gene in UM1 and UM2 cells is wild type or mutant remains to be a question. By using PCR to amplify the TP53 gene and subsequently sequence the amplified DNA, we found that the TP53 gene in UM1 and UM2 cells has single point mutation at codon 72 on exon 4, where a Guanine replaces Cytidine in the wild type TP53 sequence (Figure 3). This CCC → CGC mutation causes a change from Proline to Arginine in p53 protein sequence in UM1 and UM2 cells. The single point mutations of TP53 among the four cell lines are listed in Table 2.
Sequencing of TP53 gene in UM1 (top) and UM2 (bottom) oral cancer cells. Comparison between wild-type and UM1/UM2 TP53 gene shows a C to G transversion in both UM1 and UM2 cells, occurring at codon 72. As indicated by the alignment, the single nucleotide mutation causes the change of proline (P) in wild-type TP53 gene to arginine (R) in UM1 and UM2 cells at codon 72.
TP53 mutations of head and neck cancer cell lines in this study.
| Cell line | Codon change | Amino acid change | Reference |
|---|---|---|---|
| UM1 | 72 CCC ⇒ CGC | P ⇒ R | Current study |
| UM2 | 72 CCC ⇒ CGC | P ⇒ R | Current study |
| UM-SCC5 | 157 GTC ⇒ TTC | V ⇒ F | [1] |
| UM-SCC10B | 245 GGC ⇒ TGC | G ⇒ C | [1] |
| UM-SCC6 | Wild type | N/A | [1] |
| UM-SCC17B | Wild type | N/A | [1] |
WRN binds to the NHEJ1 promoter
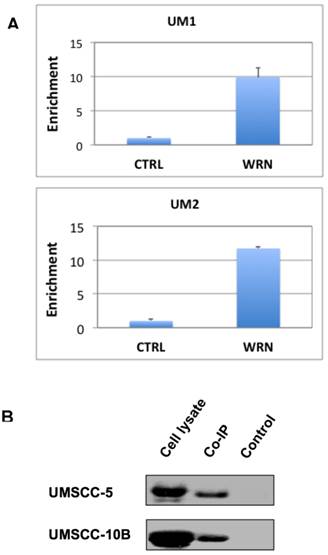
To test if WRN binds to the promoter of NHEJ1 gene, we performed the ChIP assay of UM1 and UM2 cells using anti-WRN antibody, followed by qPCR using primers for the promoter region of NHEJ1. As shown in Figure 4A, Results from the ChIP assay indicated that endogenous WRN binds to the NHEJ1 gene promoter in UM1 and UM2 cells.
(A) ChIP assay suggests that WRN may bind to the upstream promoter region of NHEJ1 in UM1 and UM2 oral cancer cells. Shown here is qPCR of the pull-down products from ChIP assays. (B) Co-IP assay suggests that mutant p53 binds to WRN in UM-SCC5 and UM-SCC10B cells. Western blotting was used to detect p53 in whole cell lysates, anti-WRN pull-down and negative control samples.
Binding of mutant p53 to WRN
To verify if mutant p53 protein interacts with WRN in HSNCC cells, we performed Co-IP assay with the protein lysates from UMSCC-5 and UMSCC-10B. Both UMSCC-5 and UMSCC-10B express high levels of mutant p53. As shown in Figure 4B, mutant p53 was co-immunoprecipitated by the anti-WRN antibody.
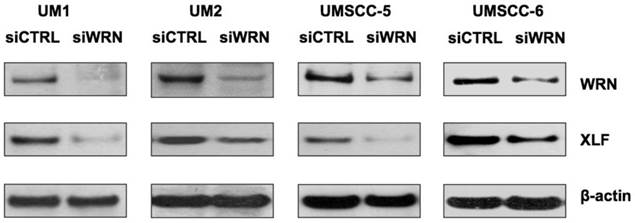
siRNA knockdown of WRN leads to down-regulation of XLF
To further explore if WRN has a potential role in regulating XLF expression, we knocked down WRN using siRNA and confirmed the reduced expression of WRN in the cancer cells. As shown in Figure 5, siRNA inhibition of WRN expression led to the down-regulation of XLF in the head and neck cancer cells.
siRNA knockdown of WRN leads to the down-regulation of XLF in HNSCC cells. WRN and XLF were measured by Western blot analysis.
Discussion
In this study, we first compared tissue protein expression between HPV(+) and HPV(-) HNSCCs using 2-DGE and then identified the proteins that are altered in HPV(+) HNSCCs with LC-MS/MS. As shown in Table 1, a number of proteins are differentially expressed between the two cancer tissue phenotypes suggesting that HPV(+) HNSCC cells may contain unique proteome profile characteristic of viral oncogenic transformation. Particularly, we found that XLF, an important factor of the NHEJ repair process, is over-expressed in HPV(+) HNSCCs when compared to HPV(-) HNSCCs. It is well known that HPV(+) HNSCCs harbor wild-type TP53 gene and the expression of p53 is low due to E6-associated degradation [19, 20]. Therefore, we compared the expression levels of XLF and p53 between wild type (UM-SCC6 and UM-SCC17B) and mutant TP53 (UM-SCC5 and UM-SCC10B) HNSCC cell lines. As expected, p53 expression is substantially low in the cancer cells with wild type TP53 gene. However, mutant TP53 HNSCC lines displayed a dramatic over-expression of p53 when compared to wild-type HNSCC lines (Figure 2A). The TP53 is one of the most frequent mutated genes in tumors, with more than half of all human cancers displaying some forms of mutations at this gene locus, and most of the TP53 mutations are missense forms that mainly reside in the region coding for the DNA binding domain [21]. The relationship between tobacco use and insurgence of HNSCC strongly suggest that mutation of TP53 is one hallmark genetic alteration of these tumors [22]. Unlike wild-type p53, which is generally a highly controlled, short-lived protein under stressed conditions, mutated forms of TP53 often lead to the accumulation of full length mutant p53 molecules with a prolonged half-life [23]. In contrast to a majority of tumor suppressor genes, which are mostly inactivated as a result of a mutation, mutant p53 has been shown to obtain gain-of-function oncogenic activity aside from the normal role of wild-type p53, and may promote metastatic potential of tumor cells. From a variety of studies, it becomes apparent mutations in TP53 are connected to the poor response of HNSCC to anticancer therapy [24-27]. Mice knocked in with p53R270H or R172H, corresponding to the human hotspot mutants p53R273H and p53R175H, respectively, developed highly metastatic tumors [28, 29]. Recent studies also demonstrated that mutant p53 can augment cell migration and invasion in in vitro assays [21, 30, 31]. Both UM-SCC5 and UM-SCC10B cell lines used in our study carry TP53 point mutations within the DNA-binding domain [18]. These mutations caused significant accumulation of mutant p53 with prolonged half-life in the UM-SCC5 and UM-SCC10B cells, which explains why p53 levels are dramatically high in these cells. On the other hand, XLF expression was found to be significantly lower in mutant TP53 HNSCC lines (UM-SCC5 and UM-SCC10B) versus wild-type TP53 HNSCC lines (UM-SCC6 and UM-SCC17B), suggesting that there is an inverse correlation between the expression of mutant p53 and XLF.
We also compared the expression of p53 and XLF between highly invasive UM1 and low invasive UM2 oral cancer cell lines. Both p53 and XLF are similarly expressed between the two cell lines, which presumably is due to that the two cell lines were derived from a same patient's tumor. Our sequencing analysis indicates that there is a C→G transversion at codon position 72 of TP53 gene in both UM1 and UM2 cells, which caused the replacement of proline with arginine for the protein. This single nucleotide polymorphism is located in the proline rich region but not within the central region of DNA-binding domain (codon 101-306), which is the target of 90% of p53 mutations found in human cancers. Many studies have investigated the genetic link between this variation and cancer susceptibility; however, the results have been controversial. For instance, TP53 codon 72 polymorphism was associated with an increased risk of lung cancer [32] and renal cell carcinoma [33]. However, meta-analyses failed to show significant associations between TP53 codon 72 polymorphisms with cervical cancer [34], colorectal cancer [35] and endometrial cancer [36]. Nevertheless, this single variation may not be sufficient to completely impair p53 function in UM1 and UM2 cells, and certainly not as oncogenic as mutant TP53 found in UM-SCC5 and UM-SCC10B cells which contain mutations within the DNA-binding domain.
Our studies suggest that WRN may be involved in the regulation of XLF expression in HNSCC cells. The ChIP assay results indicate that WRN binds to the NHEJ1 gene promoter in UM1 and UM2 cells. This is not surprising considering that WRN is a RecQ-like helicase which unwinds double-stranded DNA to facilitate DNA replication or gene transcription. In addition, knockdown of WRN in head and neck cancer cells led to the down-regulation of XLF expression. These findings seem to imply that WRN may be involved in the regulation of NHEJ1 gene expression in HNSCC cells. In fact, previous studies have shown that WRN has a role in RNA polymerase II-dependent gene transcription. WRN functionally interacts with rRNA and unwind not only a duplex DNA but also an RNA-DNA heteroduplex [37-39]. Furthermore, DNA microarray analysis indicates that WRN protein, by virtue of its helicase and transcription-activating activities, as well as its protein interactions, is directly or indirectly involved in the transcription of genes upstream in the network of aging pathways [40]. Collectively, these previous findings indicate involvement of WRN in transcription and suggest that decreased transcription rate might be a cause of premature aging phenotypes observed in WS patients [40].
Although there has been no evidence on its direct regulation of XLF expression, p53 may be involved in this regulatory process through its interaction with WRN. The binding of mutant p53 to WRN has been well demonstrated in previous studies and shown to inhibit WRN exonuclease/ATPase activity [41, 42]. Our Co-IP results further confirm that mutant p53 protein (V157F and G245C, UM-SCC5 and UM-SCC10B cells) binds to WRN and may downplay the functional role of WRN. In UM-SCC5 and UM-SCC10B cells, the mutant p53 expression levels are substantially high, which may partially deactivate the function of WRN due to its strong binding to WRN. Consequently, the expression of XLF is significantly inhibited, which might explain the inverse relationship between p53 and XLF expression levels in the HNSCC cells.
As a summary, we have found WRN may be involved in the regulation of XLF, an important DSB repair factor, in HNSCC cells. We have also found that XLF is over-expressed in HPV(+) versus HPV(-) HNSCCs and significantly down-regulated in HNSCC cells expressing high level of mutant p53 when compared to the HNSCC cells harboring wild-type TP53. This may be due to the binding of mutant p53 to WRN protein, partially inactivating the function of WRN and therefore down-regulating the expression of XLF. However, in HNSCC cells harboring wild-type TP53, p53 protein expression is very low and therefore may not affect the activity of WRN. In the future, we plan to knock out or knock down the expression of mutant p53 in the HNSCC cell lines harboring mutant TP53 gene, and further verify if down-regulation of mutant p53 leads to the over-expression of XLF in the HNSCC cells. Nevertheless, the physiologic role of XLF in NHEJ pathway has been well established, and its down-regulation is known to cause a defect in DSB repair. Our findings suggest there is a possible interplay of WRN/p53 on the regulation of XLF expression, and provide additional insight regarding the role of mutant p53 and WRN in NHEJ process.
Acknowledgements
This study was supported in part by the TRDRP and Wendy Case Cancer Fund.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Weterings E, Chen DJ. The endless tale of non-homologous end-joining. Cell Res. 2008;18:114-24
2. Buck D, Malivert L, de Chasseval R, Barraud A, Fondanèche M-C, Sanal O. et al. Cernunnos, a Novel Nonhomologous End-Joining Factor, Is Mutated in Human Immunodeficiency with Microcephaly. Cell. 2006;124:287-99
3. Callebaut I, Malivert L, Fischer A, Mornon J-P, Revy P, de Villartay J-P. Cernunnos Interacts with the XRCC4·DNA-ligase IV Complex and Is Homologous to the Yeast Nonhomologous End-joining Factor Nej1. Journal of Biological Chemistry. 2006;281:13857-60
4. Hammel M, Rey M, Yu Y, Mani RS, Classen S, Liu M. et al. XRCC4 Protein Interactions with XRCC4-like Factor (XLF) Create an Extended Grooved Scaffold for DNA Ligation and Double Strand Break Repair. Journal of Biological Chemistry. 2012;286:32638-50
5. Yano K, Morotomi-Yano K, Akiyama H. Cernunnos/XLF: a new player in DNA double-strand break repair. Int J Biochem Cell Biol. 2009;41:1237-40
6. Shirodkar P, Fenton AL, Meng L, Koch CA. Identification and functional characterization of a Ku-binding motif in aprataxin polynucleotide kinase/phosphatase-like factor (APLF). J Biol Chem. 2013;288:19604-13
7. Yano K, Morotomi-Yano K, Lee KJ, Chen DJ. Functional significance of the interaction with Ku in DNA double-strand break recognition of XLF. FEBS Lett. 2011;585:841-6
8. Multani AS, Chang S. WRN at telomeres: implications for aging and cancer. Journal of Cell Science. 2007;120:713-21
9. Rossi ML, Ghosh AK, Bohr VA. Roles of Werner syndrome protein in protection of genome integrity. DNA Repair (Amst). 2010;9:331-44
10. Lee JW, Harrigan J, Opresko PL, Bohr VA. Pathways and functions of the Werner syndrome protein. Mech Ageing Dev. 2005;126:79-86
11. Li B, Comai L. Functional Interaction between Ku and the Werner Syndrome Protein in DNA End Processing. Journal of Biological Chemistry. 2000;275:28349-52
12. Cooper MP, Machwe A, Orren DK, Brosh RM, Ramsden D, Bohr VA. Ku complex interacts with and stimulates the Werner protein. Genes & Development. 2000;14:907-12
13. Li B, Comai L. Requirements for the Nucleolytic Processing of DNA Ends by the Werner Syndrome Protein-Ku70/80 Complex. Journal of Biological Chemistry. 2001;276:9896-902
14. Karmakar P, Snowden CM, Ramsden DA, Bohr VA. Ku heterodimer binds to both ends of the Werner protein and functional interaction occurs at the Werner Nâ€terminus. Nucleic Acids Research. 2002;30:3583-91
15. Sidorova JM. Roles of the Werner syndrome RecQ helicase in DNA replication. DNA Repair. 2008;7:1776-86
16. Mahaney BL, Meek K, Lees-miller SP. Repair of ionizing radiation-induced DNA double-strand breaks by non-homologous end-joining. Biochemical Journal. 2009;417:639-50
17. Blander G, Kipnis J, Leal JF, Yu CE, Schellenberg GD, Oren M. Physical and functional interaction between p53 and the Werner's syndrome protein. J Biol Chem. 1999;274:29463-9
18. Bradford CR, Zhu S, Ogawa H, Ogawa T, Ubell M, Narayan A. et al. P53 mutation correlates with cisplatin sensitivity in head and neck squamous cell carcinoma lines. Head & Neck. 2003;25:654-61
19. Wiest T, Schwarz E, Enders C, Flechtenmacher C, Bosch FX. Involvement of intact HPV16 E6/E7 gene expression in head and neck cancers with unaltered p53 status and perturbed pRb cell cycle control. Oncogene. 2002;21:1510-7
20. van Houten VMM, Snijders PJF, van den Brekel MWM, Kummer JA, Meijer CJLM, van Leeuwen B. et al. Biological evidence that human papillomaviruses are etiologically involved in a subgroup of head and neck squamous cell carcinomas. International Journal of Cancer. 2001;93:232-5
21. Rivlin N, Brosh R, Oren M, Rotter V. Mutations in the p53 Tumor Suppressor Gene: Important Milestones at the Various Steps of Tumorigenesis. Genes Cancer. 2011;2:466-74
22. Strano S, Dell'Orso S, Mongiovi AM, Monti O, Lapi E, Di Agostino S. et al. Mutant p53 proteins: between loss and gain of function. Head Neck. 2007;29:488-96
23. Freed-Pastor WA, Prives C. Mutant p53: one name, many proteins. Genes Dev. 2012;26:1268-86
24. Koch WM, Brennan JA, Zahurak M, Goodman SN, Westra WH, Schwab D. et al. p53 mutation and locoregional treatment failure in head and neck squamous cell carcinoma. J Natl Cancer Inst. 1996;88:1580-6
25. Tassone P, Old M, Teknos TN, Pan Q. p53-based therapeutics for head and neck squamous cell carcinoma. Oral Oncol. 2013;49:733-7
26. Skinner HD, Sandulache VC, Ow TJ, Meyn RE, Yordy JS, Beadle BM. et al. TP53 disruptive mutations lead to head and neck cancer treatment failure through inhibition of radiation-induced senescence. Clin Cancer Res. 2012;18:290-300
27. Peltonen JK, Helppi HM, Paakko P, Turpeenniemi-Hujanen T, Vahakangas KH. p53 in head and neck cancer: functional consequences and environmental implications of TP53 mutations. Head Neck Oncol. 2010;2:36
28. Lang GA, Iwakuma T, Suh YA, Liu G, Rao VA, Parant JM. et al. Gain of function of a p53 hot spot mutation in a mouse model of Li-Fraumeni syndrome. Cell. 2004;119:861-72
29. Heinlein C, Krepulat F, Lohler J, Speidel D, Deppert W, Tolstonog GV. Mutant p53(R270H) gain of function phenotype in a mouse model for oncogene-induced mammary carcinogenesis. Int J Cancer. 2008;122:1701-9
30. Muller PA, Caswell PT, Doyle B, Iwanicki MP, Tan EH, Karim S. et al. Mutant p53 drives invasion by promoting integrin recycling. Cell. 2009;139:1327-41
31. Adorno M, Cordenonsi M, Montagner M, Dupont S, Wong C, Hann B. et al. A Mutant-p53/Smad complex opposes p63 to empower TGFbeta-induced metastasis. Cell. 2009;137:87-98
32. Piao J-M, Kim HN, Song H-R, Kweon S-S, Choi J-S, Yun W-J. et al. p53 codon 72 polymorphism and the risk of lung cancer in a Korean population. Lung Cancer. 2011;73:264-7
33. Huang C-Y, Su C-T, Chu J-S, Huang S-P, Pu Y-S, Yang H-Y. et al. The polymorphisms of P53 codon 72 and MDM2 SNP309 and renal cell carcinoma risk in a low arsenic exposure area. Toxicology and Applied Pharmacology. 2011;257:349-55
34. Klug SJ, Ressing M, Koenig J, Abba MC, Agorastos T, Brenna SMF. et al. TP53 codon 72 polymorphism and cervical cancer: a pooled analysis of individual data from 49 studies. The Lancet Oncology. 2009;10:772-84
35. Wang J-J, Zheng Y, Sun L, Wang L, Yu P-B, Dong J-H. et al. TP53 codon 72 polymorphism and colorectal cancer susceptibility: a meta-analysis. Mol Biol Rep. 2011;38:4847-53
36. Jiang D-K, Yao L, Ren W-H, Wang W-Z, Peng B, Yu L. TP53 Arg72Pro polymorphism and endometrial cancer risk: a meta-analysis. Med Oncol. 2011;28:1129-35
37. Rossi ML, Ghosh AK, Bohr VA. Roles of Werner syndrome protein in protection of genome integrity. DNA Repair. 2010;9:331-44
38. Balajee AS, Machwe A, May A, Gray MD, Oshima J, Martin GM. et al. The Werner Syndrome Protein Is Involved in RNA Polymerase II Transcription. Molecular Biology of the Cell. 1999;10:2655-68
39. Suzuki N, Shimamoto A, Imamura O, Kuromitsu J, Kitao S, Goto M. et al. DNA helicase activity in Werner's syndrome gene product synthesized in a baculovirus system. Nucleic Acids Research. 1997;25:2973-8
40. Kyng KJ, May A, Kølvraa S, Bohr VA. Gene expression profiling in Werner syndrome closely resembles that of normal aging. Proceedings of the National Academy of Sciences. 2003;100:12259-64
41. Brosh RM, Karmakar P, Sommers JA, Yang Q, Wang XW, Spillare EA. et al. p53 Modulates the Exonuclease Activity of Werner Syndrome Protein. Journal of Biological Chemistry. 2001;276:35093-102
42. Yang Q, Zhang R, Wang XW, Spillare EA, Linke SP, Subramanian D. et al. The Processing of Holliday Junctions by BLM and WRN Helicases Is Regulated by p53. Journal of Biological Chemistry. 2002;277:31980-7
Author contact
![]() Corresponding author: Shen Hu, Ph.D., University of California, Los Angeles, CA 90095-1668; Phone: 310-206-8834;E-mail: shenhuedu
Corresponding author: Shen Hu, Ph.D., University of California, Los Angeles, CA 90095-1668; Phone: 310-206-8834;E-mail: shenhuedu