Impact Factor ISSN: 1837-9664

 Global reach, higher impact
Global reach, higher impactJ Cancer 2016; 7(11):1388-1395. doi:10.7150/jca.15274 This issue Cite
Research Paper
Notch Signaling Activation in Cervical Cancer Cells Induces Cell Growth Arrest with the Involvement of the Nuclear Receptor NR4A2
Lichun Sun1,2,4, ![]() , Mingqiu Liu3,
, Mingqiu Liu3, ![]() , Guang-Chun Sun1, Xu Yang1, Qingqing Qian1, Shuyu Feng3, L. Vienna Mackey4, David H. Coy4
, Guang-Chun Sun1, Xu Yang1, Qingqing Qian1, Shuyu Feng3, L. Vienna Mackey4, David H. Coy4
1. Department of Pharmacy, The Fifth People's Hospital of Shanghai, Fudan University; 801 He-Qing Rd., Shanghai 200240, China;
2. Hunan Province Cooperative Innovation Center for Molecular Target New Drug Study, Institute of Pharmacy & Pharmacology, University of South China, Hengyang, 421001, China;
3. State Key Laboratory of Genetic Engineering, Institute of Genetics, School of Life Science, Fudan University, Shanghai 200433, China;
4. Department of Medicine, School of Medicine, Tulane Health Sciences Center, New Orleans, LA 70112-2699, USA.
Received 2016-2-15; Accepted 2016-5-24; Published 2016-6-30
Abstract
Cervical cancer is a second leading cancer death in women world-wide, with most cases in less developed countries. Notch signaling is highly conserved with its involvement in many cancers. In the present study, we established stable cervical cell lines with Notch activation and inactivation and found that Notch activation played a suppressive role in cervical cancer cells. Meanwhile, the transient overexpression of the active intracellular domain of all four Notch receptors (ICN1, 2, 3, and 4) also induced the suppression of cervical cancer Hela cell growth. ICN1 also induced cell cycle arrest at phase G1. Notch1 signaling activation affected the expression of serial genes, especially the genes associated with cAMP signaling, with an increase of genes like THBS1, VCL, p63, c-Myc and SCG2, a decrease of genes like NR4A2, PCK2 and BCL-2. Particularly,
The nuclear receptor NR4A2 was observed to induce cell proliferation via MTT assay and reduce cell apoptosis via FACS assay. Furthermore, NR4A2's activation could reverse ICN1-induced suppression of cell growth while erasing ICN1-induced increase of tumor suppressor p63. These findings support that Notch signaling mediates cervical cancer cell growth suppression with the involvement of nuclear receptor NR4A2. Notably, Notch/NR4A2/p63 signaling cascade possibly is a new signling pathway undisclosed.
Keywords: cervical cancer, cell proliferation, cell apoptosis, Notch signaling, nuclear receptor NR4A2, tumor suppressor p63.
Introduction
As the second leading cause of cancer death, cervical cancer is a major health concern for women world-wide (1, 2), with most cases occurring in less developed countries. The highest incidences are happening in Latin America, the Caribbean and Africa (World Cancer Research: www.wcrf.org). Cervical cancer is known to be involved with multiple signaling pathways (3-6). The human papillomavirus (HPV) has been shown to be an essential component in cervical cancer progression (1-4, 7). However, many other factors, such as Notch, Wnt, COX2, NF-KB, p53 and RhoC, are also critical elements associated with the development of cervical cancer (4, 6-12).Notch signaling especially has been found to play a critical role in cervical cancer development.
Notch signaling is highly conserved and is critical for human development. Importantly, Notch signaling is found aberrantly expressed in many types of cancers and is involved in cancer progression. The up-regulation of Notch1 and the cognate ligand, Jagged1, have been demonstrated in cervical cancer cells (2, 13-15). This prompted an investigation into the possible role of Notch signaling in this cancer progression. Notch signaling activation in cervical cancer further displays suppression on cell proliferation and tumor growth (7, 14, 16, 17). The strategy of targeting Notch signaling provides a potential and effective therapeutic alternative in the treatment of cervical cancer (18-20). However, Notch signaling is much more complicated. Notch signaling acts differently in different stages of cervical cancer development, with an up-regulation of this signaling in early stages and a down-regulation in its late stage. The controversial effects of Notch signaling, in the same cervical cancer cell models, also are reported by other independent investigators(5, 21). Thus, the role of Notch signaling and its precise molecular mechanisms are not completely known.
Notch1 activation was also observed to stimulate the signaling of G protein-coupled somatostatin receptors xxx. This characteristic has been applied in combination therapy by combining a Notch signaling activator with a SSTR2-targeting peptide-drug and using them together as a conjugate. The combination therapy displays enhanced anti-tumor activity when compared to each used alone (19, 20, 22). However, the Notch-mediated mechanism is not yet clear. Also, Notch activation could enhance for skolin-induced cAMP production (19). We hypothesized that genes associated with cAMP signaling may be involved in Notch-mediated signaling networks. In the present study, we further investigated and determined the effects of the active forms of all four Notch receptors on cervical cancer cells. Particularly, we evaluated the effects of Notch1 activation on certain genes associated with cAMP/Ca2+ signaling via PCR array. We found that the nuclear receptor NR4A2 was down-regulated by Notch activation. NR4A2 activation increased cervical cancer cell growth via acting in an oncogenic role. Targeting crosstalk events of Notch and NR4A2 is likely to provide valuable paradigms around which to develop highly specific chemotherapeutic interventions.
Materials and Methods
Materials
The plasmids expressing the intracellular domains of the four Notch receptors (ICN, ICN2, ICN3, and ICN4) and dnMAML (DNL) were gifts from Dr. Wu (University of Florida). The plasmids with the genes NR4A2 (Nurr1), Vinculin (VCL), THBS1 (TSP1), p63 and Twist were obtained from Addgene (www.addgene.org), including pCCL-NR4A2 (plasmid 35000) and the controls pCCL (10881), pmEN1-VCL (54304), pmEN1 (53976), pBabe-Twist1 (1783), pBabe (10668), pcDNA-THBS1 (12405) and pcDNA-p63 (27008). Antibodies of p21 (Cat. No.: sc-756), p63 (sc-8343), c-Myc (sc-788), NR4A2 (sc-990), HES1 (sc-25392) and β-actin (sc-1616-HRP) were purchased from Santa Cruz Biotechnology (Santa Cruz, CA).
Cell culture and cell transfection
Human cervical cancer Hela cells were purchased from ATCC (American Type Culture Collection, Manassas, VA). The stable cancer cell lines Hela-ICN1 (over-expressing ICN1, Notch1 activation) and Hela-DNL1 (over-expressing dominant-negative mastermind-like1 (DNL1, or dnMAML1), Notch inactivation) along with Hela-GFP cells were established as described previously (19). These cells were maintained in MEM medium supplemented with 10% fetal bovine serum (FBS), 1% penicillin/streptomycin and 0.5% kanamycin. For transient transfection, 0.5ml of a Hela cell suspension with 2x105 cells/ml was plated in each well of 24-well plates and cultured overnight. Two μl of LipofectamineTM 2000 (Lipo-2000) and a total amount (0.4μg) of DNA were added separately into each vial with 50 μl Opti-MEM transfection medium, and combined together after a 5-10 minute incubation. The DNA-Lipo-2000 complexes were mixed well and incubated for 20-30 minutes and then added to each well. Growth medium was replaced 4-5 hours later and cells were incubated for 72-hours.
Real-time PCR and PCR array
Total RNA was isolated from tumor cells and RT-PCR was performed as described in the protocols (Invitrogen, Carlsbad, CA). The primers and conditions for RT-PCR analyses were as described previously (19). Real-time PCR assays and PCR arrays (cAMP/Ca2+ signaling pathway (PAHS-066), GPCR signaling pathway (PAHS-071)) (SABiosciences) were performed as described (19). β-actin was used as the internal control and data were analyzed by applying the 2-ΔΔCT methods.
Western blot analysis
This assay was employed as described in the protocol (Santa Cruz). Briefly, cells were harvested, re-suspended in RIPA buffer with cocktail inhibitors, homogenized with a 21-gauge needle, mixed with the loading buffer and heated for 7 min at 95oC. Supernatants were loaded to run on a 8-16% Tris-glycine gel after centrifugation at 13,000xg. Protein was transferred to a nitrocellulose membrane and blocked with 5% fat-free milk, washed and incubated with primary antibody. The membrane was washed again and incubated with second antibody (Santa Cruz). Films were developed according to the ECL system protocol (Amersham Biosciences, England). The experiments were separately done three times.
Cell proliferation
The cell proliferation assay (Promega, Madison, WI) was performed as described (19). Briefly, 50 μl aliquots of medium with different concentrations of compounds were added to 96-well plates. All compound concentrations were tested in triplicate. Another 50 μl of the BON cell stock (1x105cells/ml of media) was dispensed into each test well and the plates were incubated at 37oC in a CO2 incubator for 3 days. Following the incubation period, 15 μl of the dye solution was added to each well and the plates were then incubated at 37oC for 4 hours, followed by the addition of 100 µl per well of the solubilization solution. The plates were incubated at 37oC until the contents in each well became a uniform-colored solution. The absorbance was measured and recorded at 570 nm by a Victor Plate Reader (PerkinElmer, Boston, MA).
Cell cycle
Cells were plated in 6-well plates and incubated overnight. The test compounds were added. The Coulter DNA Prep reagents kit (Cat. No.: PN 6607055) from Beckman Coulter (Fullerton, CA) was used and assayed as described previously (23). Analysis was done on a Beckman-Coulter Epics FC500 analyzer using CXP software for acquisition and the ModFit LT v3.1 (Verity Software) for Cell Cycle Modeling. Experiments were repeated three times.
Cell attachment
The cell attachment assay (ECM205) was used to study the effects of VPA on cell attachment to different components, including fibronectin, vitronectin, laminin, collagen I and collagen IV (ECM205, Chemicon). The assay was performed after a 72-hour incubation according to the kit instructions (Chemicon). One ml of cells (1x105 cells/ml) was plated in 24-well plates and treated with or without the test compound-VPA. The treated cells were harvested with enzyme-free cell dissociation buffer and added to each well of strips that were pre-rehydrated with PBS buffer. The strips were incubated for 45-60 minutes at 37oC in the incubator. The strips were washed 3 times, stained with 0.2% crystal violet for 5 minutes and washed 3-5 times. One hundred μl of solubilization buffer was added to each well and the absorbance at 570 nm was determined after the cell-bound stain was completely solubilized. The assays were normalized by cell proliferation assays.
Results
Cervical cancer is the leading cause of cancer death in women. Notch signaling is of importance in cervical cancer progression, with this signaling being up-regulated in its early stage and down-regulated in late stage. The Notch-mediated mechanism in this cancer is not clear yet. Our previous data showed that Notch activation could enhance for skolin-induced cAMP production (19). In this study, we evaluated the effects of Notch activation on cell growth and analyzed the involvement of cAMP/Ca2+ signaling in Notch-mediated growth suppression.
Activated Notch signaling acts as a tumor suppressor
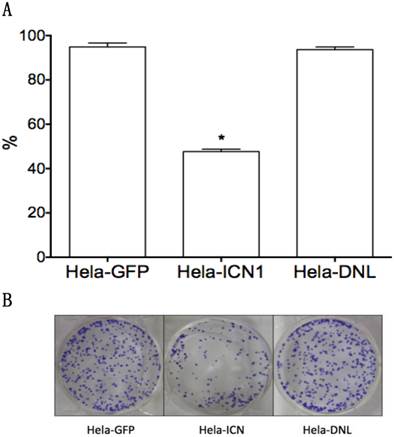
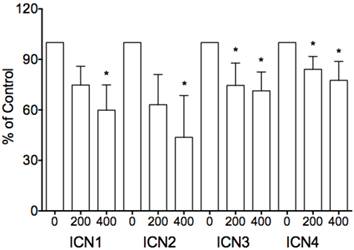
We investigated the effects of Notch1 signaling on cervical cancer Hela cells. Via MTT assay, Hela-ICN1 cells with Notch1 activation were found to grow slowly compared to control Hela-GFP cells, with Notch-induced inhibitory rates being 52.4%, and with no significant difference in Hela-DNL1cells (knockdown of Notch signaling) (Fig. 1). Notch signaling inhibitors DBZ and DAPT had no effect at the tested concentrations (data not shown). Furthermore, we did transient transfection with the active forms of all four Notch receptors (ICN1, ICN2, ICN3, ICN4) and found that all four ICNs could induce cell growth suppression, with ICN1 and ICN2 displaying more significant effects on Hela cell growth. As shown in Fig. 2, the effects are dose-dependent, with the inhibitory rates being 25.3% (ICN1: 200ng), 40.2% (ICN1: 400ng), 36.9% (ICN2: 200ng), 50.3 % (ICN2, 400ng), 25.5% (ICN3: 200ng), 28.6% (ICN3, 400ng), 15.9% (ICN4: 200ng) and 22.5% (ICN4, 400ng).These data support that Notch signaling serves as a tumor suppressor in cervical cancer Hela cells.
The effects of Notch signaling on cervical cancer Hela cell growth. Compared to the control (Hela-GFP), ICN1's over-expression induced cell growth arrest in Hela-ICN1 cells (Notch signaling activation), with an inhibitory rate of 52.4%, but there is no significant effect in Hela-DNL cells (Notch signaling knockdown). A, cell proliferation assay; B, cell colony assay. Asterisk (*) shows P value being less than 0.05.
The inhibitory effects of transient transfection of four Notch active forms (ICN1 to ICN4) on cell proliferation. The inhibition is dose-dependent, with the inhibitory rates being 25.3% (ICN1: 200ng), 40.2% (ICN1: 400ng), 36.9% (ICN2: 200ng), 50.3 % (ICN2, 400ng), 25.5% (ICN3: 200ng), 28.6% (ICN3, 400ng), 15.9% (ICN4: 200ng) and 22.5% (ICN4, 400ng), respectively. Asterisk (*) shows P value being less than 0.05.
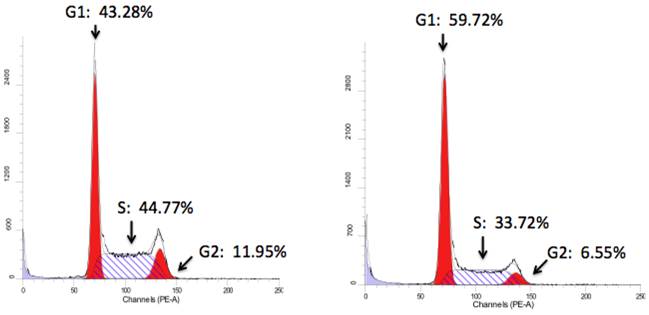
Further, FACS analysis was used to evaluate Notch1-mediated cell cycle progression. Notch1 induced cell cycle arrest at phase G1 (Hela-ICN1), with a rate of 62% compared to 47% of the control (Hela-GFP) (Fig. 3). Meanwhile, FACS analysis showed that the Hela-ICN1 cell debris was 11% compared to 4% of Hela-GFP cell debris.
Cell cycle analysis. Notch1 activation in Hela-ICN1cells (right) results in 59.72% (Phase G1), 6.55% (G2) and 33.72% (S), with 43.28% (G1), 11.95% (G2) and 44.77% (S) in the control Hela-GFP(left).
Cell attachment is a critical step for cell migration and tumor metastasis. Also in our previous study, we observed Notch signaling activation could induce cervical cancer cell morphological changes in Hela-ICN1 cells. Thus, we did cell attachment assays to assess the effects of Notch signaling on cell attachment. We first tested the attachment of cervical cancer Hela cells to the extracellular matrix (ECM) components that included fibronectin, vitronectin, laminin, collagen I and collagen IV and found that cells strongly attached to laminin, but had little or no attachment to the others. Then, we found that Notch1 signaling activation, via transient transfection of ICN1, showed no significant effect on the attachment of cervical cancer Hela cells to the ECM component laminin (ECM103), but THBS1, upregulated by ICN1, was observed to increase cell attachment (Fig. 4).
The effects of gene over-expression on cell-laminin adhesion (ECM103). There are no significant effects of genes such as ICN1, NR4A2, vinculin, THBS1, Twist1 on ECM103. However, THBS1 enhanced cell adherence to laminin, with a rate of 58.1%, and asterisk (*) shows P value < 0.05.
Notch1 signaling activation regulates gene expression
Our previous study showed that Notch1 activation extremely enhanced for skolin-induced production of the second messenger cAMP that transduces intracellular signaling, and it also regulated somatostatin signaling that belongs to G protein-coupled receptor signaling. This implied that Notch signaling might be involved in signaling pathways associated with cAMP and GPCRs. Thus, via PCR arrays, we investigated the effects of Notch1 activation on the genes associated with cAMP/Ca2+ signaling and GPCR signaling. We found that Notch1 activation in Hela-ICN1 cells regulated the expressional changes of certain genes that are related to CRE (cAMP responsive element), SRE(serum response element), Ca2+and other cAMP-coupled G protein signaling.
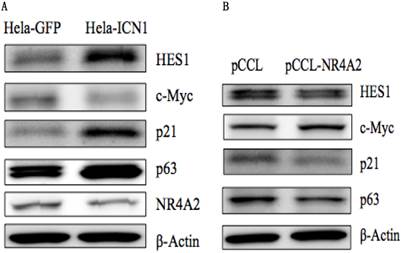
As shown in Table 1, ICN1 induced an increase in such genes as vinculin (VCL) (5.2 fold), SCG2 (8.8 fold), ADRB2 (4.1 fold), SST (203 fold), SSTR2 (20.8 fold) and THBS1(TSP1) (60.7 fold), and a decrease of such genes as NR4A2 (-5 fold), SGK1(-1.5 fold), NPY(-3.8 fold), ENO2 (-1.7 fold), CALR (-1.3 fold), PCK2 (-2.8 fold), CREB1 (-1.6 fold), CGA (-15.6 fold), NF1(-1.4 fold), BRCA1(-1.5 fold), VEGFA (-1.82 fold) and RB1 (-2.8 fold), (Table 1) and had no effect on genes with CRE/SRE sequences in the promoters or belonging to GPCRs such as FGF6, IL-2, IL-6, GCG2, DRD1, PDPK1 andOPRD1 (data not shown). Certain other genes (and some cancer-related genes) were observed to be modulated by Notch1 activation, with an ICN1-induced increase of Notch target genesHES1, p63, p21 and Twist1, and a decrease of c-Myc. The expression of certain of these genes such as NR4A2, HES1, p63, p21 and c-Mycin Hela-ICN1 cells was confirmed at the protein level by Western blot analysis (Fig. 5A). Next, we investigated the effects of some chosen genes on cell growth and their involvement in Notch1-mediation signaling pathways (discussion below).
PCR array shows the effects of Notch1 signaling activation on the expression of certain genes at the mRNA level.
| Symbol | Gene description | Unique sequenceor signaling | Fold | Change |
|---|---|---|---|---|
| THBS1 | Thrombospondin 1 | SRE/SRE-like sequence | 60.67 ± 15.32 | ↑ |
| VCL | Vinculin | SRE/SRE-like sequence | 5.18 ± 1.32 | ↑ |
| SCG2 | Secretogranin II | CRE enhancer sequence SRE/SRE-like sequence | 8.82 ± 3.75 | ↑ |
| SST | Somatostatin | CRE enhancer sequence | 202.5 ± 46* | ↑ |
| SSTR2 | SST receptor 2 | CRE enhancer sequence | 20.77 ± 3.46* | ↑ |
| p63 | Tumor suppressor | 10.03 ± 1.28 | ↑ | |
| Twist1 | Cell differentiation | 8.67 ± 0.57 | ↑ | |
| ADRB2 | Adrenergic receptor, beta-2 | cAMP-coupled G protein signaling | 4.1 ± 0.09 | ↑ |
| NR4A2 | Nuclear receptor subfamily 4, group A, member 2 (Nurr1) | CRE enhancer sequence SRE/SRE-like sequence | -2.82 ± 0.17 | ↓ |
| SGK1 | Serum/glucocorticoid regulated kinase 1 | CRE enhancer sequence | -1.51 ± 0.14 | ↓ |
| NPY | Neuropeptide Y | Ca2+ responsive element | -3.78 ± 0.33 | ↓ |
| ENO2 | Enolase 2 | CRE enhancer sequence | -1.67 ± 0.21 | ↓ |
| CALR | Calreticulin | Ca2+ responsive element | -1.29 ± 0.05 | ↓ |
| PCK2 | Phosphoenolpyruvatecarboxykinase 2 | CRE enhancer sequence | -2.80 ± 1.09 | ↓ |
| BCL-2 | B-cell CLL/lymphoma 2 | CRE enhancer sequence | -6.95± 1.30* | ↓ |
| PCNA | Proliferating cell nuclear antigen | CRE enhancer sequence | -2.16 ± 0.02* | ↓ |
| STAT3 | Signal transducer and activator of transcription 3 | CRE enhancer sequence | -3.58± 0.88* | ↓ |
| NF1 | Neurofibromin 1 | CRE enhancer sequence | -1.36 ± 0.34 | ↓ |
| CREB1 | cAMP responsive element binding protein 1 | CRE enhancer sequence | -1.55 ± 0.15 | ↓ |
| CGA | Glycoprotein hormone, alpha polypeptide | CRE enhancer sequence | -15.67 ± 0.99 | ↓ |
| BRCA1 | Breast cancer 1, early onset | CRE enhancer sequence | -1.47 ± 0.09 | ↓ |
| RB1 | Retinoblastoma 1 | CRE enhancer sequence | -2.83 ± 0.98 | ↓ |
| JUND | Jun D proto-oncogene | CRE enhancer sequence | -1.73 ± 0.69 | ↓ |
| JUNB | Jun B proto-oncogene | SRE/SRE-like sequence | -4.65 ± 0.06 | ↓ |
| VEGFA | Vascular endothelial growth factor A | cAMP-coupled G protein signaling, | -1.82 ± 0.22 | ↓ |
* note: data are cited from ref.(19).
Western blot analysis. (A), Notch1 activation (Hela-ICN1) induced an increase of the Notch target genes HES1, p21 and p63, and a decrease of c-Myc and NR4A2. (B), NR4A2 over-expression induced a decrease of HES1, p21 and p63, and an increase of c-Myc.
NR4A2 increases cell proliferation and THBS1 increases cell attachment
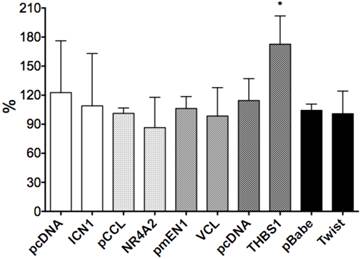
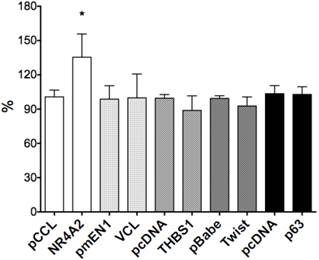
We selected six Notch1-mediated genes including NR4A2, Vinculin, THBS1, p63 and Twist, and did transient transfection with the plasmids expressing those genes. We assessed the effects of these genes on cell growth by cell proliferation assay and found stimulative effects by NR4A2, with a rate of 34.7%, but with little or no significant effects on VCL, p63, THBS1 or Twist1 (Fig. 6).
The effects of gene over-expression on cell proliferation. NR4A2 induced cell proliferation, with an increased rate of 34.7% and the asterisk (*) shows P values< 0.05. There are no significant effects of the genes VCL, THBS1, Twist1 and p63.
As described above, we observed that Notch1 activation, by over-expressing ICN1, reduced cell-laminin attachment. We also investigated the effects of NR4A2, VCL, THBS1 and Twist1 on Hela cell attachment. By cell attachment assay (ECM103), we found that THBS1 induced an increase of the cell-laminin attachment, with a rate of 58.1%, with the other three, NR4A2, VCL and Twist1, having no significant effects (Fig. 4). These findings indicate that different mechanism are involved in cell proliferation and cell attachment.
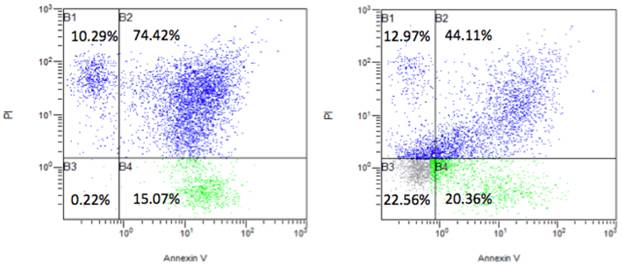
Via FACS analysis, we further found that NR4A2 reduced cell apoptosis. Cells were continuously cultured for 2 days without changing medium after transfection with the transfection agent Lipo-2000. This would result in some cell death. We further did apoptosis assays and found that there was an apoptotic rate 89.4% (apoptosis and necro-apoptosis together) in the control group with the control pCCL vector, but the apoptotic rate dropped to 64.4% in the NR4A2-treated group and with an increased rate of visible cells (22.3%) (Fig. 7).
The effects of the nuclear receptor NR4A2 on cell apoptosis via FACS analysis. The transient transfection of NR4A2 could reverse cell apoptosis and increase visible cells, with a rate of 22.56% (right), compared to the visible cell rate of 0.22% in control (left).
Based on the results above, we hypothesized that NR4A2 might play an oncogenic role and thus further analyzed the effects of NR4A2 activation on the oncogene c-Myc, tumor suppressors p21 and p63, and the Notch target gene HES1. We observed thatNR4A2 activation induced a decrease of p21, p63, and HES1, with a slight increase of c-Myc (Fig. 5B), indicating Notch signaling and NR4A2 signaling reversely co-modulate downstream signaling while Notch plays the tumor-suppressive role and NR4A2 plays the oncogenic role.
NR4A2 reverses ICN1-induced cell growth suppression via regulating p63 signaling
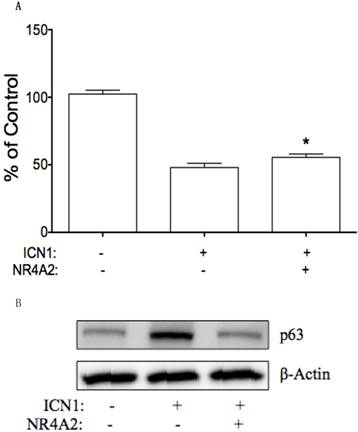
NR4A2 may be involved in ICN1-mediated effects on cervical cancer cells. We further tested this hypothesis by transient transfection of NR4A2 in Hela-ICN1cells (Notch1 activation), with Hela-GFP cells as the control. We found that NR4A2reversed ICN1-induced cell growth arrest. As shown in Fig. 8A, ICN1 decreased cell growth with an inhibition rate of 52%, NR4A2 reduce the ICN1-induced inhibitory rate to 44%. Also, via Western blot analysis, we found that NR4A2 reversed an ICN1-induced increase of p63 (Fig. 8B), but not of p21 and c-Myc, indicating that NR4A2 and Notch signaling share a common signaling involvement with p63 and that Notch1 induces cell growth arrest partly via regulating the downstream NR4A2/p63 signaling pathway.
NR4A2 reversed Notch1-induced cell growth arrest. (A), MTT assay showed that Notch1 activation in Hela-ICN1 reduced cell growth, with an inhibitory rate of 52%. The co-expression of NR4A2 and ICN1 reduced the Notch1-induced inhibitory rate to 44%, Asterisk (*) shows P values< 0.05. (B), NR4A2 via wetsern blot assay reduced the increase of Notch1-induced p63, with no effects on p21 and c-Myc.
Discussion
The role of Notch signaling in cervical cancer is not clearly determined. The levels of Notch signaling in cervical cancer seem to be stage-specific. Activated Notch1 at high levels, induces apoptosis and cell growth arrest through increasing the expression of p52, p21 and others (6, 12). However, Notch signaling in this cancer is dysfunctional and controversial (16, 17, 21). Thus, a more complete understanding of the molecular mechanisms of this Notch pathway in cervical cancer will lead to the development of novel therapeutic strategies. In our previous study in cervical cancer, Notch1 activation could induce cell growth arrest and tumor suppression, identical to other reports.
In the present study, we did assays to confirm the function of Notch in cervical cancer cells. Our assays showed that the knockdown of Notch signaling via over-expressing dominant-negative mastermind-like1 (DNL1, or dnMAML1) did not significantly affect cervical cancer Hela cell growth. However, we confirmed the inhibitory effects of Notch1 activation (16, 19, 20), and identified the similar inhibitory effects via the activation of the other three Notch receptors (ICN2, 3 and 4). We also found that activated Notch signaling stimulated the somatostatin signaling that blocks cAMP signaling, and enhanced for skolin-induced cAMP production (19). Thus, we hypothesized that cAMP signaling might be involved in Notch-mediated signaling networks.
cAMPis one well-known second messenger and is involved in various biological activities. cAMP signaling modulates cell functions and the associated signaling pathways. Our previous studies showed that Notch induced an increase of SST and SSTR2, with promotors' containing a CRE (cAMP responsive element) site (19). Meanwhile, we found that Notch modulated the expression of certain genes associated with cAMP/Ca2+ signaling, especially the genes with promotors containing CRE, SRE and Ca2+ elements and being responsive to cAMP or Ca2+. As shown in Table 1, Notch induced an increase of such genes as SST, SSTR2, THBS1 and VCL, and a decrease of such genes as NR4A2, STAT3 and CGA. Further, we evaluated the three CRE-containing genes VCL, THBS1 and NR4A2, tumor suppressor p63 and cell differentiation factor Twist1, for their effects on cell growth and cell adhesion. The nuclear receptor NR4A2 was observed to stimulate cell proliferation with others having no significant effects. In the cell adhesion assay, NR4A2 did not work, but the angiogenesis regulator THBS1increased Hela cells adhesion to laminin. NR4A2, acting as a transcription factor, induced cell differentiation and maintained dopaminergic neurons (24, 25). THBS1 is an adhesive glycoprotein and affects cell adhesion and cell migration (26, 27). Activated Notch signaling attenuates HPV-induced cervical cell transformation while its inactivation promotes cell transformation and carcinogenesis (2, 7, 28). Notch might partly suppress cell growth via attenuating NR4A2 and affect cell transformation and tumor metastasis via activating THBS1.
In summary, the identification of the correlation of Notch signaling and these factors in cervical cancer cells may support these findings indicating that targeting these crosstalk events likely will provide valuable paradigms to currently available clinical interventions targeting cervical cancer. Meanwhile, cAMP signaling may play a critical role in Notch-mediated cell functions via modulating the pathways involved with such genes as NR4A2 and THBS1. The precise mechanisms need to be identified and could be helpful in developing targeted drugs against cervical cancer.
Acknowledgements
The authors gratefully acknowledge the financial supports from Shanghai Science and Technology Committee of China (Grant No.14411972700), the financial supports from Tulane Peptide Research Fund and Open Project Funding from State Key Laboratory of Genetic Engineering of Fudan University Open Project Funding.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Walboomers JM, Jacobs MV, Manos MM, Bosch FX, Kummer JA, Shah KV. Human papillomavirus is a necessary cause of invasive cervical cancer worldwide. J Pathol. 1999;189(1):12-9
2. Wu L, Griffin JD. Modulation of Notch signaling by mastermind-like (MAML) transcriptional co-activators and their involvement in tumorigenesis. Seminars Can Biol. 2004;14(5):348-56
3. Duenas-Gonzalez A, Lizano M, Candelaria M, Cetina L, Arce C, Cervera E. Epigenetics of cervical cancer. An overview and therapeutic perspectives. Mol Cancer. 2005;4:38
4. Perez-Plasencia C, Duenas-Gonzalez A, Alatorre-Tavera B. Second hit in cervical carcinogenesis process: involvement of wnt/beta catenin pathway. Intl Arch Med. 2008;1(1):10
5. Weijzen S, Zlobin A, Braid M, Miele L, Kast WM. HPV16 E6 and E7 oncoproteins regulate Notch-1 expression and cooperate to induce transformation. J Cell Physiol. 2003;194(3):356-62
6. Yousif NG, Sadiq AM, Yousif MG, Al-Mudhafar RH, Al-Baghdadi JJ, Hadi N. Notch1 ligand signaling pathway activated in cervical cancer: poor prognosis with high-level JAG1/Notch1. Arch GynecolObstet. 2015;292(4):899-904
7. Talora C, Sgroi DC, Crum CP, Dotto GP. Specific down-modulation of Notch1 signaling in cervical cancer cells is required for sustained HPV-E6/E7 expression and late steps of malignant transformation. Genes Development. 2002;16(17):2252-63
8. Ramdass B, Maliekal TT, Lakshmi S, Rehman M, Rema P, Nair P. et al. Coexpression of Notch1 and NF-kappaB signaling pathway components in human cervical cancer progression. Gynecol Oncol. 2007;104(2):352-61
9. Ma YY, Wei SJ, Lin YC, Lung JC, Chang TC, Whang-Peng J. et al. PIK3CA as an oncogene in cervical cancer. Oncogene. 2000;19(23):2739-44
10. Kulkarni S, Rader JS, Zhang F, Liapis H, Koki AT, Masferrer JL. et al. Cyclooxygenase-2 is overexpressed in human cervical cancer. Clin Can Res. 2001;7(2):429-34
11. Rincon-Arano H, Rosales R, Mora N, Rodriguez-Castaneda A, Rosales C. R-Ras promotes tumor growth of cervical epithelial cells. Cancer. 2003;97(3):575-85
12. Yun J, Espinoza I, Pannuti A, Romero D, Martinez L, Caskey M. et al. p53 Modulates Notch Signaling in MCF-7 Breast Cancer Cells by Associating With the Notch Transcriptional Complex Via MAML1. J Cell physiol. 2015;230(12):3115-27
13. Allenspach EJ, Maillard I, Aster JC, Pear WS. Notch signaling in cancer. Cancer Biol Therap. 2002;1(5):466-76
14. Maliekal TT, Bajaj J, Giri V, Subramanyam D, Krishna S. The role of Notch signaling in human cervical cancer: implications for solid tumors. Oncogene. 2008;27(38):5110-4
15. Zagouras P, Stifani S, Blaumueller CM, Carcangiu ML, Artavanis-Tsakonas S. Alterations in Notch signaling in neoplastic lesions of the human cervix. PNAS. 1995;92(14):6414-8
16. Wang L, Qin H, Chen B, Xin X, Li J, Han H. Overexpressed active Notch1 induces cell growth arrest of HeLa cervical carcinoma cells. Intl J Gynecol Cancer. 2007;17(6):1283-92
17. Talora C, Cialfi S, Segatto O, Morrone S, Kim Choi J, Frati L. et al. Constitutively active Notch1 induces growth arrest of HPV-positive cervical cancer cells via separate signaling pathways. Exp Cell Res. 2005;305(2):343-54
18. del Campo JM, Prat A, Gil-Moreno A, Perez J, Parera M. Update on novel therapeutic agents for cervical cancer. Gynecol Oncol. 2008;110(3 Suppl 2):S72-6
19. Franko-Tobin LG, Mackey LV, Huang W, Song X, Jin B, Luo J. et al. Notch1-mediated tumor suppression in cervical cancer with the involvement of SST signaling and its application in enhanced SSTR-targeted therapeutics. The Oncologist. 2012;17(2):220-32
20. Tsai C, Leslie JS, Franko-Tobin LG, Prasnal MC, Yang T, Vienna Mackey L. et al. Valproic acid suppresses cervical cancer tumor progression possibly via activating Notch1 signaling and enhances receptor-targeted cancer chemotherapeutic via activating somatostatin receptor type II. Arch Gynecol Obstet. 2013;288(2):393-400
21. Yu H, Zhao X, Huang S, Jian L, Qian G, Ge S. Blocking Notch1 signaling by RNA interference can induce growth inhibition in HeLa cells. Intl J Gynecol Cancer. 2007;17(2):511-6
22. Sun L, Qian Q, Sun G, Mackey LV, Fuselier JA, Coy DH. et al. Valproic acid induces NET cell growth arrest and enhances tumor suppression of the receptor-targeted peptide-drug conjugate via activating somatostatin receptor type II. J Drug Targeting. 2015;27:1-9
23. Sun G, Mackey LV, Coy DH, Yu CY, Sun L. The Histone Deacetylase Inhibitor Vaproic Acid Induces Cell Growth Arrest in Hepatocellular Carcinoma Cells via Suppressing Notch Signaling. J Cancer. 2015;6(10):996-1004
24. Paillasse MR, de Medina P. The NR4A nuclear receptors as potential targets for anti-aging interventions. Med Hypotheses. 2015;84(2):135-40
25. Alavian KN, Jeddi S, Naghipour SI, Nabili P, Licznerski P, Tierney TS. The lifelong maintenance of mesencephalic dopaminergic neurons by Nurr1 and engrailed. Journal of biomedical science. 2014;21:27
26. Yafai Y, Eichler W, Iandiev I, Unterlauft JD, Jochmann C, Wiedemann P. et al. Thrombospondin-1 is produced by retinal glial cells and inhibits the growth of vascular endothelial cells. Ophthal Res. 2014;52(2):81-8
27. Feng N, Wang Z, Zhang Z, He X, Wang C, Zhang L. miR-487b promotes human umbilical vein endothelial cell proliferation, migration, invasion and tube formation through regulating THBS1. Neurosci Lett. 2015;591:1-7
28. Sakamoto K, Fujii T, Kawachi H, Miki Y, Omura K, Morita K. et al. Reduction of NOTCH1 expression pertains to maturation abnormalities of keratinocytes in squamous neoplasms. Lab Invest. 2012;92(5):688-702
Author contact
![]() Corresponding author: Lichun Sun: Tel:504-988-1179 Email: lsunedu Mingqiu Liu: Tel: 86-21-6564-2504 Email: liumqedu.cn
Corresponding author: Lichun Sun: Tel:504-988-1179 Email: lsunedu Mingqiu Liu: Tel: 86-21-6564-2504 Email: liumqedu.cn