Impact Factor ISSN: 1837-9664

 Global reach, higher impact
Global reach, higher impactJ Cancer 2016; 7(11):1441-1451. doi:10.7150/jca.15556 This issue Cite
Review
Genomic/Epigenomic Alterations in Ovarian Carcinoma: Translational Insight into Clinical Practice
Anliang Dong1, Yan Lu1, Bingjian Lu2 ![]()
1. Women's Hospital & Institute of Translational Medicine, School of Medicine, Zhejiang University, China;
2. Department of Surgical Pathology, Women's Hospital, School of Medicine, Zhejiang University, China.
Received 2016-3-16; Accepted 2016-5-24; Published 2016-7-5
Abstract
Ovarian carcinoma is the most lethal gynecological malignancy worldwide. Recent advance in genomic/epigenomic researches will impact on our prevention, detection and intervention on ovarian carcinoma. Detection of germline mutations in BRCA1/BRCA2, mismatch repair genes, and other genes in the homologous recombination/DNA repair pathway propelled the genetic surveillance of most hereditary ovarian carcinomas. Germline or somatic mutations in SMARCA4 in familial and sporadic small cell carcinoma of the ovary, hypercalcemia type, lead to our recognition on this rare aggressive tumor as a new entity of the atypical teratoma/rhaboid tumor family. Genome-wide association studies have identified many genetic variants that will contribute to the evaluation of ovarian carcinoma risk and prognostic prediction. Whole exome sequencing and whole genome sequencing discovered rare mutations in other drive mutations except p53, but demonstrated the presence of high genomic heterogeneity and adaptability in the genetic evolution of high grade ovarian serous carcinomas that occurs in cancer progression and chemotherapy. Gene mutations, copy number aberrations and DNA methylations provided promising biomarkers for the detection, diagnosis, prognosis, therapy response and targets of ovarian cancer. These findings underscore the necessity to translate these potential biomarkers into clinical practice.
Keywords: ovarian carcinoma, mutation, hereditary, whole genome sequencing, whole exome sequencing, methylation.
Background
Ovarian carcinoma (OC) is the most lethal gynecological malignancy worldwide. In the United States, OC is the fifth leading cause of cancer death in women, with estimated 14,240 deaths in 2016 [1]. Most OCs are high grade serous ovarian carcinomas (HGSOC). They are commonly diagnosed at an advanced stage with peritoneal dissemination and massive ascites. The 5-year survival rate in patients with advanced OC remains approximately 30% despite current standard combined therapy of debulking surgery and neoadjuvant chemotherapy of paclitaxel and carboplatin.
Our recent knowledge of molecular biology suggests that OCs can be divided into two groups, type I and type II [2]. Type I OC encompasses slow growing tumors and harbors mutations in KRAS, BRAF, PTEN, and CTNNB1, etc. Type II OC is clinically aggressive and pathologically high grade. They are characterized by TP53 mutations. Nearly one half of type II OCs have BRCA1/BRCA2 mutations, predisposing a considerable part of these women to hereditary breast and ovarian cancer (HBOC) [3]. Additional susceptibility genes in hereditary ovarian carcinomas are known to alter the homologous recombination (HR) /DNA repair pathway [3-5]. A recent surge in genomics/epigenomics studies has indicated that OC represents a genetically heterogeneous and complex group of diseases and does not conceive as a single entity [3-6]. The genetic heterogeneity may reflect the exerted clonal selections in the progression of OCs, such as preserving cancer cells with chemoresistance and metastatic capacity. These findings indicate the urgent requirement of personalized regimen or target therapy in treating OCs. They provide useful biomarkers for the assessment of diagnosis, detection, intervention, and prognosis in OCs. This review will give a glimpse on the genomic advances in OC. Particular interest will be focused on the translating insight of these genomic alterations into clinical and pathological practice [Table 1].
Summary of selected genetic alterations and clinical significance in ovarian carcinomas.
| Catalogues | Major genetic alterations | Clinical implications | Ref(s). |
|---|---|---|---|
| Detection/diagnosis | |||
| Germline mutations in BRCA1 or BRCA2 | Increased lifetime risk of developing OC; routine surveillance for early OC; indications for risk-reducing salpino-oorphorectomy | 9, 10 | |
| Germline mutations in mismatch repair genes | A 8%-10% risk of OC (Lynch syndrome); routine clinical surveillance for early OC | 13 | |
| Novel germline mutations in BARD1, BRIP1, PALB2, RAD50, RAD51C, RAD51D, TP53, ASXL1, MAP3K1, and SETD2, etc | Conferred a subset of familial OCs with high and moderate penetrance or a moderate OC susceptibility that may warrant their use in routine clinical genetic testing | 15-19 | |
| An integrated analysis of germline and somatic exome variants in OC | The candidate variants and genes have important implications for OC susceptibility and the development of screening strategies. | 20 | |
| Germline mutation in SMARCA4 (BRG1) | Improvements in genetic counseling and early detection for SSCHOT | 32-34 | |
| Mutations/loss of expression in SMARCA4 (BRG1) | Essential for precision diagnosis and potential novel treatment for SSCHOT | 35-37 | |
| Mutations of PIK3CA, RB1, and MED1 in plasma of OC patients following therapy | Applicable to monitor OC patients with high systemic tumor burden, metastasis and therapy response | 61 | |
| Risk assessment | |||
| rs7651446(3q25), rs9303542 (17q21), rs11782652 (8q21), rs1243180 (10p12), rs757210 (17q12) | Predicting OC risks | 41 | |
| rs8170 and rs2363956 at 19p13.11 | Predicting survival and genome-wide serous OC risks | 42 | |
| rs2072590 (2q31), rs2665390 (3q25), rs10088218 (8q24), rs9303542 (17q21) | Predicting OC risks | 43 | |
| rs3814113 (9p22.2) | OC risks, strongest for serous OC risks | 44 | |
| rs752590 (2q13), rs711830 (2q31.1), rs688187 (19q13.2) | Risk associations with mucinous OC | 45 | |
| Mutations in BRIP1 (c.2040_2041insTT, c.1702_1703del) | An increase in OC risks | 46 | |
| rs3814113 (9p22.2) | A reduced OC risk in BRCA1/BRCA2 mutation carriers | 49 | |
| Chemotherapy response/prognosis evaluation | |||
| Germline/somatic mutations in BRCA1, BRCA2 and other genes in the HR pathway | 1) Predictive of platinum sensitivity and longer survival in women with HGSOC; 2) Benefit from PARP inhibitors. | 3, 29-31 | |
| rs7874043 in TTC39B | The minor allele is strongly associated with PFS in patients with serous carcinoma following first-line chemotherapy. | 50 | |
| rs4910232(11p15.3), rs2549714(16q23), and rs6674079 (1q22) | The rare alleles were significantly associated with poorer outcomes in OC patients who underwent first-line treatment of cytoreductive surgery and chemotherapy. | 51 | |
| rs1649942 | Associated with PFS and OS in OC patients with carboplatin-based chemotherapy | 52 | |
| Reactivation of HR genes in platinum-resistant versus primary OCs; Increased platinum score of 13 CNAs in recurrent tumors | Treatment options should be tailored to the changing genetic profiles. All primary platinum-sensitive HGSOCs are qualified for second-line PARP inhibitor treatment. | 56 | |
| Clonal escape in chemotherapy; Novel mutations in the Golgi and ECM pathways | Target therapy towards the persistent mutations may be effective for tumor relapse while novel mutations may offer new therapeutic targets for recurrent tumors. | 57 | |
| Mutations from 8 members of the ADAMTS family | Helpful molecular markers for predicting chemotherapy response and prognosis in OC. | 58 | |
| Gains on 1q, 5q14~q23, and 13q21~q32, and losses of 8p and 9q | Clinical carboplatin resistance | 62 | |
| Gains on 1q25.2 and 1q32.2 | Clinical carboplatin resistance | 63 | |
| Loss on 13q32.1 and 8p21.1 | Predictive markers of chemoresistant serous carcinoma | 64 | |
| Gain in 3q26.2, and losses in 6q11.2-12, 9p22.3, 9p22.2-22.1, 9p22.1-21.3, Xp22.2-22.12, Xp22.11-11.3, and Xp11.23-11.1. | Potential predictive markers of chemotherapy resistance | 65 | |
| Gains in 9p13.2-13.1, 9q21.2-21.32, 9q21.33, 9q22.2-22.31, 9q22.32-22.33 and 9q33.1-34.11 | Potential predictive markers of docetaxel/carboplatin resistance | 66 | |
| Losses of 4p, 4q31.1- qter, 5q12-q22, 8p, 16q, and X | Poor survival in stage III OC | 68 | |
| CCNE1 amplification | Poor prognosis in postoperative OCs | 4, 69 | |
| High-level amplifications at 8q24, loss of 5q | Favorable prognosis for serous OC | 70 | |
| Gain in 5p or gain in 1p and loss in 5q | A higher or significantly decrease risk of recurrence | 72 | |
| Two distinct hierarchical clusters of CNA | Patients from cluster-1 had a significantly shorter median PFS than those from cluster-2. | 73 | |
| Met amplification in ovarian clear cell carcinoma | Worse survival | 74 | |
| Target therapy/individualized therapy | |||
| Various molecular subtypes of OC signature associated with survival | They provide an opportunity to improve OC outcomes through subtype-stratified care. | 3 | |
| Few point mutations in low grade serous carcinomas and borderline tumors | Target therapeutic agents against BRAF and KRAS might be particularly effective for the recurrent inoperative cases. | 53,54 | |
| Recurrent mutations in ELF3, RNF43, GNAS, ERBB3 and KLF5 in mucinous OC | Potential novel targeted therapy for some high grade mucinous carcinomas | 55 | |
| The heterogeneity in the genome of HGSOC under the selective pressure of chemotherapy | Overcoming resistance to conventional chemotherapy will require a diversity of approaches, such as use of new inhibitors MDR1 and PARP. | 4 | |
| PPM1D amplification | A potential therapeutic target for a subgroup of ovarian clear cell carcinomas | 75 | |
| HER2 amplification | A potential therapeutic target | 76-78 | |
Abbreviations: OC=ovarian carcinoma; PFS= poor progression-free survival; HR= homologue recombinant; OS=overall survival; CNA=copy number aberration; HGSOC= high grade serous ovarian carcinoma.
Germline Mutations in Hereditary Ovarian Carcinoma
Approximately 15% to 20% of OCs occur in a familial context with a highly penetrant, autosomal dominant genetic predisposition. These hereditary OCs have three major types including hereditary breast/OC (HBOC), site-specific OC, and Lynch syndrome II (hereditary non-polyposis colorectal cancer, HNPCC). The modified strategy for surveillance and clinical management of these hereditary OCs were listed in Figure 1. The detection of germline mutations in susceptibility genes remains the backbone in this protocol.
The modified strategy for surveillance and clinical management of hereditary ovarian cancers. Abbreviations: HBOC, hereditary breast/ovarian cancer; FA, Fanconi anemia; RRSO, risk-reducing salpino-oorphorectomy
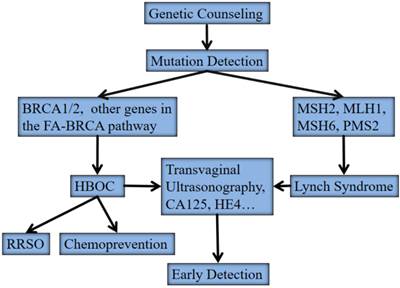
HBOC is predominantly associated with germline mutations in two major susceptibility genes, BRCA1 and BRCA2 [7, 8]. Site-specific OC is believed to be an “ovarian-specific” variant of HBOC since no other susceptibility genes other than BRCA1 and BRCA2 have been identified yet. Women with a germline mutation in either BRCA1 or BRCA2 have a 45%-60% or 27% lifetime risk of developing OC by the age of 70 years, respectively [9, 10]. OC patients without familial history may also have germline BRCA1/BRCA2 mutations [3-5, 11]. A recent randomized controlled trial showed that population-based genetic testing on BRCA1/BRCA2 mutations in Ashkenazi Jews identified additional 56% carriers compared with the family history based testing [12]. Women with Lynch syndrome predispose to colorectal carcinoma, endometrial carcinoma, and OC, etc. The cumulative risk of OC is estimated to be 8%-10%. Germline mutations in mismatch repair genes, predominantly in MSH2, MLH1, MSH6 and PMS2, account for women with Lynch syndrome. These mutations have a much smaller contribution to hereditary OC than those of BRCA1/2. Germline mutation testing of mismatch repair genes can be considered directly for families that meet the Amsterdam criteria or tumors, and women with OC that harbor microsatellite instability with high frequency (MSI-H) or negative immunostaining for mismatch repair gene products in tumor tissues [12, 13]. A genome-wide array comparative genomic hybridization (aCGH) analysis showed that HBOC and Lynch syndrome associated OC had different genetic profiles and represented distinct genetic pathways [14]. Losses of 4q34, 13q12-q32 and 19p13 were overrepresented in the HBOC whereas gains on chromosomes 17 and 19 characterized the Lynch syndrome tumors.
Owing to the rapid development in DNA sequencing technology, a substantial number of novel germline mutations have also been identified in familial OCs or patients with early-onset OCs, which had no mutations in BRCA1/BRCA2 and mismatch repair genes. The reported genes included BARD1, BRIP1, CHEK2, MRE11A, NBN, PALB2, RAD50, RAD51C, RAD51D, TP53, NF1, MAP3K4, CDKN2B, and MLL3, etc [3-5, 15-19]. RAD51C and RAD51D are mutated in 1.5%-4% and 0.9% HBOC families with high and moderate penetrance, respectively [15, 16]. A recent case control study [19] in a large population found that the occurrence of RAD51C (0.41%) and RAD51D (0.35%) germline mutation was higher than that of RAD51B (0.06%). RAD51C and RAD51D mutations conferred a moderate OC susceptibility that may warrant their use alongside BRCA1 and BRCA2 in routine clinical genetic testing. PALB2 mutations have been found in 33 of 972 families (3.4%) with high risk of breast cancer, and 18 of these 33 families (55%) had a family member with OC [17]. A study of 1915 OC patients found that 347 (18%) carried pathogenic germline mutations in genes associated with OC risk, including PALB2, BARD1, BRIP1, RAD51C, RAD51D, BRCA1, BRCA2, MSH2, MLH1, PMS2, and MSH6[5]. Most mutations occur in BRCA1 (n=182) and BRCA2 (n=98). A whole exome sequencing (WES) study on 429 OCs identified that germline truncations and large deletions across Fanconi pathway genes occurred in 20% of cases [20]. The novel OC susceptibility genes included ASXL1, MAP3K1, and SETD2, etc. The targeted capture and massively parallel sequencing may simultaneously detect all these mutations with high coverage, sensitivity, and cost efficiency. This technique has been successfully found that 24% of 360 women with ovarian, fallopian tube, and peritoneal carcinoma carried germline loss-of-function mutations including 18% in BRCA1 or BRCA2, and 6% in BARD1, BRIP1, CHEK2, MRE11A, MSH6, NBN, PALB2, RAD50, RAD51C, or TP53[18]. These germline mutations will lead to significant improvements in genetic counseling and early detections of familiar OCs [Table 1].
The mutation spectrum in the susceptible genes is broad including small deletions, insertions, point mutations, and gene rearrangements that typically lead to prematurely truncated or non-functional protein products. Genetic tests on these germline mutations is critical for OC screening, but they were limited by the laborious and expensive traditional sequencing-based approach due to the large size, the wide distribution of the mutations, the absence of hotspots, and the lower prevalence of mutations in these genes. Genetic counseling has to be provided before genetic tests, such as the family cancer history for HBOC and the Amsterdam criteria for Lynch syndrome. The strong associations between morphology and genotype in OCs are also helpful in the identification of patients for genetic tests. Both BRCA1- and BRCA2- associated HGSOC had more frequent Solid, pseudoEndometrioid, and Transitional cell carcinoma-like morphology (SET features) [21]. OCs in Lynch syndrome are invariably associated the histotypes of endometrioid (in pure or mixed form) carcinoma and clear cell carcinoma [22, 23].
There are two main clinical options for germline mutation carriers: increased surveillance for the detection of stage I OC, or risk-reducing salpino-oophorectomy (RRSO). Currently transvaginal sonography and serum CA125 remains the mainstay for OC detection, but lack sensitivity and specificity for early stage OC. In addition to CA125, other serum biomarkers such as human epididymis protein 4 (HE4) may improve the screening efficiency for early detection of OC to a certain degree [24]. RRSO has increasingly been shown to be an effective strategy for reducing cancer risk by up to 96%, but it has the disadvantages of hormonal deprivation and potential peritoneal carcinoma. Occult carcinomas are frequently found in the fallopian tube (57%-100%) at the time of RRSO [25]. Tubal epitheliums in RRSO samples have increased proliferating index, the likelihood of p53 mutation, and progression into serous tubal intraepithelial carcinoma [26]. These findings support that tumor cells from the fallopian tube are one of the major origin of OC [2]. Therefore, the removal of both fallopian tubes can theoretically replace RRSO to reduce the OC risk although more compelling evidence should be provided in the future. Oral contraceptive provides an alternative for chemoprevention of OC in women with high OC risks who do not accept RRSO.
Several investigations have indicated that gene mutations may have therapeutic significance [Table 1]. BRCA1- or BRCA2-functionally deficient cell lines are more sensitive to platinum [27, 28]. BRCA1/BRCA2 mutations are predictive of platinum sensitivity and longer survival in HGSOC patients [29]. A recent study also showed that OC patients with a BRCA2 mutation had longer progression-free survival and overall survival compared with those without mutations [30]. For breast and OC patients with pathogenic BRCA mutations, PARP inhibitors might be one of the most promising targeted substances since the efficacy of this monotherapy reached a response rate of approximately 40% over an average of six months. A phase II clinical trial of Olaparib (AZD2281), an oral PARP inhibitor, showed an objective responsive rate of 33% (14/42) in OCs who received multiple prior treatments. Most of these responsive cases (11/14) had a germline BRCA1/BRCA2 mutation [31].
Small cell carcinoma of the ovary, hypercalcemic type (SCCOHT) is the most common undifferentiated malignancy in women under 40 years. The diagnosis of SCCOHT is challenging even for the expert pathologists due to its overlapping morphological features with juvenile granulosa cell tumor and other malignancies in the ovary. Recent advance on genomic alterations has substantially changed our recognition on this rare ovarian cancer. Familial cases have been suggested to show an autosomal dominant transmission. A WES study on six individuals from three families discovered germline mutations in SMARCA4 (BRG1) [32]. Germline or somatic mutations were frequently found and correlated with loss of protein expression in sporadic SCCOHT cases [33, 34]. Subsequent immumohistochemical studies have indicated that loss of SMARCA4 and SMARCA2 protein expression is sensitive and specific for SCCOHT, that make them valuable in the differential diagnosis of SCCOHT [35-37]. These findings suggest that SCCOHT is a new entity of the atypical teratoma/rhabdoid tumor (AT/RT) family. Genetic testing and consideration of the novel chemotherapeutic regimens used to AT/RT will help to improve the prognosis of SCCOHT.
Genetic Variants and Somatic Mutations in OC
It is well established that two distinct pathways of pathogenesis exist in OC, type I and type II [2]. Type I OC encompasses histotypes of low-grade serous carcinoma, endometrioid carcinoma, mucinous carcinoma and some clear cell carcinomas while type II contains HGSOC and other poorly differentiated carcinomas. Both types of OC are known to harbor mutations in different genes [2, 3]. ARIDA1 mutations are present in 46% and 30% of endometrioid carcinoma and clear cell carcinoma, respectively [38]. Moreover, consistent ARIDA1and PIK3CA mutations in ovarian clear cell carcinoma and concurrent endometriosis support that benign endometriotic lesions are clonally related to clear cell carcinomas [39]. The next-generation sequencing technology and bioinformatics approaches enable new high-throughput genomic screening, such as genome-wide association studies (GWAS), whole genome sequencing (WGS), and whole exome sequencing (WES), RNA-sequencing and other targeted methods. So far, these methods have been widely applied in cancer research. The Cancer Genome Atlas (TCGA) has been developed to generate and analyze molecular profiles at the DNA, mRNA, protein and epigenetic levels for various cancers and their subtypes including OC [40]. The surge of deep sequencing uncovered a substantial number of cancer susceptibility genetic variants and somatic mutations in OC tissues. These genetic data give us more significant insights on the pathogenesis and clinical intervention on OCs.
Genome-wide association studies (GWAS)
GWAS is designed to compare genotype frequencies of the common variants throughout the genome between cases and controls. GWAS studies identified many genetic variants, most single nucleotide polymorphisms (SNP), that show strong associations with OC risks [Table 1, 41-45]. A study on 457 Icelanders using SNP chips found a rare (0.41% allelic frequency) frameshift mutation, c.2040_2041insTT, in the BRIP1 (FANCJ) gene that confers an increase in OC risk with a moderate penetrance [46]. Recent investigations have been focused on the association between OC risks and SNPs in susceptible chromosomal regions (such as 2q31, 3q25, 5p15, 8q21, 8q24, 10p12, 17q12, 17q21.31, and 19p13) or in a specific functional group (such as epithelial-mesenchymal transition genes) in a large sample size [47, 48]. However, these variants only showed weak effects on OC risk with the odds ratios around 0.8 or 1.2 in general. In the evaluation of cancer risk, these low-penetrant loci have little clinical utility in the general population, but may be useful for counseling women with BRCA1/BRCA2 mutations [Fig. 1]. BRCA1 mutation carriers with the TT genotype at SNP rs3814113 (at the 9p22.2 locus) had a predictive OC risk to age at 80 years of 48% while those with the CC genotype had a risk of 33% [49].
GWAS is also helpful to identify genetic predictors of treatment outcome in OC patients [Table 1, 50-52]. A recent GWAS study on 1244 patients with ovarian serous carcinoma given carboplatin- and paclitaxel-based chemotherapy, identified that the minor alleles of two SNPs (rs7874043 and rs72700653) were associated with progression-free survival [50]. Functional analysis showed that both SNPs act as a transcriptional enhancer on PSIP1 and CCDC171 promoters. PSIP1 is a known oncogene that controls a caspase-independent lysosomal cell death pathway. Successful inhibition of PSIP1 may provide a novel approach to target OC. A GWAS study from Ovarian Cancer Association Consortium identified five SNPs significantly associated with poorer outcomes in OC patients underwent first-line treatment of cytoreductive surgery and chemotherapy [51]. Three of them, rs4910232 (11p15.3), rs2549714 (16q23), and rs6674079 (1q22), were located in long noncoding RNAs (lncRNAs) RP11-179A10.1, RP11-314O13.1, and RP11-284F21.8, respectively. Huang RS et al. [52] also found that SNP rs1649942 was significantly associated with decreased progression free survival and overall survival in OC patients received at least four cycles of carboplatin-based chemotherapy. The potential mechanism of this SNP may be through its association with the expression of 18 target genes, ten of which are correlated with carboplatin sensitivity in lymphoblastoid cell lines. Taken together, genetic variations in chemotherapy response appears to be partly responsible for the different clinical outcomes in OC patients following chemotherapy.
Whole-exome sequencing (WES)
WES allowed the discovery of low-frequency variants in individuals with familial, highly penetrant diseases, or those with certain complex quantitative traits by using exome array-captured sequencing. In 2011, TCGA Network provided a large-scale integrative view of mRNA expression, miRNA expression, promoter methylation, DNA copy number and exome sequencing in HGSOCs [3]. That study found frequent TP53 mutation (96%), infrequent but statistically recurrent mutations in other genes including BRCA1, CSMD3, NF1, CDK12, FAT3, GABRA6, GRCA2 and RB1, and additionally 113 significant focal DNA copy number aberrations (CNAs) and promoter methylations of 168 genes. The integrative analysis of various genetic alterations generated four transcriptional subtypes, three microRNA subtypes, four promoter methylation subtypes and a 193-gene transcriptional signature, which were correlated with overall survival in OC. A later WES study with 429 OC cases from TCGA [20] observed novel significantly mutated genes including NRAS, NF2, ATR, ATP, and APC, etc.. Moreover, the combined analysis of germline-somatic changes revealed several pathways with significant enrichment of variants, such as Fanconi/DNA repair pathway, MAPK pathway and histone methyltransferases in HGSOCs[20]. These discoveries from the TCGA project provided molecular targets and related pathways, for potential therapeutic interventions on OC in the future.
Two WES studies with a small sample size demonstrated that point mutations were very few in low grade serous carcinoma (70 somatic mutations in 64 genes in 7 cases) and serous borderline tumors of the ovary (16 somatic mutations in 2 cases) [53, 54]. These data indicates that low grade serous carcinoma and serous borderline tumor do not require many mutations to achieve malignancy or malignant potential. Therefore, target therapeutic agents, such as those against BRAF and KRAS, might be particularly effective for the recurrent inoperative cases. Ryland GL, e al. [55] performed WES on 24 mucinous tumors including 5 benign cystadenomas, 8 borderline tumors, and 11 carcinomas. They found a high percentage of TP53 mutations and recurrent mutations in RNF43, ELF3, GNAS, ERBB3 and KLF5. These diversified mutations suggest genetic heterogeneity in the pathogenesis of ovarian mucinous carcinoma and confer to a novel targeted therapy for some high grade mucinous carcinomas considering of the limited success in treating advanced mucinous carcinomas.
Most HGSOCs benefit from platinum- and taxol-based chemotherapy initially, but progressive chemoresistance will occur subsequently and result in tumor recurrence and metastasis eventually. Little is known about the underlying mechanisms of chemoresistance. Lanbrechts S et al. [56] investigated on the WES and SNP profiling in 31 before and after first-line chemotherapy-paired OC tissues. They found frequent HR-deficiency (loss-of-heterozygosity and mutation in HR genes) in primary tumors, relapse tumors and tumors resistant to second-line platinum, implicating the persistent qualification for second-line PARP inhibitor treatment in recurrent and chemoresistant tumors. A platinum score of 13 regions with copy number alterations including MECOM, CCNE1 and ERBB2 increased in recurrent tumors. The copy number alterations contribute to the acquired resistance after a single line of platinum therapy. About 58% (623/1074) of somatic mutations were identical between primary and recurrent tumors. Consistent with this finding, another WES study showed that most mutations in primary tumors persisted in tumor cells from ascites at three time points (primary, first recurrence and second recurrence) [57]. Both studies suggested that most clones could escape current chemotherapy treatments. In addition, novel mutations in the Golgi and ECM pathways were also found in recurrent tumors [57]. Both WES studies suggested the genome evolution in the progression of HGSOC from primary to recurrent or chemoresistant diseases. Biopsies at relapse are prerequisite for personalized genomic evaluation to tailor treatment. The persistent mutations in the recurrent tumors indicate that improved target therapy towards these mutations may be ultimately effective for the prevention and intervention of tumor relapse while novel mutations may offer new therapeutic targets for recurrent tumors. Recently, Liu, et al. [58] analyzed WES data from 512 OC patients and found that mutations from 8 members of the ADAMTS family were significantly associated with a higher chemotherapy sensitivity, a longer platinum-free duration, a better overall survival and progression-free survival than ADAMTS wild-type cases, even after adjustment by BRCA1/BRCA2 mutations, surgical stage, residual tumor, and patient age. ADAMTS mutations appeared to be helpful molecular markers for predicting chemotherapy response in OC patients.
The necrotic or apoptotic cells can release DNA into the bloodstream [59]. The term, cell-free DNA (cfDNA), is referred to the dead cell-derived DNA that is selectively isolated from the plasma. In cancer patients, such cfDNA from tumor cells is called circulating tumor DNA (ctDNA) [60].The majority of plasma cfDNA in cancer patients come from cancer cells. ctDNA is a reflection of genomic alterations in solid cancers and offer a unique non-invasive “liquid biopsy” for serially monitoring cancers. Theoretically, all genomic alterations in cancer tissues, such as gene mutations, microsatellite instability, gene amplifications, and promoter hypermethylation, etc., can be detected in ctDNA. The potential application of high-throughput sequencing, such as WES, in plasma ctDNA constitutes a new paradigm for the detection and monitoring of various cancers. Murtaza M [61] undertook exome sequencing to track genomic evolution of metastatic cancers in response to therapy in serial plasma samples from six patients with advanced breast, ovarian and lung cancers over 1-2 years. They found a functional mutation in PIK3CA following treatment with paclitaxel, a truncating mutation in RB1 following treatment with cisplatin, a truncating mutation in MED1 following treatment with tamoxifen and trastuzumab, and following subsequent treatment with lapatinib, a splicing mutation in GAS6 in the same patient, and a resistance-conferring mutation in EGFR following treatment with gefitinib. This non-invasive approach characterized plasma cancer exomes is applicable to monitor OC patients with high systemic tumor burden, metastasis and therapy response.
Whole-genome sequencing (WGS)
WGS is technically propelled by next generation sequencing. It can detect rare and novel genetic variations, and may become the most promising approach in precision (personalized) medicine with the decreasing sequencing cost and expanding bioinformatics resources in the future. The first WGS analysis of tumor and germline DNA samples from 92 patients with HGSOC was reported recently [4]. The tumors included primary refractory, resistant, sensitive and matched acquired resistant disease. This study confirmed previous findings including prevalent TP53 mutations, infrequent somatic mutations in other driver genes, deficiency in HR pathway (50% cases including germline and somatic mutations of related genes and BRCA1 methylation), and CCNE1 gain/amplification (19% cases). CCNE1 gain/amplification, which was largely exclusive of HR pathway dysfunction, was common in primary resistant and refractory tumors. Tumors with BRCA1 mutations had more structural variants and genome-wide coding small mutations compared to HR-intact samples. They had a favorable response to treatment. Disruption of transcription unit (gene breakage), and commonly inactivated tumor suppressor genes NF1, RB1, PTEN, RAD51B, FOXO1, and BCL2L11, also contributed to the acquired chemoresistance. Other molecular events associated with acquired chemoresistance included reversions of germline mutations in BRCA1/BRCA2 in individual patients, loss of promotor methylation in BRCA1, transition of C2/immune molecular subtype to C1 subtype, and recurrent promoter fusions involving ABCB1, resulting in overexpression of the drug efflux pump MDR1. These findings clearly demonstrate the heterogeneity and adaptability in the genome of HGSOC with the pressure of chemotherapy. Personalized genomic evaluation should be urgently prompted for patients with recurrent HGSOC. A diversity of novel precision solutions should critically be encouraged to overcome the resistance to current chemotherapy.
DNA CNAs in OC
Genomic instability is a hallmark of cancer, and genomic aberrations in the form of DNA copy number alterations are important in tumorigenesis of OC [4]. HGSOC is genetically unstable by showing highly rearranged karyotypes with numerical and structural chromosome aberrations. Conventional comparative genome hybridization (CGH) and aCGH have been used to scan genome-wide genetic alterations. Knowledge about the acquired genomic CNAs of OCs remains unsatisfactory in general because of small sample sizes in most studies and the insufficient genomic mapping resolution (5~10 Mb by CGH and 0.2~2Mb by aCGH) to delineate precise patterns of gene amplifications and losses. The identification of these chromosomal aberrations and the genes rearranged behind the aberrations may ultimately have a clinical application in predicting chemotherapy response, prognosis, and recurrence risks, etc.
Resistance to chemotherapy in OC is not well understood yet. Specific CNAs are potential biomarkers for chemoresistance in OC [Table 1] [56, 62-66]. Osterberg L, et al.[62] found that gains of 1q, 5q14~q23, and 13q21~q32, and losses of 8p and 9q were clinically associated with carboplatin resistance in 63 early-stage OCs. Their later study using high solution aCGH further specified the genomic regions of carboplatin resistance [63]. The regions with copy number changes might contain candidate genes involved in carboplatin resistance, which are essential for improving the prognosis and treatment of women with OC. The candidate genes in these regions include RASAL2, GPR52, RGL1, FAIM3, RAP2A, and EXTL3, etc [63, 64]. The extensive non-linear divergence of chemo-sensitive and resistant sub-clones on cell line series before and after platinum resistance supported the presence of a profound intra-tumor genetic heterogeneity in genomically unstable HGSOCs [67]. Identifying different genetic sub-clones has important clinical implications for developing novel therapy targeting a common or specific genetic alteration, and personalized, combined therapies targeting different subpopulations.
Some CNAs may be associated with tumor relapse and survival in OCs [Table 1, 68-75].Amplifications of some candidate genes, such as CCNE1 and PPM1D, were potential indicators for poor outcomes in ovarian serous carcinomas and clear cell carcinomas [4, 69, 75]. These genes are possible therapeutic targets for a specific subgroup of OCs.
Trastuzumab, a humanized monoclonal antibody targeting the human epidermal growth factor receptor 2 (HER2) has impacted on the treatment of breast cancer. Effort has been taken to investigate HER2 amplification and related target therapy in OCs. The observed rates of HER2 overexpression and/or amplification in OCs ranged from 2% to 66%. Tuefferd et al. [76] found that the rate of HER2 overexpression was 12.8% (42/320) in patients with advanced ovarian or primary peritoneal carcinomas. HER2 overexpression did not demonstrate a prognostic value in this study. In contrast, a tissue microarray study on 401 serous OCs showed that HER2 amplification was detected in 7% of the cancers, and was associated with a poor prognosis, a shorter disease-free survival and overall survival [77]. HER2 amplification was observed in about 19% ovarian mucinous carcinomas, but was not indicative for the assessment of overall survival [78]. Whether HER2 amplification is of prognostic significance is yet to be determined, HER2-directed therapies remain a potential target considering the dismal prognosis of patients in late stages of OCs.
Epigenetic alterations in OC
Epigenetic modifications, which unlike genetic alterations, does not alter the primary DNA sequence, but influence the transcriptional process by DNA methylation, histone modifications and dysregulations of nucleosomes. DNA hypermethylation typically occurs in gene-associated CpG islands and hyomethylation in repetitive genomic DNA. Core histone proteins (H2A,-2B, -3 and -4) can be modified by methylation, acetylation, and phosphorylation, etc. Many tumor suppressor genes can by silenced by DNA hypermethylation. The well characterized genes include BRCA1, hMLH1, MGMT, HOXA9, RASSF1A, OPCML and CCBE1, etc [79, 80]. Emerging data have implicated that the inactivation of tumor suppressor genes by epigenetic alterations should be regarded as “the second hit” in the Knudson's two-hit hypothesis in cancer biology. Novel epigenetic therapies, such as DNA methyltransferase inhibitors and histone deacetylase inhibitors, have become potential therapeutic options for various cancers including OC.
Epigenetic biomarkers, particularly DNA methylations, have potential clinical utility for detection/diagnosis, chemotherapy response and prognosis in OC [Table 2, 81-95]. Epigenetic regulation of Wnt and Akt/mTOR pathways may serve as biomarkers for prognosis and/or treatment response in OC [91, 92, 96, 97]. Dai et al. [92] examined promoter methylation at 302 loci in a panel of 137 Wnt pathway genes in 111 screening cases and 61 validation cases. They demonstrated that methylations at 7 loci (FZD4, DVL1, NFATC3, ROCK1, LRP5, AXIN1, and NKD1) were associated with poor progression-free survival. Furthermore, hypermethylations of DVL1 and NFATC3 were involved in poor response to platinum chemotherapy. Patients with progressive or stable disease had increased methylation than those with partial or complete response. The authors recently analyzed the progression free survival-related DNA methylation at the CpG island promoter of genes in other pathways including Akt/mTOR, p53, redox, and HR DNA repair [97]. They identified that methylations of promoter regions of VEGFB, VEGFA, HDAC11, FANCA, E2F1, GPX4, PRDX2, RAD54L and RECQL4 were associated with increased hazard of disease progression, independently from conventional clinical prognostic factors both in the screening cohort (n=150) and the TCGA validation cohort (n=311). Moreover, methylations at VEGFB and GPX4 were more likely to have poor response to chemotherapy. In combination with methylation profile in the previous Wnt pathway [92], they constructed a diagnostic model including the methylations of NKD1, VEGFB and PRDX2, from which, methylation index was calculated to identify two distinct prognostic groups. The patients with increased methylation index were more likely to have poor response to chemotherapy.
Selected genes with promotor hypermethylation and their clinical correlations in ovarian carcinomas.
| Genes | Clinical correlations | Ref(s). |
|---|---|---|
| RASSF1A | Detection of OC | 81, 82 |
| BRCA1 | Detection of OC; poor prognosis; improved chemotherapy response | 81,83,84 |
| APC | Serum/ascites diagnosis of OC | 81,83 |
| MGMT | Detection of OC; improved chemotherapy response | 83,85 |
| hMLH1 | Poor prognosis; improved chemotherapy response | 86-88 |
| HOXA9 | Detection of OC | 89 |
| OPCML | Detection of OC | 90 |
| SFRP-1, -2, -4, -5 | Detection of OC; Cancer recurrence; Poor prognosis | 91 |
| FZD4, DVL1, NFATC3, ROCK1, LRP5, AXIN1, and NKD1 | Poor prognosis | 92 |
| FBXO32 | Poor prognosis | 93 |
| HOXA11 | Poor clinical outcome. | 94 |
| FANCF | Cisplatin resistance | 95 |
Abbreviations: OC=ovarian carcinoma.
Some recent studies have focused on genome-wide identification of methylated biomarkers in OCs. The methylated DNA immunoprecipitation microarray (MeDIP-chip) identified 367 CpG islands specifically methylated in OC versus normal ovaries [98]. A publication from the TCGA data found 168 epigenetically silenced genes [3]. AMT, CCL21, and SPARCL1 showed promtor hypermethylation in most cancers and may serve as biomarkers for the presence of OC. Consensus clustering of these methylations across the tumors generated four subtypes that were associated with prognostic differences. Other genome-wide studies in OC have produced methylation signatures associated with progression-free survival [99, 100], and response to platinum-based chemotherapy [101]. Genome-wide methylation analyses can reliably identify markers of potentially prognostic value. Häfner et al. [102] detected 220 differentially methylated regions in tumor tissue of patients with short vs. long progression-free survival (106 hypo- and 114 hypermethylated regions) by genome-wide array analyses. Validation experiments proved that patients harboring methylation at the CpG island of RUNX3/CAMK2N1 had a significantly lower progression free survival. These potential biomarkers identified by genome screens merits further investigation in large cohorts with well clinical documentation.
Non-invasive assay has been successfully applied to detect specific methylation profiles in the blood or ascites from OC patients in several studies [81, 82, 90, 103]. Liggett TE, et al. [82] found that different methylation patterns of three promoters (RASSF1A, CALCA, and EP300) in cell-free plasma DNA can discriminate between OC vs. healthy controls with a sensitivity of 90.0% and a specificity of 86.7%. Similarly, a combined analysis of methylations at five genes (BRCA1, HIC1,PAX5, PGR and THBS1) in patient plasma DNA generated 85% sensitivity and 61% specificity for cancer detection [103]. A recent study from China [90] showed that OPCML hypermethylation for serum samples was detected in 80.0% OC patients and not in healthy individuals. Tumor-specific hypermethylations in serum DNA appeared to be valuable biomarkers for the early detection of OC.
Conclusions and future perspectives
A rich resource of information has been generated from molecular and genetic studies on OCs. Identification of germline mutations of BRCA1/BRCA2 and other genes have substantially propelled the surveillance and prevention of hereditary OCs. Germline/somatic mutations of SMARCA4 are now essential for the precision diagnosis of SCCOHT, and provide potential novel therapy for this rare aggressive ovarian cancer. The application of new high-throughput genomic techniques has uncovered a substantial number of genetic susceptible variants, somatic mutations, copy number alterations and epigenetic modifications in OC. Theoretically, the tumor specific genetic alterations correlate with the presence of OC. As a collar, they are precious biomarkers with potential clinical utility for risk assessment, diagnosis/detection, prognosis and response to chemotherapy, and target/precision therapy in OC patients [Table 1; Table 2]. Genetic susceptible variants are important tools for molecular test in high-risk populations. Biomarkers from early staging OC cases are helpful for early cancer detection and that from recurrent tumors for monitoring systemic tumor burden, metastasis and therapy response. Fortunately, it is clear that some genetic changes, such as gene mutations and DNA methylations, have been detected by non-invasive methods in the blood and ascites from women with OC. Moreover, even detection of most genomic and epigenomic alterations in tumor tissue can provide valuable information for precision medicine and prognostic prediction at present. Finally, the presence of great heterogeneity and apparent chemotherapy-exerted clonal evolution in OC underscore the necessity of genomic evaluation for different tumor tissues, such as primary and metastasis/recurrence, or low and high grade, and adjust therapy accordingly.
Before translating these genomic and epigenomic alterations into clinical utility, several key points should be considered: 1) Large, well annotated prospective cohorts are required to further validate the clinical significance of the potential biomarkers. The performance of these biomarkers should be reproducible and consistent among various laboratories. 2) The biological samples for research should be obtained, stored and processed properly following the required protocols and standards. The related clinicopathological documentations should be clear and precise including longitudinal follow up and other details. 3) Novel non-invasive assays should be prompted for early detection, diagnosis, prognosis and therapy response. Cell free plasma/serum DNA provides a hopeful source for these molecular tests, but more compelling evidence should be accumulated to support the identical genetic/epigenetic patterns between samples from blood and tumor tissues. 4) For target/precision therapy, extensive molecular biological experiments should be performed to clarify the functions of potential targets, and more robust and stable models should be developed to identify molecular signatures with various clinical phenotypes. A combination of target therapy, epigenetic therapy and conventional therapy, will holds great promise for the treatment of OCs in the future.
Abbreviations
OC: ovarian carcinoma; HGSOC: high grade serous ovarian carcinomas; HBOC: hereditary breast/ovarian cancer; RRSO: risk-reducing salpino-oorphorectomy; GWAS: genome-wide association studies; WGS: whole genome sequencing; WES: whole exome sequencing; TCGA: The Cancer Genome Atlas; cfDNA: cell-free DNA; CGH: comparative genomic hybridization; aCGH: array comparative genomic hybridization; CNA: copy number aberration; SNP: single nucleotide polymorphism; HR: homologous recombination; SCCOHT: Small cell carcinoma of the ovary, hypercalcemic type
Acknowledgements
This work was supported by a natural science foundation of Zhejiang Province, PR China (No: Y211071).
Competing interests
The authors have declared that no competing interest exists.
References
1. Siegel R, Miller K, Jemal A. Cancer statistics, 2016. CA. Cancer J Clin. 2016;66:7-30
2. Kurman RJ, Shih IeM. The Dualistic Model of Ovarian Carcinogenesis: Revisited, Revised, and Expanded. Am J Pathol. 2016;186:733-747
3. Cancer Genome Atlas Research Network. Integrated genomic analyses of ovarian carcinoma. Nature. 2011;474:609-615
4. Patch AM, Christie EL, Etemadmoghadam D. et al. Whole-genome characterization of chemoresistant ovarian cancer. Nature. 2015;521:489-494
5. Norquist BM, Harrell MI, Brady MF. et al. Inherited Mutations in Women With Ovarian Carcinoma. JAMA Oncol. 2016;2:482-490
6. Vaughan S, Coward JI, Bast RC Jr. et al. Rethinking ovarian cancer: recommendations for improving outcomes. Nat Rev Cancer. 2011;11:719-725
7. Miki Y, Swensen J, Shattuck-Eidens D. et al. A strong candidate for the breast and ovarian cancer susceptibility gene BRCA1. Science. 1994;266:66-71
8. Wooster R, Neuhausen SL, Mangion J. et al. Localization of a breast cancer susceptibility gene, BRCA2, to chromosome 13q12-13. Science. 1994;265:2088-2090
9. Levy-Lahad E, Catane R, Eisenberg S. et al. Founder BRCA1 and BRCA2 mutations in Ashkenazi Jews in Israel: frequency and differential penetrance in ovarian cancer and in breast-ovarian cancer families. Am J Hum Genet. 1997;60:1059-1067
10. Moslehi R, Chu W, Karlan B. et al. BRCA1 and BRCA2 mutation analysis of 208 Ashkenazi Jewish women with ovarian cancer. Am J Hum Genet. 2000;66:1259-1272
11. Manchanda R, Loggenberg K, Sanderson S. et al. Population testing for cancer predisposing BRCA1/BRCA2 mutations in the Ashkenazi-Jewish community: a randomized controlled trial. J Natl Cancer Inst. 2014;107:379
12. Umar A, Boland CR, Terdiman JP. et al. Revised Bethesda Guidelines for hereditary nonpolyposis colorectal cancer (Lynch syndrome) and microsatellite instability. J Natl Cancer Inst. 2004;96:261-268
13. Walsh CS, Blum A, Walts A. et al. Lynch syndrome among gynecologic oncology patients meeting Bethesda guidelines for screening. Gynecol Oncol. 2010;116:516-521
14. Domanska K, Malander S, Staaf J. et al. Genetic profiles distinguish different types of hereditary ovarian cancer. Oncol Rep. 2010;24:885-895
15. Meindl A, Hellebrand H, Wiek C. et al. Germline mutations in breast and ovarian cancer pedigrees establish RAD51C as a human cancer susceptibility gene. Nat Genet. 2010;42:410-414
16. Loveday C, Turnbull C, Ramsay E. et al. Germline mutations in RAD51D confer susceptibility to ovarian cancer. Nat Genet. 2011;43:879-882
17. Casadei S, Norquist BM, Walsh T. et al. Contribution of inherited mutations in the BRCA2-interacting protein PALB2 to familial breast cancer. Cancer Res. 2011;71:2222-2229
18. Walsh T, Casadei S, Lee MK. et al. Mutations in 12 genes for inherited ovarian, fallopian tube, and peritoneal carcinoma identified by massively parallel sequencing. Proc Natl Acad Sci U S A. 2011;108:18032-18037
19. Song H, Dicks E, Ramus SJ. et al. Contribution of Germline Mutations in the RAD51B, RAD51C, and RAD51D Genes to Ovarian Cancer in the Population. J Clin Oncol. 2015;33:2901-2907
20. Kanchi KL, Johnson KJ, Lu C. et al. Integrated analysis of germline and somatic variants in ovarian cancer. Nat Commun. 2014;5:3156
21. Soslow RA, Han G, Park KJ. et al. Morphologic patterns associated with BRCA1 and BRCA2 genotype in ovarian carcinoma. Mod Pathol. 2012;25:625-636
22. Chui MH, Ryan P, Radigan J. et al. The histomorphology of Lynch syndrome-associated ovarian carcinomas: toward a subtype-specific screening strategy. Am J Surg Pathol. 2014;38:1173-1181
23. Chui MH, Gilks CB, Cooper K. et al. Identifying Lynch syndrome in patients with ovarian carcinoma: the significance of tumor subtype. Adv Anat Pathol. 2013;20:378-386
24. Escudero JM, Auge JM, Filella X. et al. Comparison of serum human epididymis protein 4 with cancer antigen 125 as a tumor marker in patients with malignant and nonmalignant diseases. Clin Chem. 2011;57:1534-1544
25. Powell CB, Chen LM, McLennan J. et al. Risk-reducing salpingo-oophorectomy (RRSO) in BRCA mutation carriers: experience with a consecutive series of 111 patients using a standardized surgical-pathological protocol. Int J Gynecol Cancer. 2011;21:846-851
26. Norquist BM, Garcia RL, Allison KH. et al. The molecular pathogenesis of hereditary ovarian carcinoma: alterations in the tubal epithelium of women with BRCA1 and BRCA2 mutations. Cancer. 2010;116:5261-5271
27. Samouëlian V, Maugard CM, Jolicoeur M. et al. Chemosensitivity and radiosensitivity profiles of four new human epithelial ovarian cancer cell lines exhibiting genetic alterations in BRCA2, TGFbeta-RII, KRAS2, TP53 and/or CDNK2A. Cancer Chemother Pharmacol. 2004;54:497-504
28. Husain A, He G, Venkatraman ES. et al. BRCA1 up-regulation is associated with repair-mediated resistance to cis-diamminedichloroplatinum (II). Cancer Res. 1998;58:1120-1123
29. Pennington KP, Walsh T, Harrell MI. et al. Germline and somatic mutations in homologous recombination genes predict platinum response and survival in ovarian, fallopian tube, and peritoneal carcinomas. Clin Cancer Res. 2014;20:764-775
30. Harter P, Johnson T, Berton-Rigaud D. et al. BRCA1/2 mutations associated with progression-free survival in ovarian cancer patients in the AGO-OVAR 16 study. Gynecol Oncol. 2016;140:443-449
31. Ledermann J, Harter P, Gourley C. et al. Olaparib maintenance therapy in patients with platinum-sensitive relapsed serous ovarian cancer: a preplanned retrospective analysis of outcomes by BRCA status in a randomised phase 2 trial. Lancet Oncol. 2014;15:852-861
32. Witkowski L, Carrot-Zhang J, Albrecht S. et al. Germline and somatic SMARCA4 mutations characterize small cell carcinoma of the ovary, hypercalcemic type. Nat Genet. 2014;46:438-443
33. Jelinic P, Mueller JJ, Olvera N. et al. Recurrent SMARCA4 mutations in small cell carcinoma of the ovary. Nat Genet. 2014;46:424-426
34. Ramos P, Karnezis AN, Craig DW. et al. Small cell carcinoma of the ovary, hypercalcemic type, displays frequent inactivating germline and somatic mutations in SMARCA4. Nat Genet. 2014;46:427-429
35. Conlon N, Silva A, Guerra E. et al. Loss of SMARCA4 Expression Is Both Sensitive and Specific for the Diagnosis of Small Cell Carcinoma of Ovary, Hypercalcemic Type. Am J Surg Pathol. 2016;40:395-403
36. Jelinic P, Schlappe BA, Conlon N. et al. Concomitant loss of SMARCA2 and SMARCA4 expression in small cell carcinoma of the ovary, hypercalcemic type. Mod Pathol. 2016;29:60-66
37. Karnezis AN, Wang Y, Ramos P. et al. Dual loss of the SWI/SNF complex ATPases SMARCA4/BRG1 and SMARCA2/BRM is highly sensitive and specific for small cell carcinoma of the ovary, hypercalcaemic type. J Pathol. 2016;238:389-400
38. Wiegand KC, Shah SP, Al-Agha OM. et al. ARID1A mutations in endometriosis-associated ovarian carcinomas. Engl J Med. 2010;363:1532-1543
39. Anglesio MS, Bashashati A, Wang YK. et al. Multifocal endometriotic lesions associated with cancer are clonal and carry a high mutation burden. J Pathol. 2015;236:201-209
40. Cancer Genome Atlas Research Network. The Cancer Genome Atlas Pan-Cancer analysis project. Nat Genet. 2013;45:1113-1120
41. Pharoah PD, Tsai YY, Ramus SJ. et al. GWAS meta-analysis and replication identifies three new susceptibility loci for ovarian cancer. Nat Genet. 2013;45:362-370 370e1-2
42. Bolton KL, Tyrer J, Song H. et al. Common variants at 19p13 are associated with susceptibility to ovarian cancer. Nat Genet. 2010;42:880-884
43. Goode EL, Chenevix-Trench G, Song H. et al. A genome-wide association study identifies susceptibility loci for ovarian cancer at 2q31 and 8q24. Nat Genet. 2010;42:874-879
44. Song H, Ramus SJ, Tyrer J. et al. A genome-wide association study identifies a new ovarian cancer susceptibility locus on 9p22.2. Nat Genet. 2009;41:996-1000
45. Kelemen LE, Lawrenson K, Tyrer J. et al. Genome-wide significant risk associations for mucinous ovarian carcinoma. Nat Genet. 2015;47:888-897
46. Rafnar T, Gudbjartsson DF, Sulem P. et al. Mutations in BRIP1 confer high risk of ovarian cancer. Nat Genet. 2011;43:1104-1107
47. Earp M, Winham SJ, Larson N. et al. A targeted genetic association study of epithelial ovarian cancer susceptibility. Oncotarget. 2016;7:7381-7389
48. Amankwah EK, Lin HY, Tyrer JP. et al. Epithelial-Mesenchymal Transition (EMT) Gene Variants and Epithelial Ovarian Cancer (EOC) Risk. Genet Epidemiol. 2015;39:689-697
49. Ramus SJ, Kartsonaki C, Gayther SA. et al. Genetic variation at 9p22.2 and ovarian cancer risk for BRCA1 and BRCA2 mutation carriers. J Natl Cancer Inst. 2011;103:105-116
50. French JD, Johnatty SE, Lu Y. et al. Germline polymorphisms in an enhancer of PSIP1 are associated with progression-free survival in epithelial ovarian cancer. Oncotarget. 2016;7:6353-6368
51. Johnatty SE, Tyrer JP, Kar S. et al. Genome-wide Analysis Identifies Novel Loci Associated with Ovarian Cancer Outcomes: Findings from the Ovarian Cancer Association Consortium. Clin Cancer Res. 2015;21:5264-5276
52. Huang RS, Johnatty SE, Gamazon ER. et al. Platinum sensitivity-related germline polymorphism discovered via a cell-based approach and analysis of its association with outcome in ovarian cancer patients. Clin Cancer Res. 2011;17:5490-5500
53. Boyd J, Luo B, Peri S. et al. Whole exome sequence analysis of serous borderline tumors of the ovary. Gynecol Oncol. 2013;130:560-564
54. Jones S, Wang TL, Kurman RJ. et al. Low-grade serous carcinomas of the ovary contain very few point mutations. J Pathol. 2012;226:413-420
55. Ryland GL, Hunter SM, Doyle MA. et al. Mutational landscape of mucinous ovarian carcinoma and its neoplastic precursors. Genome Med. 2015;7:87
56. Lambrechts S, Smeets D, Moisse M. et al. Genetic heterogeneity after first-line chemotherapy in high-grade serous ovarian cancer. Eur J Cancer. 2016;53:51-64
57. Castellarin M, Milne K, Zeng T. et al. Clonal evolution of high-grade serous ovarian carcinoma from primary to recurrent disease. J Pathol. 2013;229:515-524
58. Liu Y, Yasukawa M, Chen K. et al. Association of Somatic Mutations of ADAMTS Genes With Chemotherapy Sensitivity and Survival in High-Grade Serous Ovarian Carcinoma. JAMA Oncol. 2015;1:486-494
59. Jahr S, Hentze H, Englisch S. et al. DNA fragments in the blood plasma of cancer patients: quantitations and evidence for their origin from apoptotic and necrotic cells. Cancer Res. 2001;61:1659-1665
60. Sidransky D. Emerging molecular markers of cancer. Nat Rev Cancer. 2002;2:210-219
61. Murtaza M, Dawson SJ, Tsui DW. et al. Non-invasive analysis of acquired resistance to cancer therapy by sequencing of plasma DNA. Nature. 2013;497:108-112
62. Osterberg L, Levan K, Partheen K. et al. Cytogenetic analysis of carboplatin resistance in early-stage epithelial ovarian carcinoma. Cancer Genet Cytogenet. 2005;163:144-150
63. Osterberg L, Levan K, Partheen K. et al. High-resolution genomic profiling of carboplatin resistance in early-stage epithelial ovarian carcinoma. Cytogenet Genome Res. 2009;125:8-18
64. Kim SW, Kim JW, Kim YT. et al. Analysis of chromosomal changes in serous ovarian carcinoma using high-resolution array comparative genomic hybridization: Potential predictive markers of chemoresistant disease. Genes Chromosomes Cancer. 2007;46:1-9
65. Osterberg L, Levan K, Partheen K. et al. Potential predictive markers of chemotherapy resistance in stage III ovarian serous carcinomas. BMC Cancer. 2009;9:368
66. Osterberg L, Levan K, Partheen K. et al. Specific copy number alterations associated with docetaxel/carboplatin response in ovarian carcinomas. Anticancer Res. 2010;30:4451-4458
67. Cooke SL, Ng CK, Melnyk N. et al. Genomic analysis of genetic heterogeneity and evolution in high-grade serous ovarian carcinoma. Oncogene. 2010;29:4905-4913
68. Partheen K, Levan K, Osterberg L. et al. Analysis of cytogenetic alterations in stage III serous ovarian adenocarcinoma reveals a heterogeneous group regarding survival, surgical outcome, and substage. Genes Chromosomes Cancer. 2004;40:342-348
69. Mayr D, Kanitz V, Anderegg B. et al. Analysis of gene amplification and prognostic markers in ovarian cancer using comparative genomic hybridization for microarrays and immunohistochemical analysis for tissue microarrays. Am J Clin Pathol. 2006;126:101-109
70. Staebler A, Karberg B, Behm J. et al. Chromosomal losses of regions on 5q and lack of high-level amplifications at 8q24 are associated with favorable prognosis for ovarian serous carcinoma. Genes Chromosomes Cancer. 2006;45:905-917
71. Hu J, Khanna V, Jones MW. et al. Comparative study of primary and recurrent ovarian serous carcinomas: comparative genomic hybridization analysis with a potential application for prognosis. Gynecol Oncol. 2003;89:369-375
72. Bruchim I, Israeli O, Mahmud SM. et al. Genetic alterations detected by comparative genomic hybridization and recurrence rate in epithelial ovarian carcinoma. Cancer Genet Cytogenet. 2009;190:66-70
73. Tan DS, Iravani M, McCluggage WG. et al. Genomic analysis reveals the molecular heterogeneity of ovarian clear cell carcinomas. Clin Cancer Res. 2011;17:1521-1534
74. Yamashita Y, Akatsuka S, Shinjo K. et al. Met is the most frequently amplified gene in endometriosis-associated ovarian clear cell adenocarcinoma and correlates with worsened prognosis. PLoS One. 2013;8:e57724
75. Tan DS, Lambros MB, Rayter S. et al. PPM1D is a potential therapeutic target in ovarian clear cell carcinomas. Clin Cancer Res. 2009;15:2269-2280
76. Tuefferd M, Couturier J, Penault-Llorca F. et al. HER2 status in ovarian carcinomas: a multicenter GINECO study of 320 patients. PLoS One. 2007;2:e1138
77. Lassus H, Leminen A, Vayrynen A. et al. ERBB2 amplification is superior to protein expression status in predicting patient outcome in serous ovarian carcinoma. Gynecol Oncol. 2004;92:31-39
78. Anglesio MS, Kommoss S, Tolcher MC. et al. Molecular characterization of mucinous ovarian tumours supports a stratified treatment approach with HER2 targeting in 19% of carcinomas. J Pathol. 2013;229:111-120
79. Gloss BS, Samimi G. Epigenetic biomarkers in epithelial ovarian cancer. Cancer Lett. 2014;342:257-263
80. Maldonado L, Hoque MO. Epigenomics and ovarian carcinoma. Biomark Med. 2010;4:543-570
81. Ibanez de Caceres I, Battagli C, Esteller M. et al. Tumor cell-specific BRCA1 and RASSF1A hypermethylation in serum, plasma, and peritoneal fluid from ovarian cancer patients. Cancer Res. 2004;64:6476-6481
82. Liggett TE, Melnikov A, Yi Q. et al. Distinctive DNA methylation patterns of cell-free plasma DNA in women with malignant ovarian tumors. Gynecol Oncol. 2011;120:113-120
83. Teodoridis JM, Hall J, Marsh S. et al. CpG island methylation of DNA damage response genes in advanced ovarian cancer. Cancer Res. 2005;65:8961-8967
84. Chiang JW, Karlan BY, Cass L. et al. BRCA1 promoter methylation predicts adverse ovarian cancer prognosis. Gynecol Oncol. 2006;101:403-410
85. Makarla PB, Saboorian MH, Ashfaq R. et al. Promoter hypermethylation profile of ovarian epithelial neoplasms. Clin Cancer Res. 2005;11:5365-5369
86. Gifford G, Paul J, Vasey PA. et al. The acquisition of hMLH1 methylation in plasma DNA after chemotherapy predicts poor survival for ovarian cancer patients. Clin Cancer Res. 2004;10:4420-4426
87. Plumb JA, Strathdee G, Sludden J. et al. Reversal of drug resistance in human tumor xenografts by 2'-deoxy-5-azacytidine-induced demethylation of the hMLH1 gene promoter. Cancer Res. 2000;60:6039-6044
88. Strathdee G, MacKean MJ, Illand M. et al. A role for methylation of the hMLH1 promoter in loss of hMLH1 expression and drug resistance in ovarian cancer. Oncogene. 1999;18:2335-2341
89. Montavon C, Gloss BS, Warton K. et al. Prognostic and diagnostic significance of DNA methylation patterns in high grade serous ovarian cancer. Gynecol Oncol. 2012;124:582-588
90. Zhou F, Ma M, Tao G. et al. Detection of circulating methylated opioid binding protein/cell adhesion molecule-like gene as a biomarker for ovarian carcinoma. Clin Lab. 2014;60:759-765
91. Su HY, Lai HC, Lin YW. et al. An epigenetic marker panel for screening and prognostic prediction of ovarian cancer. Int J Cancer. 2009;124:387-393
92. Dai W, Teodoridis JM, Zeller C. et al. Systematic CpG islands methylation profiling of genes in the wnt pathway in epithelial ovarian cancer identifies biomarkers of progression-free survival. Clin Cancer Res. 2011;17:4052-4062
93. Chou JL, Su HY, Chen LY. et al. Promoter hypermethylation of FBXO32, a novel TGF-beta/SMAD4 target gene and tumor suppressor, is associated with poor prognosis in human ovarian cancer. Lab Invest. 2010;90:414-425
94. Fiegl H, Windbichler G, Mueller-Holzner E. et al. HOXA11 DNA methylation-a novel prognostic biomarker in ovarian cancer. Int J Cancer. 2008;123:725-729
95. Taniguchi T, Tischkowitz M, Ameziane N. et al. Disruption of the fanconi anemia-BRCA pathway in cisplatin-sensitive ovarian tumors. Nat Med. 2003;9:5668-5674
96. Zhu J, Zhang S, Gu L. et al. Epigenetic silencing of DKK2 and Wnt signal pathway components in human ovarian carcinoma. Carcinogenesis. 2012;33:2334-2343
97. Dai W, Zeller C, Masrour N. et al. Promoter CpG island methylation of genes in key cancer pathways associates with clinical outcome in high-grade serous ovarian cancer. Clin Cancer Res. 2013;19:5788-5797
98. Michaelson-Cohen R, Keshet I, Straussman R. et al. Genome-wide de novo methylation in epithelial ovarian cancer. Int J Gynecol Cancer. 2011;21:269-279
99. Wei SH, Chen CM, Strathdee G. et al. Methylation microarray analysis of late-stage ovarian carcinomas distinguishes progression-free survival in patients and identifies candidate epigenetic markers. Clin Cancer Res. 2002;8:2246-2252
100. Bauerschlag DO, Ammerpohl O, Bräutigam K. et al. Progression-free survival in ovarian cancer is reflected in epigenetic DNA methylation profiles. Oncology. 2011;80:12-20
101. Li M, Balch C, Montgomery JS. et al. Integrated analysis of DNA methylation and gene expression reveals specific signaling pathways associated with platinum resistance in ovarian cancer. BMC Med Genomics. 2009;2:34
102. Häfner N, Steinbach D, Jansen L. et al. RUNX3 and CAMK2N1 hypermethylation as prognostic marker for epithelial ovarian cancer. Int J Cancer. 2016;138:217-228
103. Melnikov A, Scholtens D, Godwin A. et al. Differential methylation profile of ovarian cancer in tissues and plasma. J Mol Diagn. 2009;11:60-65
Author contact
![]() Corresponding author: Bingjian Lu, M.D., Department of Surgical Pathology, the Affiliated Women's Hospital, School of Medicine, Zhejiang University, 1 Xueshi Road, Hangzhou, Zhejiang Province, PR China, 310006. Email: lbjedu.cn. Tel: 86-571-89991702.
Corresponding author: Bingjian Lu, M.D., Department of Surgical Pathology, the Affiliated Women's Hospital, School of Medicine, Zhejiang University, 1 Xueshi Road, Hangzhou, Zhejiang Province, PR China, 310006. Email: lbjedu.cn. Tel: 86-571-89991702.