Impact Factor ISSN: 1837-9664

 Global reach, higher impact
Global reach, higher impactJ Cancer 2016; 7(11):1557-1564. doi:10.7150/jca.15566 This issue Cite
Research Paper
Bleomycin (BLM) Induces Epithelial-to-Mesenchymal Transition in Cultured A549 Cells via the TGF-β/Smad Signaling Pathway
Kui-Jun Chen1,2*, Qing Li3*, Cang-mei Wen1,2, Zhao-Xia Duan1,2, Jie yuan Zhang1, Chuan Xu1, Jian-Min Wang1,2 ![]()
1. Research Institute of Surgery, Daping Hospital, Third Military Medical University, Chongqing , 400042, PR China;
2. State Key Laboratory of Trauma, Burn and Combined Injury, Chongqing 400042 PR China;
3. Cancer Center, Daping Hospital and Research Institute of Surgery, Third Military Medical University, Chongqing, 400042, China.
* Kui-Jun Chen and Qing Li have contributed equally to this work.
Received 2016-3-17; Accepted 2016-5-18; Published 2016-7-9
Abstract
The epithelial-to-mesenchymal transition (EMT) is a crucial cellular event in wound healing, tissue repair, and cancer progression in adult tissues, with the interactions with numerous signals. In this study, we aimed to determine whether bleomycin (BLM), an agent that causes pulmonary fibrosis, induces the EMT of the alveolar epithelial cell line A549 and investigated the possible mechanisms. We examined the EMT involved changes in cell morphology, isoform switching of the fibroblast growth factor receptor 2 (FGFR2) by alternative splicing, and expression of the phenotypic markers including E-cadherin, vimentin, and α-SMA using RT-PCR, Western blotting, and immunofluorescence assays. A TGF-β/Smad inhibitor was used to determine whether coculture with BLM would inhibit the EMT of A549 cells. The results showed that BLM induced the EMT of A549 cells possibly via the TGF-β/Smad signaling pathway, evident from the decrease in the expression of E-cadherin and increase in the expression on vimentin.
Keywords: Bleomycin, Epithelial-to-Mesenchymal Transition, Pulmonary fibrosis, TGF-β.
Introduction
Idiopathic pulmonary fibrosis (IPF) is the most common fibrosing interstitial lung disease with a characteristics of the histopathological pattern of usual interstitial pneumonia; IPF has a median survival time of three to four years [1, 2]. IPF is an important and devastating lung disease with a prevalence of 2-29/100,000 persons and an incidence of approximately 10/100,000 persons per year, and its incidence continues to rise [3]. Drug-induced interstitial lung disease (ILD), particularly pulmonary fibrosis, is a serious clinical concern. It has been suggested that myofibroblasts play a major role in the onset and development of ILD, and it was recently revealed that some of these myofibroblasts were derived from lung epithelial cells through the epithelial-mesenchymal transition (EMT) [4]. Another hypothesis suggests that impaired re-epithelialization, following lung injury, triggers the process of pulmonary fibrosis. However, the mechanism for the impaired re-epithelialization in pulmonary fibrosis is not well understood.
Exposure to the cytotoxic agent bleomycin (BLM) can be harmful due to the induction of potentially fatal pulmonary toxicity, so called bleomycin-induced pneumonitis (BIP) that can progress to lung fibrosis. [5]. The pathogenesis of BIP has been extensively studied in animals [6], and BLM-induced pulmonary fibrosis in rodents has often been used as a model for the investigation of human pulmonary fibrosis [7, 8]. Bleomycin-induced apoptosis of epithelial and non-epithelial cells plays an important role in pulmonary fibrogenesis [5]. However, whether the EMT is truly involved in BLM-induced lung fibrogenesis remains unclear.
Transforming growth factor (TGF)-β is one of the central mediators in the process of collagen production by fibroblasts, as has been shown by a study in animals [9]. TGF-β [10] is known to be the main cytokine involved in the pathogenesis of IPF that induces EMT in the human alveolar epithelial cell line A549 [11]. It has been suggested that targeting TGF-β/Smad signaling pathway is an important approach to the prevention and treatment of bleomycin-induced pulmonary fibrosis in rats [12]. Interestingly, we detected significantly higher TGF-β1 expression in BLM-treated cell culture supernatants of A549 cells than that in normal control cells.
In this study, we validated the selected biomarkers of the EMT in BLM-treated A549 cells, and showed that BLM could activate the EMT with the involvement of the Smad2/3 signaling pathway.
Materials and Methods
Cell culture
The cell line A549 was purchased from the Shanghai Institute of Cell Biology, China. A549 cells were grown in DMEM containing 1.5 mg/ml of sodium bicarbonate (ATCC, Manassas, VA) and 10% calf serum (N-10) in a 5% CO2 incubator. Cells were seeded at 0.7 × 105 cells per well in six-well plates, and the medium was replaced with 2 ml serum-free medium with or without 20 μg/mL BLM for 48 h.
RT-PCR
Total RNA was purified using the E.Z.N.A.TM Total RNA Kit Ⅱ (OMEGA, CA, U.S.A.). The complementary DNA was obtained from 500 ng/g of total RNA using the SuperscriptTM III First-Strand Synthesis system and Platinum PCR SuperMix (Invitrogen Carlsbad, CA, U.S.A.). The sequences of the primers used in this study are shown in Table 1.
Primers used in this study. (Invitrogen).
| E-cadherin | Fw: 5' ACG CCT GGG ACT CCA CCT A 3' |
|---|---|
| Rv: 5' CCT TGC CTT CTT TGT CTT TGT T 3' | |
| Vimentin | Fw: 5' TGA CCG CTT CGC CAA CTA 3' |
| Rv: 5' CCC GCA TCT CCT CCT CGT A 3' | |
| GAPDH | Fw: 5' GGT CGG AGT CAA CGG ATT TGG TCG 3' |
| Rv: 5' CCT CCG ACG CCT GCT TCA CCA C 3' | |
| FGFR2 | Fw: 5' TGG ATC AAG CAC GTG GAA AAG A 3' |
| Rv: 5' GGC GAT TAA GAA GAC CCC TAT GC 3' |
Antibodies and immunoblotting
Cell lysates were collected and fractionated by sodium docecylsulfate-polyacrylamide gel electrophoresis, transferred to a polyvinylidene difluoride membrane, and probed using rabbit polyclonal anti-E-cadherin antibody (1:1000; Abcam), mouse monoclonal anti-vimentin antibody (1:1000; Abcam), and rabbit polyclonal anti-actin antibody (1:5000; Abcam). The primary antibodies were detected using a horseradish peroxidase (HRP)-conjugated antibody, which was visualized by enhanced chemiluminescence (SuperSignal®, West Pico Chemiluminescent Substrate, USA).
ELISA
The ELISA kits for measuring TGF-β1 were all purchased from Abcam® (Human: ab100647; mouse: ab119557). All reagents, samples, and standards were prepared following the manufacturer's instructions. In brief, the standards or samples were added to each well. Following the incubation, the plate was washed, and then biotin-conjugated anti-mouse TGF beta 1 antibody was added to the appropriate wells. The plate was then incubated and washed, and then prepared streptavidin HRP conjugate was added to the appropriate wells. The plate was incubated at room temperature, washed, and then TMB substrate was added to each well. Finally, stop solution was added to each well. Plates were read at 450 nm immediately.
Immunofluorescence assay
Briefly, cells for the immunofluorescence assay were seeded onto 22-mm round cover slips (Thermo Fisher Scientific) and treated with BLM or TGF-β for 48 h. Then, the cells were washed with PBS for 5 min, fixed in ice-cold 4% paraformaldehyde for 15 min, and permeabilized with 1% Triton X-100 in PBS for 15 min. Following the blocking with 10% goat serum in PBS for 2 h at room temperature, the cells were incubated with primary antibodies overnight at 4℃ and fluorescent secondary antibodies for 1 h at room temperature. The cell nuclei were stained with 10 µM DAPI for 5 min. The fluorescent images were obtained using a fluorescence microscope (OLYMPUS BX50). The rabbit polyclonal anti-E-cadherin antibody (1:1000; Abcam) and the mouse monoclonal anti-vimentin antibody (1:1000; Abcam) were used as primary antibodies. The Alexa Fluor® 488 goat anti-mouse IgG (H+L) antibody and the Alexa Fluor® 594 goat anti-rabbit IgG (H+L) antibody were used as secondary antibodies.
Statistical analysis
The data are expressed as the mean ± standard deviation (SD), from three independent experiments. Statistical analyses were performed by two-tailed Student's t test. P values less than 0.05 were considered statistically significant.
Results
Changes in cell morphology and expression of EMT-related markers
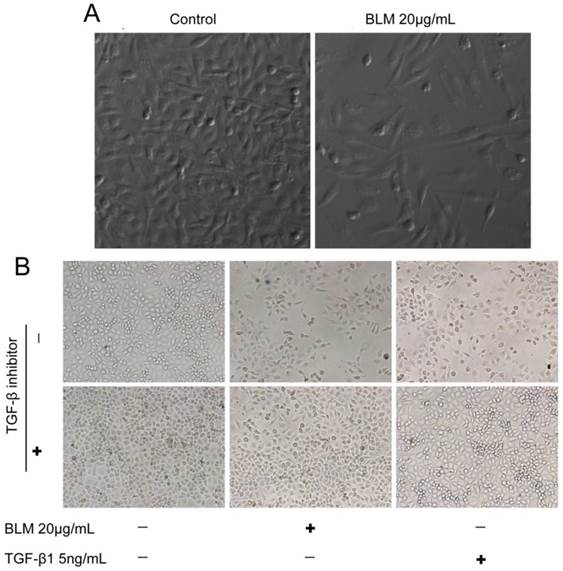
To detect whether bleomycin (BLM) induces EMT in vitro, we first determined the concentrations of BLM and TGF-β1 required for the induction of EMT in cultured A549 cells. Changes in cell morphology were assessed under phase contrast light microscopy (Fig. 1). The cells were changed to more fibroblast-like cells after treatment with 20 μg/mL BLM for 4 days in serum-free medium (Fig. 1 A). Then, we changed the treatment time to 48 h, and TGF-β1 (5 ng/mL) was added based on the study by Ju Hee Kim [13]. The control group of A549 cells maintained the classic cobblestone epithelial morphology and growth pattern; however, after treatment with 20 μg/mL BLM for 4 days, loss of E-cadherin and reorganization of vimentin from the epithelial cells to fibroblastic stress fibers were observed by RT-PCR (Fig. 2 A). Table 2 shows the changes in area (μm2), perimeter (μm), and roundness of A549 cells after incubation with BLM at various concentrations in serum-free medium for 24 h or 48 h.
The morphology of the cells was changed after BLM or TGF-β1 treatment. (A) Cells became more fibroblast-like after incubation with 20 μg/mL BLM for 4 days (200×). (B) Cells were less fibroblast-like after treatment for a shorter duration of time, approximately 48 h, and were normal when cocultured with the inhibitor (40×).
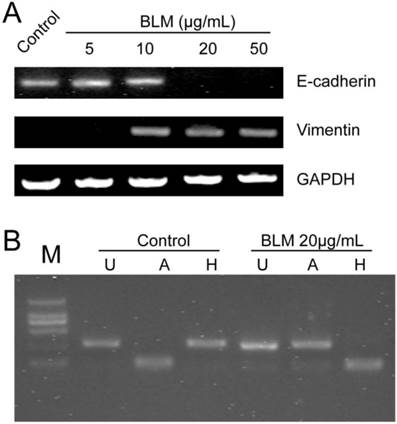
Expression levels of EMT-related markers and the FGFR2 splice variant analysis after BLM treatment. (A) A549 cells were incubated with BLM at various concentrations for 4 days. RT-PCR was used to detect the expression of the epithelial marker E-cadherin and the mesenchymal marker vimentin. E-cadherin disappeared after treatment with 20 μg/mL or 50 μg/mL BLM. (B) RT-PCR products were digested with AvaI [A] or HincII [H], which specifically cut exon IIIb- and exon IIIc-containing products, respectively. [U] Indicates undigested PCR products; [M] DL2000.
Morphology changing of A549 cells after incubating with BLM at various concentrations in serum-free medium for 24 h or 48 h. Every cell saple chose six images in same scale. (200×) SPOT software was used to measure about 20 cells per image randomly. And the mean values ± SD are presented. (*P≤0.05), (**P≤0.01).
| Sample | Time | Area (μm2) | Perimeter (μm) | Roundness | |
|---|---|---|---|---|---|
| Control | 24h | 112.56±26.15 | 187.91±32.66 | 0.65±0.12 | |
| 48h | 106.64±24.75 | 182.21±9.68 | 0.64±0.12 | ||
| BLM(μg/mL) | 10 | 24h | 184.71±28.97* | 237.26±27.76* | 0.66±0.11 |
| 48h | 275.85±65.18** | 333.84±59.77** | 0.51±0.13* | ||
| 20 | 24h | 197.37±46.71* | 261.45±36.62* | 0.58±0.09* | |
| 48h | 219.94±58.99** | 338.61±78.58** | 0.41±0.12** | ||
| 50 | 24h | 225.96±72.42* | 280.46±57.39** | 0.58±0.12 | |
| 48h | 502.53±137.14** | 556.53±109.51** | 0.33±0.12** | ||
TGF-β1 level is markedly increased in BLM-treated A549 cell culture supernatants
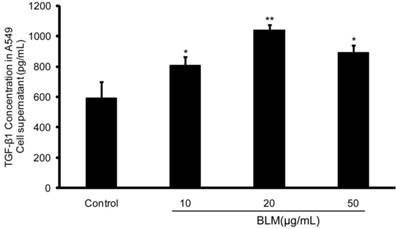
After the observation of EMT in A549 cells in the presence of BLM, ELISA was performed and the results demonstrated that the concentration of TGF-β1 in BLM-treated A549 cell culture supernatants at various concentrations for 48 h was higher than that in the normal control. Of note, there was a significant increase in the level of TGF-β1 in the 20 μg/mL treatment group (**P≤0.01) (Fig. 4). These results suggest that TGF-β1 could be a mediator of BLM-induced EMT in A549 cells.
The TGF-β1 level in BLM-treated A549 culture supernatants were significant higher than that in the normal control. A549 cells were incubated with BLM at various concentrations in serum-free medium for 48 h. Then, the culture supernatants were collected, and the concentrations of TGF-β1 were determined using an ELISA kit. The 20 µg/mL BLM treatment had the most significant effect. The experiments were performed three times, and the mean values ± SD are presented. (*P≤0.05), (**P≤0.01).
Inhibition of TGF-β1 inhibits the EMT
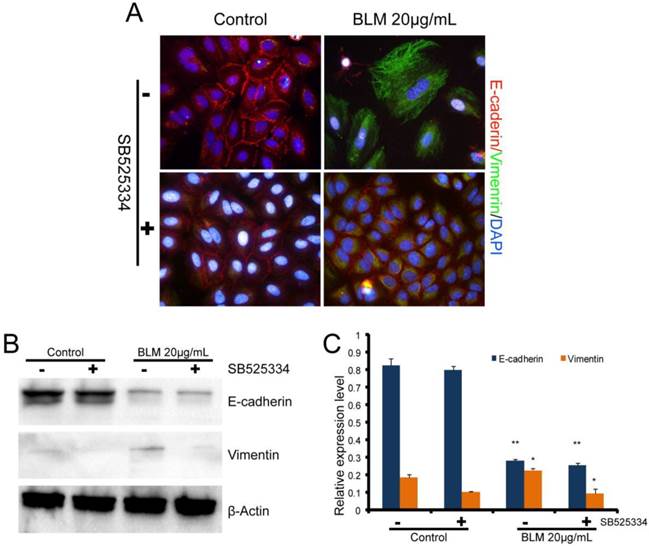
To further examine the role of TGF-β1in BLM-induced EMT, the TGF-β1 inhibitor (SB525334) was added to the cells prior to the drug treatment. The cells were then incubated with SB525334 for 1 h and then maintained in fresh serum-free medium with BLM or TGF-β1 for 48 h. Immunofluorescence analysis showed that the loss of E-cadherin and the reorganization of vimentin in A549 cells were partially inhibited (Fig. 3 A). The cells were changed to less fibroblast-like cells after treatment with BLM or TGF-β1 for short lengths of time, while the cells appeared normal when co-incubated with the TGF-β1/Smad inhibitor (Fig. 1 B). Analysis by Western blotting assay revealed the same results (Fig. 3 B, C).
Expression levels of EMT-related markers in cultured A549 cells. Cells in serum-free medium were treated with BLM or TGF-β1, respectively, for 48 h. The TGF-β inhibitor (SB525334) was used (+) 1 h prior to the drugs, then fresh serum-free medium was added to the cells. (A) Immunofluorescence analysis of EMT-related cell markers (200×). Red fluorescence indicates the epithelial marker E-cadherin, while green fluorescence indicates the mesenchymal marker vimentin. Cell nuclei are indicated in blue with DAPI. Our results show attenuated EMT that was induced by BLM when the cells were cocultured with the TGF-β inhibitor. (B) Western blot analysis of EMT-related cell markers. β-actin was used as a loading control. (C) Quantitative analysis of E-cadherin and vimentin expression in A549 cells under the indicated treatment. The experiments were performed three times, and the mean values ± SD are presented. (*P≤0.05).
BLM induces alternative splicing of FGFR2
FGFR2 splice variants contain IIIb and IIIc, which are specifically expressed in epithelial cells or mesenchymal cells, respectively, and play a key role in the EMT. To determine whether BLM induces splicing of FGFR2 during the EMT in A549 cells in vitro, total RNA from A549 cells was purified and RT-PCR was performed. Then, the products of FGFR2 fragments were digested by Ava I or Hinc II, and the result revealed differences between the control group and the treated groups (Fig. 2 B). The change in FGFR2 splice variants suggested that the EMT of A549 cells might occur after BLM treatment.
The Smad2/3 signaling pathway may participate in the BLM-induced EMT of A549 cells
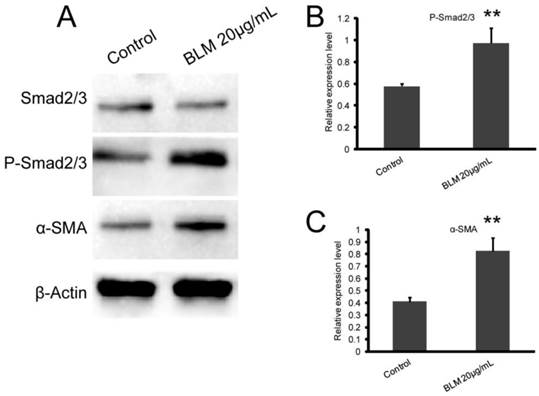
To determine whether the Smad2/3 signaling pathway plays an important role in the EMT induced by BLM, we measured the expression levels of Smad2/3, P-Smad2/3, and α-SMA. Our results showed that BLM activated the Smad2/3 signaling pathway (Fig. 5 A). After BLM treatment, the expression of phosphorylated Smad2/3 was significantly increased, and this led to the up-regulation of α-SMA expression (Fig. 5 C, D) (*P≤0.05, **P≤0.01).
BLM activates the Smad2/3 signaling pathway, which participates in the regulation of α-SMA expression. A549 cells were treated with BLM for 48 h in serum-free medium. (A) Western blot analysis of Smad2/3, P-Smad2/3, and α-SMA expression in A549 cells. Representative blots showing the up-regulation of P-Smad2/3 and α-SMA expression. β-actin was used as a loading control. (B, C) Quantitative analysis of P-Smad2/3 and α-SMA expression in A549 cells under the indicated treatment. The experiments were performed three times, and the mean values ± SD are presented. (*P≤0.05), (**P≤0.01).
Disscussion
The generation of fibroblastic foci in the lungs of patients with idiopathic pulmonary fibrosis (IPF) is the characteristics of pulmonary fibrosis [14]. The fibrosis of lung cells can originate from the following paths: the transformation of intrinsic fibroblasts and the differentiation of initial cells. The more important path is the EMT [15]. The injury and repair process of the epithelia is likely pivotal in the path mechanism of pulmonary fibrosis, and inadequate repair and injury of alveolar epithelial cells can induce the pathological process of pulmonary interstitial fibrosis [16]. Studies have shown that the EMT in alveolar epithelial cells provides a critical source of fibroblasts [17]. The EMT is a process in which fully differentiated epithelial cells undergo phenotypic transition to fully differentiated mesenchymal cells, often myofibroblasts and fibroblasts [18]. The biomarkers of epithelial cell transdifferentiation include disappearance of cell polarity, loss of expression of the epithelial marker E-cadherin, increased expression of α-SMA and vimentin, and an elongated shape [19]. Studies have indicated that transforming growth factor1 (TGF-β1) plays an important role in the EMT [20]. In the study of numerous pulmonary fibrosis models, the most common and successful model was bleomycin-induced pulmonary fibrosis [21]. However, the specific mechanism for the EMT in pulmonary fibrosis and bleomycin-induced pulmonary fibrosis were uncertain or undetermined in current reports.
TGF-β1 is a multifunction cytokine that plays a pivotal role in the EMT and has been implicated as a “master switch” in the induction of fibrosis in the lungs. It is involved in the regulation of processes such as tissue morphogenesis and differentiation through effects on cell differentiation, apoptosis, proliferation, and extracellular matrix production [20]. In pulmonary fibrosis, TGF-β1 has been regarded as the most important factor. Previous studies showed that increased expression levels of α-SMA and vimentin were found in primary type Ⅱ alveolar epithelial cells treated with TGF-β1, while biomarkers of epithelial cells such as E-cadherin and cytokeratin were down-regulated, and the cell shape became elongated [22]. In addition, reports showed that TGF-β1 can directly induce the EMT in alveolar epithelial cells [20], A549 cells [13], and bronchial epithelial cells [23].
The FGFR2 splice variant may participate in the EMT in A549 cells induced by BLM. Moreover, the alternative splicing events of FGFR2 are also critical in the EMT. The spliceosomes of FGFR2 contain two types: one is KGFR (FGFR2-Ⅲb) Ⅲa/Ⅲb, which is encoded by exons 7 and 8 and found in epithelial cells, while the other is BEK (FGFR2-Ⅲc) Ⅲa/Ⅲc, which is encoded by exons 7 and 9 and found in mesenchymal cells. FGFR2 alternative splicing plays a crucial role in various diseases such as in the growth and development of tissue or tumor fibrosis [24]. The previous studies have shown that the alteration of FGFR2-Ⅲc to FGFR2-Ⅲb can induce the transformation of mesenchymal cells to epithelial cells, and the expression of FGFR2-Ⅲb can be decreased during the process of EMT which is induced by TGF-β1 [25, 26].
The induction mechanism of TGF-β in the EMT is primarily due to two signaling pathways. The first is the β-integrin signal transduction pathway (also referred to as the Smads signaling pathway), in which TGF-β binds to the subtype (TGF-β type I and type II) as a ligand and becomes a complex, which leads to the activation of Smad2 and Smad3. The phosphorylated Smad2 and Smad3 compounds then form trimers with Smad4, followed by translocation into the nucleus, where they associate and cooperate with DNA binding transcription factors to regulate the transcription of target genes [27]. The second is the non-Smad signaling pathway, which includes activation of Ras and MAP kinases, Rho-like GTPases, and PI3 kinase/Akt. The relevant receptor kinases were activated by TGF-β and followed a series of responses involving the regulation of distinct processes, such as cell growth, survival, migration or invasion, and cytoskeletal organization [18, 23]. In the Smad signaling pathway, the pivotal roles of Smad2 and Smad3 were regulating the transcription of predominantly TGF-β target genes. In Smad3 knockout mice, TGF-β fails to induce the EMT and key transcriptional factors, metastasis of epithelial cells and pulmonary fibrosis are attenuated [28, 29]. Increased EMT was observed in a mammary epithelial model over-expressing Smad2 and Smad3 [27].
According to the results, alternative splicing of FGFR2 in A549 cells can be induced by BLM (Fig. 2). Along with attenuation of E-cadherin and increasing vimentin and α-SMA, the decapentaplegic P-Smad2/3 was activated. All of the results are in accordance with the EMT biomarkers described in previous studies. The 20 µg/mL BLM treatment had the most significant effect.
In this study, among the A549 cells that were treated with different concentrations of BLM for 4 days, the group treated with 20 µg/mL BLM had higher TGF-β1 levels than the control group (Fig. 4). The BLM-treated cells showed attenuated cell junctions, and the cobblestone-like cell morphology changed to a spindle or fusiform shape, leading to fibrosis compared with the control groups (Fig. 1, Table 2). The results described above indicated that the optimal concentration of BLM that led to fibrosis in A549 cells was approximately 20 µg/mL, which generated approximately 1 ng/mL of TGF-β1, similar to Kasai's study [11]. The optimal concentration treated with TGF-β1 directly was 5 ng/mL [13], which may be because the cells treated with BLM generated cytokines other than TGF-β1 that promote the EMT.
As described previously, the signal transduction pathway including Smad2/3 and Snail was involved in the mechanism of the EMT that was induced by TGF-β1 in vitro. In this study, we examined the TGF-β/Smad signaling pathway in the EMT using a TGF-β1/Smad inhibitor. The results showed that, in all of the groups (BLM and control), better suppression of the EMT process was observed in the A549 cells that contained the TGF-β1 inhibitor. Additionally, compared with the other groups, the cells in this group showed compact connections, insignificant changes in morphology (Fig. 1, Table 2), higher expression of E-cadherin and lower expression of vimentin (Fig. 3). Western blotting indicated that, in the Smad pathway, the expression of P-Smad2/3, as well as the expression of α-SMA, in BLM-treated cells was significantly higher than that of the control group (Fig. 5). The results described above revealed that the TGF-β1/Smad inhibitor can inhibit the EMT to some extent and that BLM can activate the Smad pathway. Therefore, BLM induced the EMT of A549 through the Smad pathway.
Conclusion
In summary, BLM can induce the EMT in A549, and this is related to the generated TGF-β1 concentration. The results in this study indicated that the optimal concentration of TGF-β1 that could induce the EMT of A549 was approximately 1 ng/mL, and the TGF-β1/Smad inhibitor can attenuate the EMT process. All of the results illustrated that BLM can induced the EMT of A549 via the TGF-β1/Smad signaling pathway. This may provide a basis for the theoretical foundation of the attenuation of the EMT by inhibiting the Smad signaling pathway and may provide an important basis for pulmonary fibrosis treatment.
Acknowledgements
This study was supported by the National Natural Science Foundation of China (No.81270119).We would like to thank Prof. Hong Huang and Prof. Xueqing Xu for their valuable assistance during the experimental study and data analysis.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Duck A, Pigram L, Errhalt P, Ahmed D, Chaudhuri N. Ipf care: A support program for patients with idiopathic pulmonary fibrosis treated with pirfenidone in europe. Advances in therapy. 2015;32:87-107
2. Thickett DR, Kendall C, Spencer LG, Screaton N, Wallace WA, Pinnock H, Bott J, Pigram L, Watson S, Millar AB. Improving care for patients with idiopathic pulmonary fibrosis (ipf) in the uk: A round table discussion. Thorax. 2014;69:1136-1140
3. Behr J. The diagnosis and treatment of idiopathic pulmonary fibrosis. Deutsches Arzteblatt international. 2013;110:875-881
4. Namba T, Tanaka KI, Ito Y, Hoshino T, Matoyama M, Yamakawa N, Isohama Y, Azuma A, Mizushima T. Induction of emt-like phenotypes by an active metabolite of leflunomide and its contribution to pulmonary fibrosis. Cell death and differentiation. 2010;17:1882-1895
5. Sleijfer S. Bleomycin-induced pneumonitis. Chest. 2001;120:617-624
6. Williamson JD, Sadofsky LR, Hart SP. The pathogenesis of bleomycin-induced lung injury in animals and its applicability to human idiopathic pulmonary fibrosis. Experimental lung research. 2015;41:57-73
7. Dong Y, Geng Y, Li L, Li X, Yan X, Fang Y, Li X, Dong S, Liu X, Li X, Yang X, Zheng X, Xie T, Liang J, Dai H, Liu X, Yin Z, Noble PW, Jiang D, Ning W. Blocking follistatin-like 1 attenuates bleomycin-induced pulmonary fibrosis in mice. The Journal of experimental medicine. 2015;212:235-252
8. He H, Tang H, Gao L, Wu Y, Feng Z, Lin H, Wu T. Tanshinone iia attenuates bleomycin-induced pulmonary fibrosis in rats. Molecular medicine reports. 2015;11:4190-4196
9. Shi K, Jiang J, Ma T, Xie J, Duan L, Chen R, Song P, Yu Z, Liu C, Zhu Q, Zheng J. Pathogenesis pathways of idiopathic pulmonary fibrosis in bleomycin-induced lung injury model in mice. Respiratory physiology & neurobiology. 2014;190:113-117
10. Ko H, So Y, Jeon H, Jeong MH, Choi HK, Ryu SH, Lee SW, Yoon HG, Choi KC. Tgf-beta1-induced epithelial-mesenchymal transition and acetylation of smad2 and smad3 are negatively regulated by egcg in human a549 lung cancer cells. Cancer letters. 2013;335:205-213
11. Kasai H, Allen JT, Mason RM, Kamimura T, Zhang Z. Tgf-beta1 induces human alveolar epithelial to mesenchymal cell transition (emt). Respiratory research. 2005;6:56
12. Wu J, Ling C, He H, Sun G, Gu Z, Ji B. [effect of compound panax notoginsenoside granules on tgf-beta1/smads signaling pathway in rats with bleomycin-induced pulmonary fibrosis]. Xi bao yu fen zi mian yi xue za zhi = Chinese journal of cellular and molecular immunology. 2013;29:927-930
13. Kim JH, Jang YS, Eom KS, Hwang YI, Kang HR, Jang SH, Kim CH, Park YB, Lee MG, Hyun IG, Jung KS, Kim DG. Transforming growth factor beta1 induces epithelial-to-mesenchymal transition of a549 cells. Journal of Korean medical science. 2007;22:898-904
14. Katzenstein AL, Myers JL. Idiopathic pulmonary fibrosis: Clinical relevance of pathologic classification. American journal of respiratory and critical care medicine. 1998;157:1301-1315
15. Kalluri R, Neilson EG. Epithelial-mesenchymal transition and its implications for fibrosis. The Journal of clinical investigation. 2003;112:1776-1784
16. Strieter RM. What differentiates normal lung repair and fibrosis? Inflammation, resolution of repair, and fibrosis. Proceedings of the American Thoracic Society. 2008;5:305-310
17. Willis BC, duBois RM, Borok Z. Epithelial origin of myofibroblasts during fibrosis in the lung. Proceedings of the American Thoracic Society. 2006;3:377-382
18. Zavadil J, Bottinger EP. Tgf-beta and epithelial-to-mesenchymal transitions. Oncogene. 2005;24:5764-5774
19. Zeisberg M, Neilson EG. Biomarkers for epithelial-mesenchymal transitions. The Journal of clinical investigation. 2009;119:1429-1437
20. Willis BC, Borok Z. Tgf-beta-induced emt: Mechanisms and implications for fibrotic lung disease. American journal of physiology Lung cellular and molecular physiology. 2007;293:L525-534
21. Mouratis MA, Aidinis V. Modeling pulmonary fibrosis with bleomycin. Current opinion in pulmonary medicine. 2011;17:355-361
22. Liu X. Inflammatory cytokines augments tgf-beta1-induced epithelial-mesenchymal transition in a549 cells by up-regulating tbetar-i. Cell motility and the cytoskeleton. 2008;65:935-944
23. Wu Z, Yang L, Cai L, Zhang M, Cheng X, Yang X, Xu J. Detection of epithelial to mesenchymal transition in airways of a bleomycin induced pulmonary fibrosis model derived from an alpha-smooth muscle actin-cre transgenic mouse. Respiratory research. 2007;8:1
24. Chaffer CL, Brennan JP, Slavin JL, Blick T, Thompson EW, Williams ED. Mesenchymal-to-epithelial transition facilitates bladder cancer metastasis: Role of fibroblast growth factor receptor-2. Cancer research. 2006;66:11271-11278
25. Shirakihara T, Horiguchi K, Miyazawa K, Ehata S, Shibata T, Morita I, Miyazono K, Saitoh M. Tgf-beta regulates isoform switching of fgf receptors and epithelial-mesenchymal transition. The EMBO journal. 2011;30:783-795
26. Baraniak AP, Chen JR, Garcia-Blanco MA. Fox-2 mediates epithelial cell-specific fibroblast growth factor receptor 2 exon choice. Molecular and cellular biology. 2006;26:1209-1222
27. Valcourt U, Kowanetz M, Niimi H, Heldin CH, Moustakas A. Tgf-beta and the smad signaling pathway support transcriptomic reprogramming during epithelial-mesenchymal cell transition. Molecular biology of the cell. 2005;16:1987-2002
28. Saika S, Kono-Saika S, Ohnishi Y, Sato M, Muragaki Y, Ooshima A, Flanders KC, Yoo J, Anzano M, Liu CY, Kao WW, Roberts AB. Smad3 signaling is required for epithelial-mesenchymal transition of lens epithelium after injury. The American journal of pathology. 2004;164:651-663
29. Zavadil J, Cermak L, Soto-Nieves N, Bottinger EP. Integration of tgf-beta/smad and jagged1/notch signalling in epithelial-to-mesenchymal transition. The EMBO journal. 2004;23:1155-1165
Author contact
![]() Corresponding author: Professor Jian-Min Wang, MD, PhD. State Key Laboratory of Trauma, Burns and Combined Injury, Trauma Center, Research Institute of Surgery, Daping Hospital, Third Military Medical University, Chongqing 400042, PR China.Tel:+86 023 68757461; E-mail: jmwangedu.cn.
Corresponding author: Professor Jian-Min Wang, MD, PhD. State Key Laboratory of Trauma, Burns and Combined Injury, Trauma Center, Research Institute of Surgery, Daping Hospital, Third Military Medical University, Chongqing 400042, PR China.Tel:+86 023 68757461; E-mail: jmwangedu.cn.