Impact Factor ISSN: 1837-9664

 Global reach, higher impact
Global reach, higher impactJ Cancer 2016; 7(12):1605-1609. doi:10.7150/jca.15523 This issue Cite
Short Research Communication
Differential Roles of Carboxylated and Uncarboxylated Osteocalcin in Prostate Cancer Growth
Yoshikazu Hayashi1,2, Tomoyo Kawakubo-Yasukochi1,4, Akiko Mizokami1,3, Hiroshi Takeuchi5, Seiji Nakamura2, Masato Hirata1 ![]()
1. Laboratory of Molecular and Cellular Biochemistry, Faculty of Dental Science, Kyushu University, Fukuoka 812-8582, Japan
2. Section of Oral and Maxillofacial Oncology, Faculty of Dental Science, Kyushu University, Fukuoka 812-8582, Japan
3. OBT Research Center, Faculty of Dental Science, Kyushu University, Fukuoka 812-8582, Japan
4. Department of Immunological and Molecular Pharmacology, Faculty of Pharmaceutical Science, Fukuoka University, Fukuoka 814-0180, Japan
5. Division of Applied Pharmacology, Kyushu Dental University, Kitakyushu 803-8580, Japan.
Received 2016-3-13; Accepted 2016-6-10; Published 2016-7-18
Abstract
Serum levels of osteocalcin (OC), a bone matrix non-collagenous protein secreted by osteoblasts, are correlated with pathological bone remodeling such as the bone metastasis of cancer, as well as physiological bone turnover. The pathological roles in prostate cancer growth of the two existing types of serum OC, γ-carboxylated (GlaOC) and lower- (or un-) carboxylated (GluOC), have not yet been discriminatively examined. In the present study, we demonstrate that normal prostate epithelial cell growth was promoted by both types of OC, while growth of cancer cells in the prostate was accelerated by GlaOC but suppressed by GluOC. We suggest that OC regulates prostate cancer growth depending on the γ-carboxylation, in part by triggering reduced phosphorylation of receptor tyrosine kinases.
Keywords: osteocalcin, prostate cancer
Introduction
Prostate cancer is one of the most prevalent malignant diseases of men worldwide, and it remains a challenge to distinguish and prevent lethal metastatic prostate cancers, which escape standard therapeutic intervention. Bone is a preferred organ for the metastasis of prostate cancer [1-4], and the metastasis to bone is associated with increased osteoblast activity [4]. In fact, the production of OC by osteoblasts is well correlated with the metastasis of prostate cancer progression [1], and therefore high levels of OC in the serum are assumed to be a metastatic marker of prostate cancer [2,3]. Furthermore, OC has been reported to promote prostate cancer progression [5], but previous studies did not discriminate between the two existing OC types, GlaOC and GluOC. GluOC but not GlaOC has recently been reported to improve glucose metabolism [6-10]. These findings indicate that each form of OC plays a different role depending on the pathophysiological conditions. Therefore, it is important to examine which form of OC is involved in prostate cancer progression. In the present study, GlaOC and GluOC were separately examined to determine their roles in prostate cancer cell growth.
Materials and Methods
Materials
Recombinant GluOC was prepared as described previously [7]. GlaOC was purchased from AnaSpec (Fremont, CA, USA).
Cells
A human prostate cancer cell line (PC-3) and normal prostate epithelial (ProEpi) cells were purchased from ATCC (Rockville, MD, USA) and Lonza (Basel, Switzerland), respectively. A human prostate carcinoma cell line, PPC-1, was kindly provided by Dr. J.Y. Bahk (Gyeongsang National University, Korea). The two prostate cancer cell lines and ProEpi cells were maintained in Dulbecco's Modified Eagle's Medium supplemented with 10% fetal bovine serum (Thermo Fisher Scientific, Waltham, MA, USA) and PrEGMTM Prostate Epithelial Cell Growth Medium BulletKitTM (Lonza), respectively. All cells were cultured at 37°C in humidified air containing 5% CO2.
Measurement of cell viability
Cell viability was determined with a WST-8 assay (Cell Count Reagent SF; Nakalai Tesque, Kyoto, Japan) and a BrdU uptake assay (BrdU Cell Proliferation ELISA Kit; Exalpha Biologicals, Shirley, MA, USA). Each cell seeding density was optimized before experiments. Prior to assays, cells were treated with vehicle, GluOC or GlaOC (1, 10, 100 ng/ml), in 96-well culture plates in 100 μl for 24 h or 48 h, after preculture in each maintenance medium for 24 h.
RTKs (Receptor Tyrosine Kinases) phosphorylation antibody (phosphor-RTKs) array
An RTKs phosphorylation antibody array was performed using a Human Phospho-RTK Array Kit (R&D Systems, Minneapolis, MN, USA). Following preculture for 24 h, PPC-1 or ProEpi cells were treated with GluOC (10 ng/ml) or GlaOC (10 ng/ml) for 6 h. Cells were then lysed in Lysis Buffer 17 (R&D Systems) including 3.4 μg/ml aprotinin, 10 μg/ml leupeptin, 5 μg/ml pepstatin, and 1 mM phenylmethylsulfonyl fluoride (Sigma-Aldrich, St. Louis, MO, USA). Cell lysate (73.5 μg) was analyzed using the Human Phospho-RTK Array Kit.
Statistical Analysis
Dunnett's test or Student's t-test were performed for statistical analysis where appropriate. P values < 0.05 were considered to be statistically significant. Results are expressed as mean ± standard deviation (SD).
Results and Discussion
Effects of GlaOC and GluOC on cell viability in human prostate cells
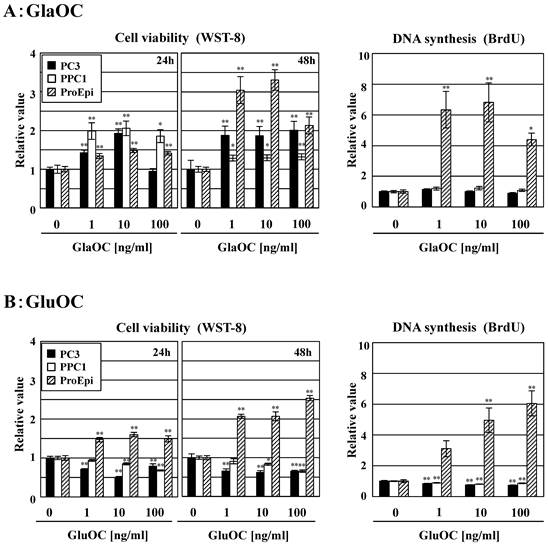
GlaOC at concentration of 1-100 ng/ml dose-dependently promoted the viability of the human prostate cancer cell lines PC-3 and PPC-1 in a WST-8 assay for 24 h, except for PC-3 cells at 100 ng/ml at 24 h (Fig 1A). We also assayed WST-8 for 48 h; more promotion than that for 24 h was observed, except for PPC1 cell line. In contrast, GluOC at the same concentrations inhibited viability of these two cell lines at 24 h and 48 h (Fig 1B). We further investigated a BrdU incorporation assay to examine the differential effects of GlaOC and GluOC on cell proliferation. GluOC dose-dependently decreased the incorporation of BrdU in the two prostate cancer cell lines in a similar manner as in the WST-8 assay, while the effect of GlaOC was not significantly manifested in this assay for unknown reasons (Fig 1AB). In contrast, both GlaOC and GluOC promoted the growth of the ProEpi cell line of normal prostate epithelial cells, as assessed by WST-8 (at 24 h and 48 h) and BrdU assays (at 24 h) (Fig 1AB). These results indicate that GlaOC and GluOC have stimulatory and inhibitory effects on prostate cancer cell growth, respectively, but that normal prostate epithelial cells respond in a similar manner to both forms.
Effects of GlaOC and GluOC on human prostate cell growth. Effect of GlaOC (A) and GluOC (B) on PC-3, PPC-1, and ProEpi cells. Each assay was performed in triplicate. Left and right panels represent WST-8 (cell viability) assays at 24 h and 48 h and BrdU uptake (DNA synthesis) assays at 24 h, respectively. The data represent mean ± SD from three experiments. Mean data are expressed as a ratio of the control. *P < 0.05 and **P < 0.01 versus the corresponding value for cells treated with vehicle.
Regulation of the phosphorylation levels of RTKs by GlaOC and GluOC in human prostate cells
Receptor tyrosine kinases (RTKs), a family of cell surface receptors for growth factors, mediate an initial signaling for cell proliferation [11]. We therefore assayed phospho-RTKs arrays using PPC-1 as cancer cells and ProEpi as normal cells to elucidate the mechanism by which GlaOC or GluOC affects cell growth. GlaOC enhanced the phosphorylation levels of nine RTKs, including FGFR1 (fibroblast growth factor receptor 1) [12], EphA4 (ephrin receptor A4) [13], EphA7 [14], EphA10 [15], EphB2 [16], EphB4 [17], EphR (ephrin receptor) [18], ALK (anaplastic lymphoma kinase) [19], and RYK [20] (Fig. 2A), which are all closely related to prostate cancer progression in PPC-1 cells [12-20]. In contrast, GluOC did not enhance any RTKs, but rather reduced phosphorylation levels of fifteen RTKs, including FGFR1 [12], EphA4 [13], EphA7 [14], EphA10 [15], EphB4 [17], EphR [18], ALK [19], RYK [20], HGFR (hepatocyte growth factor receptor) [21], FGFR3 [22], c-Ret [23], Ror1 [24], Ror2 [24], Axl [25], and EphA2 [13] (Fig. 2A), whose expression or activation are also closely related to prostate cancer progression [12-15,17-25].
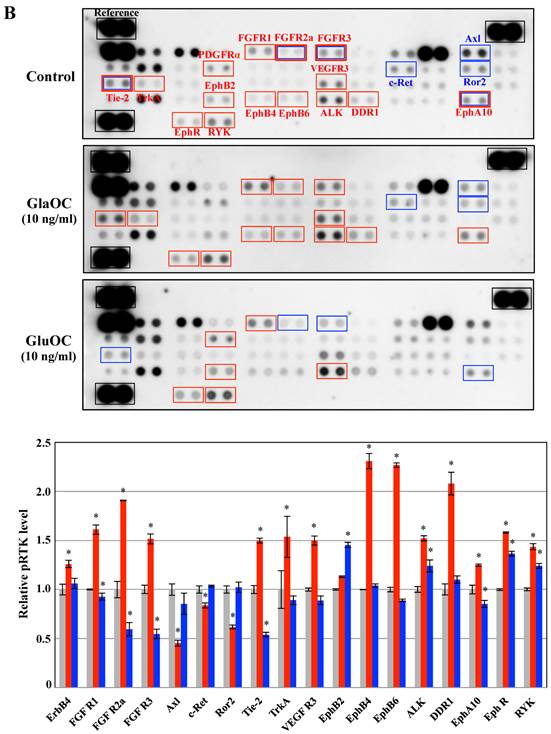
Phospho-RTKs array in PPC-1 (A) and ProEpi (B) cells. Quantitation of the dot densities of phospho-RTKs was performed using scanning images and ImageQuant LAS 4000 software (GE Healthcare UK, Buckinghamshire, England). Each pair of the kinase dots that increased (red) or decreased (blue) compared with the controls is enclosed in a square. Each assay was repeated three times. Four separate results are summarized in the graphs. *P < 0.05 versus the corresponding value for the control.
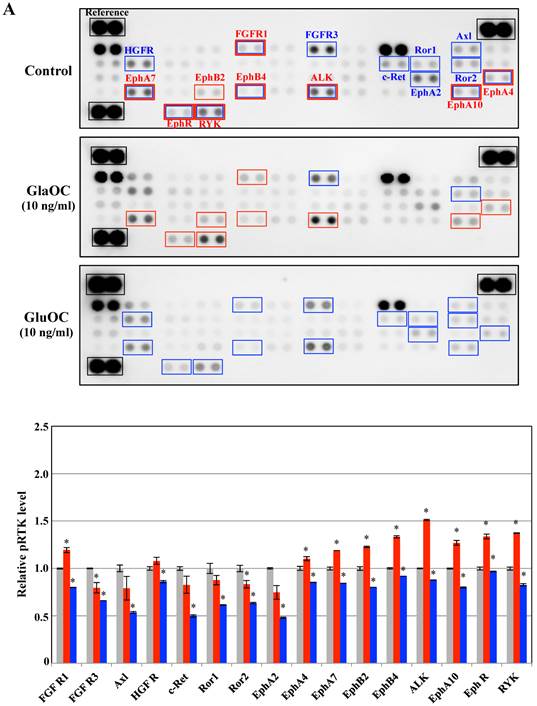
These results indicate that GlaOC promotes prostate cancer cell proliferation through the activation of nine RTKs, although it remains unclear whether GlaOC is a direct ligand for those RTKs. The mechanisms for GlaOC effect are currently unknown because the receptors for GlaOC and the signaling cascade have not yet been identified, but the tyrosine kinases are involved in the growth promotion since LY294002, an inhibitor for PI3 kinase and U0126, an inhibitor for MEK1/2 kinase, down-stream kinases after tyrosine kinase activation completely blocked the growth promotion by GlaOC (data not shown). On the contrary, GluOC is recognized by a tentative receptor, GPRC6A, followed by activation of cAMP production and protein kinase A [10], which might be implicated in the inhibition of several tyrosine kinases. We here suggest that GluOC could be a potential therapeutic agent against cancer, as it might inhibit the RTK-related signal transduction that is constitutively active in malignant cells [11].
Phosphorylation of RTKs using ProEpi cells, normal cells displayed different patterns, except for EphA10 (Fig. 2B). The most distinctive feature was that GluOC triggered increased phosphorylation of six RTKs in ProEpi cells, FGFR1 [12], EphB2 [16], EphR [18], ALK [19], RYK [20], and PDGFRα (platelet-derived growth factor receptor alpha) [26], which may be correlated with the proliferative activity triggered by GluOC in normal cells. On the other hand, GlaOC also triggered increased phosphorylation of the same RTKs as GluOC, including FGFR1 [12], EphR [18], ALK [19], and RYK [20]. Additionally, GlaOC promoted the phosphorylation of additional nine RTKs, EphA10 [15], EphB4 [17], EphB6 [27], FGFR2a [28], FGFR3 [22], DDR1 (discoidin domain receptor 1) [29], VEGFR3 [30], Tie-2 [31], and TrkA [32] (Fig 2B). The mechanisms for GluOC to promote the growth in normal cells, likely by activation of tyrosine kinases are currently unknown.
These findings indicate that serum OC may contribute to prostate tissue homeostasis by regulating the RTK phosphorylation status, and that GlaOC and GluOC are discriminatively involved in prostate cell proliferation. In order to advance future clinical studies, GlaOC and GluOC have to be considered discriminatively.
Acknowledgements
This work was supported by the Japan Society for the Promotion of Science (KAKENHI grants 24229009 to M.H., 26861554 and 16K11496 to T.K-Y., and 26861553 and 16K20421 to A.M.).
Competing Interests
The authors have declared that no competing interest exists.
References
1. Gardner TA, Lee SJ, Lee SD, Li X, Shirakawa T, Kwon DD, Park RY, Ahn KY, Jung C. Differential expression of osteocalcin during the metastatic progression of prostate cancer. Oncol Rep. 2009;21:903-908
2. Koizumi M, Yonese J, Fukui I, Ogata E. Metabolic gaps in bone formation may be a novel marker to monitor the osseous metastasis of prostate cancer. J Urol. 2002;165:1863-1866
3. Nimptsch K, Rohrmann S, Nieters A, Linseisen J. Serum undercarboxylated osteocalcin as biomarker of vitamin K intake and risk of prostate cancer: a nested case-control study in the Heidelberg cohort of the European prospective investigation into cancer and nutrition. Cancer Epidemiol Biomarkers Prev. 2009;18:49-56
4. Hagberg Thulin M, Jennbacken K, Damber JE, Welén K. Osteoblasts stimulate the osteogenic and metastatic progression of castration-resistant prostate cancer in a novel model for in vitro and in vivo studies. Clin Exp Metastasis. 2014;31:269-283
5. Pi M, Quales LD. GPRC6A regulates prostate cancer progression. Prostate. 2012;72:399-409
6. Lee NK, Sowa H, Hinoi E, Ferron M, Ahn JD, Confavreux C. et al. Endocrine regulation of energy metabolism by the skeleton. Cell. 2007;130:456-469
7. Mizokami A, Yasutake Y, Gao J, Matsuda M, Takahashi I, Takeuchi H. et al. Osteocalcin induces release of glucagon-like peptide-1 and thereby stimulates insulin secretion in mice. PLoS One. 2013;8:e57375
8. Mizokami A, Yasutake Y, Higashi S, Kawakubo-Yasukochi T, Chishaki S, Takahashi I. et al. Oral administration of osteocalcin improves glucose utilization by stimulating glucagon-like peptide-1 secretion. Bone. 2014;69:68-79
9. Kawakubo-Yasukochi T, Kondo A, Mizokami A, Hayashi Y, Chishaki S, Nakamura S. et al. Maternal oral administration of osteocalcin protects offspring from metabolic impairment in adulthood. Obesity. 2016;24:895-907
10. Otani T, Mizokami A, Hayashi Y, Gao J, Mori Y, Nakamura S. et al. Signaling pathway for adiponectin expression in adipocytes by osteocalcin. Cell Signal. 2015;27:532-544
11. Lemmon MA, Schlessinger J. Cell signaling by receptor tyrosine kinases. Cell. 2010;141:1117-1134
12. Yang F, Zhang Y, Ressler SJ, Ittmann MM, Ayala GE, Dang TD. et al. FGFR1 is essential for prostate cancer progression and metastasis. Cancer Res. 2013;73:3716-3724
13. Batson J, Maccarthy-Morrogh L, Archer A, Tanton H, Nobes CD. EphA receptors regulate prostate cancer cell dissemination through Vav2-RhoA mediated cell-cell repulsion. Biol Open. 2014;3:453-462
14. Guan M, Xu C, Zhang F, Ye C. Aberrant methylation of EphA7 in human prostate cancer and its relation to clinicopathologic features. Int J Cancer. 2009;124:88-94
15. Nagano K, Yamashita T, Inoue M, Higashisaka K, Yoshioka Y, Abe Y. et al. Eph receptor A10 has a potential as a target for a prostate cancer therapy. Biochem Biophys Res Commun. 2014;450:545-549
16. Kittles RA, Baffoe-Bonnie AB, Moses TY, Robbins CM, Ahaghotu C, Huusko P. et al. A common nonsense mutation in EphB2 is associated with prostate cancer risk in African American men with a positive family history. J Med Genet. 2006;43:507-511
17. Mertens-Walker I, Fernandini BC, Maharaj MS, Rockstroh A, Nelson CC, Herington AC. et al. The tumour-promoting receptor tyrosine kinase, Eph4B, regulates expression of integrin-β8 in prostate cancer cells. BMC Cancer. 2015;15:164
18. Lisle JE, Mertens-Walker I, Rutkowski R, Herington AC, Stephenson SA. Eph receptors and their ligands: Promising molecular biomarkers and therapeutic targets in prostate cancer. Biochim Biophys Acta. 2013;1835:243-257
19. Barreca A, Lasorsa E, Riera L, Machiorlatti R, Piva R, Ponzoni M. et al. Anaplastic lymphoma kinase in human cancer. J Mol Endocrinol. 2011;47:R11-R23
20. Leung YK, Gao Y, Lau KM, Zhang X, Ho SM. ICI 182,780-regulated gene expression in DU145 prostate cancer cells is mediated by estrogen receptor-beta/NFkappaB crosstalk. Neoplasia. 2006;8:242-249
21. Cecchi F, Rabe DC, Bottaro DP. Targeting the HGF/Met signaling pathway in cancer therapy. Expert Opin Ther Targets. 2012;16:553-572
22. Hernàndez S, de Muga S, Agell L, Juanpere N, Esgueva R, Lorente JA. et al. FGFR3 mutations in prostate cancer: association with low-grade tumors. Mod Pathol. 2009;22:848-856
23. Dawson DM, Lawrence EG, MacLennan GT, Amini SB, Kung HJ, Robinson D. et al. Altered expression of RET proto-oncogene product in prostatic intraepithelial neoplasia and prostate cancer. J Natl Cancer Inst. 1998;90:519-523
24. Rebagay G, Yan S, Lui C, Cheung NK. ROR1 and ROR2 in human malignancies: potentials for targeted therapy. Front Oncol. 2012;2:34
25. Paccez JD, Vogelsang M, Parker MI, Zerbini LF. The receptor tyrosine kinase Axl in cancer: biological functions and therapeutic implications. Int J Cancer. 2014;134:1024-1033
26. Lui Q, Jernigan D, Zhang Y, Fatatis A. Implication of platelet-derived growth factor alpha in prostate cancer skeletal metastasis. Chin J Cancer. 2011;30:612-619
27. Mohamed ER, Noguchi M, Hamed AR, Eldahshoury MZ, Hammady AR, Salem EE. et al. Reduced expression of erythropoietin-producing hepatocyte B6 receptor tyrosine kinase in prostate cancer. Oncol Lett. 2015;9:1672-1676
28. Huang Y, Hamana T, Lui J, Wang C, An L, You P. et al. Type 2 fibroblast growth factor receptor signaling preserves stemness and prevents differentiation of prostate stem cells from the basal compartment. J Biol Chem. 2015;290:17753-17761
29. Valiathan RR, Marco M, Leitinger B, Kleer CG, Fridman R. Discoidin domain receptor tyrosine kinases: new players in cancer progression. Cancer Metastasis Rev. 2012;31:295-321
30. Goodyear SM, Kheyfets SB, Garcia FU, Stearns ME. Role of the VEGFR3/VEGFD receptor axis in TGFbeta1 activation of primary prostate cell lines. Prostate. 2009;69:982-990
31. Morrissey C, Dowell A, Koreckij TD, Nguyen H, Lakely B, Fanslow WC. et al. Inhibition of angiopoietin-2 in LuCap 23.1 prostate cancer tumors decreases tumor growth and viability. Prostate. 2010;70:1799-1808
32. Festuccia C, Gravina GL, Muzi P, Millimaggi D, Dolo V, Vicentini C. et al. Her2 crosstalks with TrkA in a subset of prostate cancer cells: rationale for a guided dual treatment. Prostate. 2009;69:337-345
Author contact
![]() Corresponding author: Masato Hirata DDS, PhD, Laboratory of Molecular and Cellular Biochemistry, Faculty of Dental Science, Kyushu University, Fukuoka 812-8582, Japan. Tel: +81-92-642-6317, Fax: +81-92-642-6322, Email: hirata1kyushu-u.ac.jp
Corresponding author: Masato Hirata DDS, PhD, Laboratory of Molecular and Cellular Biochemistry, Faculty of Dental Science, Kyushu University, Fukuoka 812-8582, Japan. Tel: +81-92-642-6317, Fax: +81-92-642-6322, Email: hirata1kyushu-u.ac.jp