Impact Factor ISSN: 1837-9664

 Global reach, higher impact
Global reach, higher impactJ Cancer 2016; 7(12):1632-1644. doi:10.7150/jca.13123 This issue Cite
Research Paper
Anti-inflammatory and Antitumor Activity of a Triple Therapy for a Colitis-Related Colorectal Cancer
Gabriela Figueroa-González1,2, Verónica García-Castillo1, Jossimar Coronel-Hernández1, Eduardo López-Urrutia1, Sonia León-Cabrera1,3, Luis E Arias-Romero1, LI Terrazas1, Miriam Rodríguez-Sosa1, Alma Delia Campos-Parra2, Eduardo Zúñiga-Calzada4, Cesar Lopez-Camarillo5, Fermín Morales-González6, Nadia J Jacobo-Herrera7 ![]() , Carlos Pérez-Plasencia1,2
, Carlos Pérez-Plasencia1,2 ![]()
1. Unidad de Biomedicina, FES-Iztacala, Universidad Nacional Autónoma de México. Av. De Los Barrios, Los Reyes Iztacala, 54090 Tlalnepantla, Estado de México, México.
2. Laboratorio de Genómica, Dirección de Investigación, Instituto Nacional de Cancerología. Av. San Fernando 22, Tlalpan, Sección XVI, 14080 Ciudad de México.
3. Carrera de Médico Cirujano, FES-Iztacala, Universidad Nacional Autónoma de México. Av. De Los Barrios, Los Reyes Iztacala, 54090 Tlalnepantla, Estado de México, México.
4. Departamento de Anatomía Patológica, Instituto Nacional de Seguridad Social del Estado de México y Municipios, Avenida del trabajo s/n, 55000, Ecatepec de Morelos, Estado de México.
5. Programa de Ciencias Genómicas, UACM, Ciudad de México.
6. Dirección Médica, Instituto Jalisciense de Cancerología. Coronel Calderón 715, Guadalajara, 44280 Jalisco.
7. Bioquímica, Instituto Nacional de Ciencias Médicas y Nutrición “Salvador Zubirán”, Vasco de Quiroga 15, Tlalpan, Sección XVI, 14000, Ciudad de México.
Received 2015-7-2; Accepted 2016-5-24; Published 2016-7-25
Abstract
Colorectal cancer (CRC) is an important health issue worldwide, accounting for the third place of cancer incidence. Chronic inflammation, as seen in Crohn's disease and ulcerative colitis, is the most important risk factor for developing CRC, as it favours neoplastic transformation by enhancing epithelial cell turnover in the colonic mucosa. Treatments for CRC need to be improved; currently they are not specific and have several secondary effects in patients. The main objective of this work was to evaluate a new therapeutic strategy against a colitis-related colorectal cancer in vivo and in vitro by targeting mTOR-signaling and lactate dehydrogenase A. Together, these mechanisms directly affect tumor energetics. In this study we evaluated a better and more efficient triple therapy against a chronic inflammation-associated CRC in vivo and in vitro. After the development of tumors, mice were treated intraperitoneally during a forty-day period with single drugs or different combinations of Metformin, Sodium Oxamate and Doxorubicin.
Targeted inhibition of the mTOR pathway, lactate dehydrogenase A and the concurrent use of Doxorubicin (called in this work as triple therapy), leaded to a notable reduction in the number and size of tumors in mice, and, a significant pro-inflammatory cytokines reduction Besides, we showed that treated cells were induced to early autophagy, and apoptosis cell death.
Our results represent a novel and robust therapeutic strategy for overcoming CRC by means of targeting central molecular pathways in cancer by the combination of Metformin, Oxamate, and Doxorubicin leading to a rapid tumor growth inhibition and a dramatic colorectal crypt restoration. Besides, drug combination resulted in a notable reduction of anti-inflammatory cytokines.
Keywords: Colorectal Cancer, Chronic Inflammation, Glycolysis, mTOR, Autophagy.
Introduction
Colorectal cancer (CRC) is the third most common cancer in men (746,000 cases, 10.0% of the total) and the second in women (614,000 cases, 9.2% of the total) worldwide [1]; chronic inflammation is the most important risk factor for its development as it favors neoplastic transformation by enhancing epithelial cell turnover in the colonic mucosa. This chronic condition has emerged as the seventh hallmark of cancer affecting any stage of tumorigenesis since genetic events and inflammatory conditions facilitate cancer development because of infections as well as autoimmune diseases such as inflammatory bowel disease for colon cancer [2]. The key elements where genetic events and inflammatory conditions converge to facilitate cancer include transcription factors such as NF-κB and STAT-3, primary pro-inflammatory cytokines and chemokines, that promote the survival of cancer cells, mediate drug-resistance, and perturb their differentiation [3]. Thus, cancer-related inflammation also represents a target for innovative therapeutic strategies and prevention providing further interest for studies targeted to the inflammatory microenvironment of tumors.
Conventional therapies including surgery, current chemotherapy such as 5-Fluorouracil, Oxaliplatin, Doxorubicin, among others, and radiotherapy are frequently inadequate due to their side effects for instance cytotoxicity, chemo- and radio-resistance, failure in treatment efficacy, and potency [3]. Currently important efforts from scientific community and pharmaceutical companies are focused in searching for new treatment options targeting key hallmarks of cancer as well as in the use of combinatorial therapies or novel agents for attacking multiple target simultaneously making a more efficient therapy [4].
In the current work, we were interested to establish a new combinatorial drug therapy aimed to target DNA synthesis, aerobic glycolysis and mTOR pathway as hallmarks of cancer. Therefore, we employed doxorubicin (anthracycline antitumor antibiotic), used to treat many types of cancer, that intercalates between base pairs in the DNA helix and preventing DNA replication, as well as inhibiting topoisomerase II which leads to cell death by apoptosis [5]. Metformin (the most commonly prescribed drug for type II diabetes) has been found to inhibit cancer development and tumor growth through yet undescribed mechanisms, but some reports had suggested the activation of AMP-activated protein kinase (AMPK) which negatively regulates (Mammalian Target of Rapamycin) mTOR activation [6] both drugs, have been previously reported as inducers of apoptosis and autophagy [7]. Autophagy is an evolutionarily conserved mechanism, a multi-step lysosomal degradation process in which cells degrade long-lived proteins and damaged organelles, thus, the principal reason because autophagy occurs is a nutrient deficiency [8]. Recently, it has been proposed that Metformin also may block a metabolic stress response that stimulates the inflammatory pathway associated with a wide variety of cancers [9].
Finally, we reasoned that addition of sodium oxamate could enhance the antitumor effects of metformin and doxorubicin by affecting the ability of the tumor cells to proliferate under hypoxia with a final induction of apoptosis, autophagy and inhibition of LDHA which could lead to aerobic glycolysis inhibition as well [10] [11]. Sodium oxamate is a LDHA specific inhibitor which induces G2/M cell cycle arrest and promote apoptosis through enhancement of mitochondrial ROS generation [12]. Preventing the conversion of pyruvate to lactate can potentially produce changes in tumor microenvironment that possibly overcome cancer cell proliferation, metastasis, and inhibition of the immune response [13] [14]. LDHA (lactate dehydrogenase-A) is a key enzyme regulating this process and is over-expressed in virtually all cancer types [15]. It is now known that, despite its seemingly secondary role in the primary energetic metabolism by catalyzing the final step of aerobic glycolysis [15], the inhibition of LDHA can block tumor growth, maintenance and progression [10]. Therefore, sodium oxamate blocks aerobic glycolysis as previous reports have shown [16]. We hypothesized that the combination of Metformin - Sodium Oxamate - Doxorubicin (Triple Therapy) could enhance the efficacy of chemotherapy through an anti-cancer synergistic effect by targeting deregulation of cellular energetics, genome instability and mutation as well as tumor-promoting inflammation [4].
No previous studies have addressed the anti-cancer effect of Metformin - Sodium Oxamate - Doxorubicin in combination, so the main objective of this study was to evaluate this triple therapy as a better and more efficient antitumor treatment against a colitis-related CRC in vivo and in vitro. Our results demonstrated that the proposed triple therapy had the ability to reduce dramatically tumor growth by means of inhibition of the phosphorylated-mTOR, autophagy induction, glycolysis inhibition, and apoptosis cell death. According to inflammation-related cytokine profiles, we also found significant changes in pro-inflammatory IL-1β, TNFα, INFγ and IL-6 cytokines; taken together all this mechanisms affected the survival of cancer cells, restored the epithelial tissue structure and the anti-inflammatory environment. These results highlight the importance of targeting aberrant energetic metabolism of cancer cells and searching for novel antineoplastic drugs as promising strategies to have a clinical application in a near future.
Methods
In vivo studies
Twenty-four female Balb/c mice (Harlan Laboratories, México) aged 6 weeks were used in this study. They were maintained at Facultad de Estudios Superiores Iztacala Animal Facility according to the institutional animal care guidelines. All animals were housed in plastic cages (6 mice/cage) with drinking water and pelleted basal diet ad libitum under controlled humidity (50±10%), light (12/12 h light/dark cycles) and temperature (23±2°C). They were quarantined for the first 7 days, and then randomized by body weight into three experimental and one control groups. All groups were administered with a single intraperitoneal injection (12.5 mg/kg body weight) with the colonic carcinogen Azoxymethane (AOM) from Sigma Aldrich (USA). One week after the injection, animals received Dextran Sulfate Sodium Salt Reagent Grade (DSS) with a molecular weight of 36,000-50,000 from MP Biomedicals (México) for induction of colitis, dissolved in drinking water at a concentration of 2% (w/v) for 7 days and no further treatment for two weeks, then the DSS cycle was repeated twice.
Pharmacological treatments began after the second DSS cycle, and were administered daily during 40 days under the following scheme: Group 1: negative control, SSI (saline solution isotonic); Group 2: Doxorubicin 1 mg/Kg (positive control); Group 3: Metformin 200 mg/kg - Sodium Oxamate 15 mg/kg and Group 4: Metformin 200 mg/kg - Sodium Oxamate 15 mg/kg - Doxorubicin 1 mg/kg. Groups with individual treatments were not included due to previous experiments in our research laboratory that does not show any difference with the control groups, because of that, there were no reasonable justification to include these experimental groups according to the Guide for care and use of laboratory animals[17]. Euthanasia was carried out on each group at the end of the study and the large bowels were flushed with saline, and excised. Large bowels length were measured (from the illeocecal junction to the anal verge), and they were cut open longitudinally along the main axis, and then washed with PBS. Large bowels were macroscopically inspected and cut for selection of the medium part and stored at -80°C for further assays. The number and the size of the tumors were measured prior to the collection for the histological analyses. Tumor volume was calculated using the formula 0.52 x (width) 2 x (length). Doxorubicin was obtained commercially as Doxolem RU, Metformin and Sodium Oxamate were purchased from Santa Cruz Biotechnology (sc-202000A and sc-215880A respectively).
Histology and Immunohistochemistry
Tissue specimens were fixed in 10% formalin and histological examination was performed on paraffin-embedded sections followed by hematoxylin and eosin (H&E) as well as immunohistochemical staining. Ki67 and HER-2 overexpression analyses were conducted on 4-µm-thick tissue sections. A primary monoclonal rabbit antibodies against the Ki67 (Clone SP6; GeneTex, CA, USA) and the ErbB2/Her2 (SP3) proteins (Clone SP3; Novus Biologicals, CO, USA) were used. Slides were examined by standard light microscopy using a Standard 25 microscope (Carl Zeiss, Germany) and images were taken with a Canon G9 color camera coupled to the microscope.
The histologic classification was staged with the recommended nomenclature for intestinal neoplasia in mouse models [18]. The nomenclature parallels that used for humans. For immunohistochemical HER-2 analysis, membrane was evaluated on a score of 0 to 3+ using the score system suggested by Hoffman et.al.[19]. For Ki67, nuclear cell staining is taken as a positive reaction, and the results were expressed as percentage of positive cells of 100 counted cells. The results were grouped in 4 groups according to the percentage of stained cells: (1) <=10%; (2) 11% to 30%; (3) 31% to 50%; and (4) >50% positive cells.
Sections were examined separately by two independent pathologists, without any prior knowledge of each experimental group.
Cytokine Profiles
Protein. Large bowels were homogenized in RIPA buffer (by triplicated) using Bullet Blender and stainless steel beads (Next Advance, Inc. NY, USA). The cell debris were eliminated by centrifugation at 12,000 rpm for 15 min. Pro-inflammatory and anti-inflammatory cytokines were measured in colon homogenates using the Bio-Plex mouse Cytokine eight-plex Assay (Bio-Rad Laboratories, CA, USA) on a MagPix reader (Luminex, Bio-Rad, CA, USA). Only significant changes are shown.
mRNA. Total RNA was isolated from colonic tissues by Trizol reagent and homogenized in a Bullet Blender. cDNA was obtained by using a High Capacity cDNA Reverse Transcription kit (Applied Biosystems, CA, USA). mRNA expression for IFN-γ, TNF-α, IL-6, IL-4 and IL-10 was performed in a CFX96 Touch Real-Time PCR Detection System (BioRad) with a SYBR Select Master Mix for CFX (Applied Biosystems, CA, USA) and according to the manufacturer's instructions. Triplicate RT samples were utilized in each assay; data were normalized with ß-actin housekeeping gene. The comparative ΔΔCt method was used to quantify gene expression and the relative quantification was calculated as 2-ΔΔCt. The sequence of primers was as follows: for IL-6: forward 5´-GAGTCACAGAAGGAGTGGC-3' and reverse 5'-GACCACAGTGAGGAATGTCCA-3'; IL-4: forward 5'-TGACCTACTAGGAACCCAGGC-3' and reverse 5'-CTCGGCGCACTGACCCATCT-3'; IFN-γ: forward 5'-AGCGGCTGACTGAACTCAGATTGTAG-3', and reverse 5'-GTCACAGTTTTCAGCTGTATAGG-3'; IL-10: forward 5'-ACCTGGTAGAAGTGATGCCCCAGGCA-3', and reverse 5'-CTATGCAGTTGATGAAGATGTCAAA-3'; TNF-α: forward 5'-GGCAGGTCTACTTTGGAGTCATTGC-3', and reverse 5'-ACATTCGAGGCTCCAGTGAATTCGG-3'; β-actin: forward 5' GTGACGTTGACATCCGTAAA-3' and reverse 5'-AAAACGCAGCTCAGTAACAG-3'.
Cell Line studies
A primary culture from the mice colon cancer was developed in our laboratory through chopping the tissue finely with sharp instruments to cause minimum damage and rinse, then the pieces were seeded onto the surface of a petri dish in a small volume , this culture was maintained in RPMI medium supplemented with 10% FBS (Corning, USA) as previously reported to tumor primary culture [20]. Cells were cultured at 37 °C in an atmosphere of 5% CO2 in air (100% humidity). Treatments consisted in Metformin (25 mM), Sodium Oxamate (10 mM), Doxorubicin (0.5 μM), and no-treatment cells as negative control for 4, 8, 12 and 24h.
Western Blots
After pharmacological treatments, total proteins were extracted from primary cultured cells using RIPA buffer (Santa Cruz Biotechnology, CA, USA). From these extracts, 50 μg were separated by SDS polyacrylamide gel electrophoresis and transferred onto a polyvinylidene difluoride (PVDF) membrane (GE Healthcare, USA) in a semidry chamber Trans Blot Turbo (Bio-Rad) at 25 V 1 mA during 30 min. After blocking with 5% non-fat dry milk for 2 h, the membrane was incubated with the specific antibody overnight at 4°C on a rocking platform, washed, and then incubated with the corresponding secondary antibody for 2h at room temperature. The bolt was visualized using the SuperSignal WestFemto chemiluminescencent substrate (Pierce) in the C-DigitTM scanner (LI-COR Inc. USA).
The following antibodies used for apoptosis analysis were from Santa Cruz Biotechnology (Santa Cruz, CA): anti-PARP-1 (sc-8007), anti-caspase-3 (sc-H2777); for autophagy anti-LC3 (#2775) from Cell Signaling Technology, anti-mTOR (#AHO1232) and anti-p-mTOR (pS3448) from Invitrogen, USA; finally, regulation of inflammation was evaluated by NFKB (#8242), p-NFKB (#3033), IKBα (#4814p) and p-IKBα (#2859p) from Cell Signaling. Anti-mouse (sc-2371), anti-goat (sc-2020) and anti-rabbit (sc-2370) secondary antibodies were also purchased from Santa Cruz Biotechnology.
Statistical analysis
All values are expressed as the mean ± SEM. Data were analyzed using a one-way ANOVA analysis followed by Tukey's Multiple Comparison Test. For all statistical analyses, we used the GraphPad PRISM version 5.0 software, with a significance of p<0.05.
Results
Physiological effects on murine model: General observations
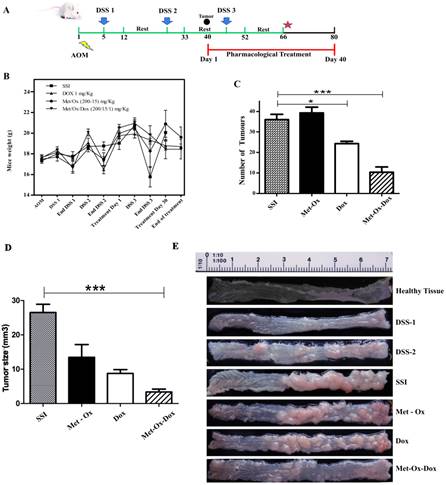
During the development of the in vivo model, bloody stools started to be observed between day 12 to 17 in all groups; those treated with Doxorubicin, Metformin - Sodium Oxamate and negative control showed more physiological changes as hair bristling, anus bleeding as well as anal prolapses due to tumor development in the distal colon. However, no such symptoms were observed in the Metformin - Sodium Oxamate - Doxorubicin group. During the course of the model (Figure 1A), body weight (Figure 1B) and tumor growth was monitored by euthanizing a representative sample of all groups at the end of each DSS cycle. The treatment started 7 days after the second DSS cycle, when tumors were observed in the middle region of the bowel sections (Figure 1A). The body weights of mice from all groups during the complete model, as well as means of the number and size of tumors counted for each experimental group are shown in Figure 1C and 1D, respectively. The mean body weights of groups 1 (SSI) and 3 (Metformin - Sodium Oxamate) were significantly smaller than those of group 2 (Doxorubicin) and 4 (Metformin - Sodium Oxamate - Doxorubicin) (P< 0.001, P< 0.001, P< 0.05 and P< 0.05 respectively).
Met-Ox-Dox treatment inhibits growth of colorectal cancer in an in vivo murine model. A, experimental strategy of the model with days of exposure to Azoxymethane (AOM) and dextran sulfate sodium (DSS), scheme of treatment with the pharmacological proposed therapy; B, C and D, Mice weight monitoring during the experimental plan, average of number and size of tumors after macroscopic inspection of the large bowels at the end of treatments; and E, macroscopic view of the large bowels at the end of the treatment. Met: Metformin; Ox: Sodium Oxamate; Dox: Doxorubicin and SSI: Sodium Saline Isotonic. Significance of * p<0.05; ***p<0.0006.
Significant suppression of chronic inflammation-associated colorectal cancer
For tumor development, all experimental groups were intraperitoneally administered first with a colonic genotoxic carcinogen (AOM) followed by an oral exposure in drinking water of a colitis inductor (DSS). In Figure 1E, the development and tumor progression can be appreciated, monitored representatively by euthanizing mice of each group during the course of the experiment.
In macroscopic comparisons with the healthy tissue, right after the first DSS cycle, little inflammation and color change of the mucosa was observed, showing slight formation of polyps after the second DSS cycle. Higher formation of polypoid-like colonic tumors in the middle and distal colon regions was also observed; which were histologically diagnosed as tubular adenocarcinoma (Figure 2D). Tumor formation increased in parallel with the progress of the model, resulting in a complete obstruction of the bowel at the end of the sixty-six day duration of the experiment with no treatment (Figure 1C-SSI). Seven days after the end of the second DSS dose, pharmacological treatments began as specified in the methods section. At the end of this study (Day 80 of the model); results showed that the group treated with Metfomin - Sodium Oxamate - Doxorubicin (Figure 1C and 1D) had a significantly decreased presence, number and size of tumors in comparison with the control groups. These data suggest that the proposed pharmacological treatment suppresses colon cancer growth.
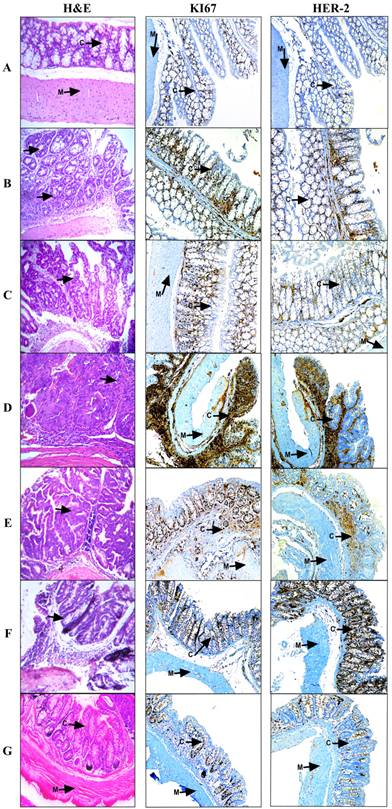
Histological and Immunohistochemical Analysis of the large bowel during the progression of the model and experimental treatments. Samples were collected at the 6th day during the resting period after each DSS dose followed by paraffin inclusion and H&E staining. A, Healthy tissue where m=muscle and c=crypts; B, DSS-1; C, DSS-2; D, SSI or Colorectal Cancer; E, Doxorubicin 1 mg/Kg; F, Metformin - Sodium Oxamate (200-15) mg/Kg and G, Metformin - Sodium Oxamate - Doxorubicin (200-15-1) mg/Kg. 100X magnification. During the curse of the model, arrows shown slight crypt distortion, branched crypts in cross section as well as epithelial hyperplasia, reduction in globet cell numbers until adenoma with intramucosal carcinoma and haphazard proliferation of malignant cells within the lamina propria.
Effects of the treatment in the epithelial tissue structure restoring
The histological and immunohistochemical analyses (Figure 2A) showed a healthy colon tissue, which displays muscle tissue and well-defined crypts lined up in parallel, with their mouths open to the lumen; in contrast, in Figures 2B and 2C chronic intestinal inflammation and slight crypt distortion with epithelial hyperplasia are showed. Tumor progression is appreciated in colon sections with repeated oral DSS cycles with the formation of adenomas (Figure 2B, 2C, 2D), composed of tubular and villous structures lined by epithelium with high grade of dysplasia. Some tumors progressed until tubular adenocarcinomas well differentiated with proliferation of malignant cells within the lamina propria (Figure 2D).
Histological analysis for tissues treated with Doxorubicin (Figure 2E) and Metformin - Sodium Oxamate (Figure 2F) still showed tubular adenomas with a high grade dysplasia, while in the Metformin - Sodium Oxamate - Doxorubicin treatment there was a significant and evident tumor regression effect (Figure 2G).
All these observations support a completely positive effect with the triple therapy against colorectal cancer that suggest an epithelial tissue structure restoring (Figure 2G). At the contrary, the treatment with doxorubicin resulted in no evidence of tumor reduction (Figure 1C and Figure 2E), while the treatment with Metformin - Oxamate presented a minimal and incomplete effect in reduction of tumor growth (Figure 1D and Figure 2F). In contrast with histological analysis, it is noticeable in the immunohistochemical staining (Figure 2A-2D) the increased expression of HER-2 and Ki67 within the CRC model, contrary to the observations after the treatment, where the triple-therapy lowered completely the expression of these proteins (IHC 1+) compared with the group treated with Doxorubicin and Metformin-Oxamate (Figure 2E-2G). For HER-2 protein, IHC 0 was found in all negative controls, IHC 1+ in the first DSS dose, IHC 2+ in the second DSS dose and IHC 3+ in the stablished colorectal cancer (third DSS dose). Different levels of Ki67 expression are showed as follow: control group (1), first DSS dose (3), second DSS dose (2) and for the third DSS dose (4). In the triple therapy group, the expression of Ki67 decreased from the maximum level expression to score 1 being comparable to the negative control. An additional picture of histological and immunohistochemical analysis of the large bowel during the progression of the model and experimental treatments are shown in more detail at 400x magnification [see Supplementary Material].
Anti-inflammatory potential activity of Metformin - Sodium Oxamate - Doxorubicin in combination
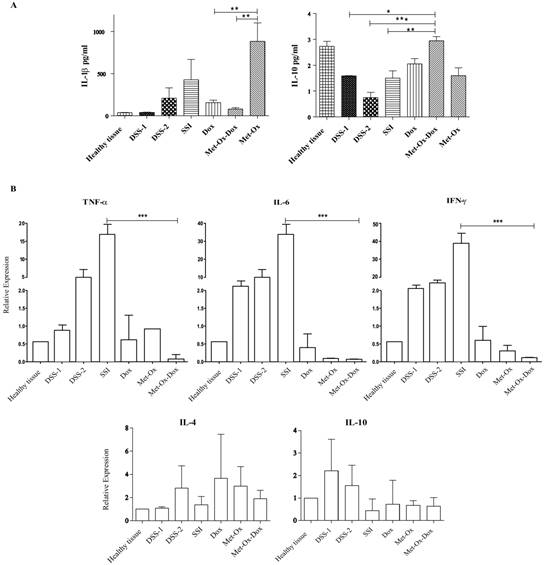
After the histological analysis and the microscopic evaluation of tissue sections; we aimed to analyze the profile of inflammatory cytokines at protein and mRNA level. Hence, tissue samples were collected and homogenized at the 6th day of the resting period after each DSS dose. Our results showed a decrease in the protein IL- 1β, and in the mRNA of genes IL-6, TNFγ, and INFγ (pro-inflammatory cytokines) with Doxorubicin (1 mg/kg); but a major decrease comparable (P<0.001) to healthy tissue and SSI group was observed in the group treated with the triple therapy (Figure 3A and 3B). On the other hand, IL-10, an anti-inflammatory cytokine, showed a significant increase at protein level with the three combined drugs compared to the doxorubicin-treated group and the healthy tissue negative control (p<0.05) (Figure 3A).
Levels of protein cytokines in the experimental groups. Samples were collected at the 6th day of the rest period after each DSS dose followed by RIPA protein and TRIZOL RNA extractions then, Bio-Plex mouse Cytokine eight-plex Assay and qRT-PCR respectively. A, IL-1β pro-inflammatory and IL-10 an anti-inflammatory cytokine. B, TNFα, IL-6, INFγ, IL-4 and IL-10 relative expression compared with the control healthy group. Each bar represents the mean ± SD from three independent experiments. Met: Metformin; Ox: Sodium Oxamate; Dox: Doxorubicin and SSI: Sodium Saline Isotonic.
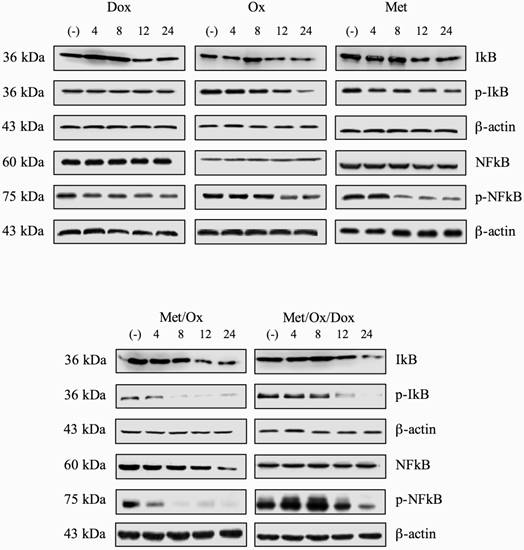
In order to know if the regulation of expression of inflammatory-related cytokine profile was related to activation of key components on inflammation pathway; we assessed the protein phosphorylation levels of NFKB and IKBα two key regulators of inflammation process. However, we were not able to show significant differences between phosphorylation levels of p-NFKB nor p-IKBα (Figure 4) using single drugs treatment. Only Met/Ox combination induced dephosphorylation of IKBα at eight hours post-treatment. Unexpectedly total protein levels for both NFKB and IKBα were unaffected at least during the evaluated period.
Total and phosphorylation protein levels for NFκB and IκB. After exposition to single drugs or combinatorial treatment, no significant change of protein levels for NFκB, p-NFκB, IκB and p-IκB was observed.
Apoptosis and autophagy induction by combinatorial therapy
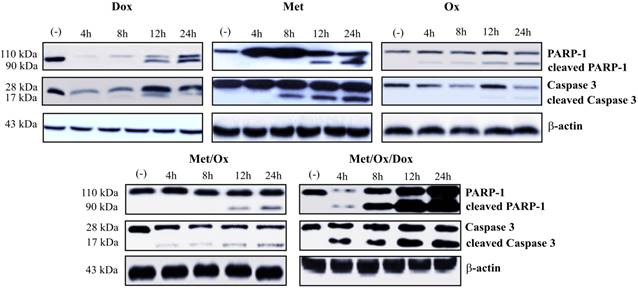
Apoptosis and autophagy protein markers were evaluated by western blot assays. Primary cultured cells were exposed to different drug combinations and Caspase-3, cleaved PARP-1 were assessed to analyze apoptosis. As shown in Figure 5, the single drug treatment with Doxorubicin, Sodium Oxamate or Metformin, did not significantly increase caspase-3 or cleaved PARP-1 detection; whereas the triple therapy showed a considerable up-regulation of cleaved PARP-1 from the 4-hour time point, which corresponds to the detection of cleaved effector caspase 3. As seen in Figure 6, the autophagy process is hyperactivated by the triple therapy; simultaneously, no phosphorylated mTOR was detected, which indicates inhibition of the mTOR pathway.
Metformin - Sodium Oxamate - Doxorubicin increased apoptosis cell death. It is shown a significant increment in the detection of caspase 3 and PARP-1 after exposure to the three drugs in combination. Met: Metformin; Ox: Sodium Oxamate; Dox: Doxorubicin.
Induction of autophagy through the combinatorial therapy. Metformin - Sodium Oxamate - Doxorubicin induced Autophagy, denoted as LC3 conversion (LC3-II) and abscence of p-mTOR from 4 h on to drug exposure. Met: Metformin; Ox: Sodium Oxamate; Dox: Doxorubicin.
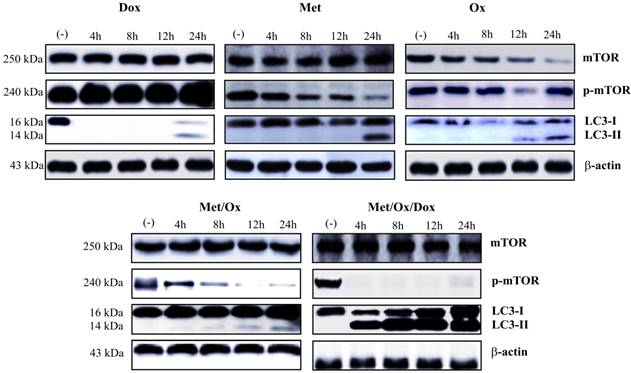
In an attempt to analyze autophagy induction through mTOR pathway inhibition, we evaluated the LC3 conversion by hyper-lipidation to LC3-II. We found a significant increase in the conversion to LC3-II. Interestingly, LC3 conversion and mTOR inhibition were found at the same time point, clearly showing induction of autophagy and an inhibition of mTOR pathway from 4 h on with the triple therapy.
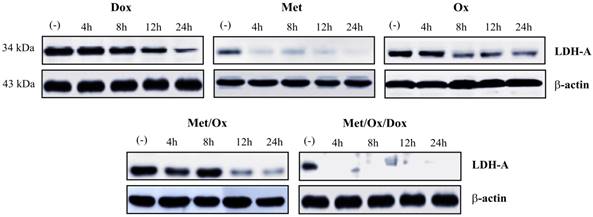
Triple-therapy inhibits LDHA expression
To investigate the expression of LDHA as a glycolysis marker we assessed western blot analysis. It was not observed the presence of LDHA protein since 4 h with the triple therapy; in contrast with the other combinations, that does not show a similar inhibition positive effect (Figure 7).
Combinatorial therapy induced glycolysis inhibition. The suppression of LDH-A enzyme since 4 h to drugs exposure revels the inhibition of the glycolysis pathway. Met: Metformin; Ox: Sodium Oxamate; Dox: Doxorubicin.
Discussion
Animal models of colitis have been developed through the years; but only a model, which develops dysplasia and cancer in a similar way to human ulcerative colitis, is useful for studying complete cancer development though all its stages. Here, we employed such a model -a modification of Tanaka and others [21]- to describe the effect of a novel triple therapy comprising a DNA intercalating agent, an mTOR pathway inhibitor and a Glycolysis (LDHA) inhibitor. We found striking results: the number and size of tumors were reduced as well as the physiological manifestations of disease, tissue regeneration, molecular evidence of enhanced cell death and a decrease of pro-inflamatory citokines.
Anticancer drugs, such as doxorubicin, are inefficient when used as monotherapy. Diverse studies have demonstrated, that there is no reduction of induced colorectal cancer [22, 23]; moreover, they cause important side effects on patients, e.g. cytotoxicity and chemotherapy resistance [24]. We hypothesized that a combination of drugs targeting simultaneously different hallmarks of cancer can enhance the efficacy of chemotherapy against colorectal cancer through a synergistic effect.
Metformin - Sodium Oxamate - Doxorubicin had a substantial effect in colorectal cancer by a significant tumor reduction as well as an epithelial tissue restoring, with a minimum of hyperplasia. After AOM and DSS administration in the animal group that received triple therapy we found remarkable histological features that suggested tumor regression of the colonic damaged mucosa that showed absence of histologically identifiable adenoma and intramucosal carcinoma (Figure 2G-H&E), the crypts also showed regenerative hyperchromatic epithelium that began at the base of the damaged glands. Respecting to lamina propria, it showed low infiltration of inflammatory cells for instance eosinophils. As it is shown in Figure 2, during the progress of the inflammation-related model (Figure 2A-Figure 2D), the distortion of the crypts and hyperplasia grade are increasing, meanwhile in the group administered with the triple therapy it was observed a tumor regression effect and a complete absence of histologically identifiable adenoma and intramucosal carcinoma (Figure 2G). Within the same context, it was observed an overexpression of Ki67 and HER-2, the first one as cell proliferation marker associated with response to therapy led us suggest the potential of lowering the proliferation capability of colorectal cancer. Meanwhile the human epidermal growth factor receptor 2 (HER-2) plays an important role in tumor proliferation, angiogenesis, differentiation and metastasis in CRC, its overexpression in different types of cancer including breast, ovarian, gastric, lung, prostate and colorectal is used as a prognostic marker of more aggressive disease [25]. In comparison with other works where an antibody against HER-2 is conjugated with doxorubicin [26], we observed that the triple therapy has a similar effect where the expression of this oncogene decrease in a considerable magnitude, therefore the proposed combination of drugs can have high impact in the relation cost-benefit for the colorectal chemotherapy.
Previous approaches to Sodium Oxamate - Metformin combinatorial therapy have been performed with acceptable results in antitumoral activity [14], however our results show that a tri-drug combinatorial therapy (Sodium Oxamate - Metformin in synergism with doxorubicin) has similar but broader effects, moreover they have an additional advantage of reducing expression of Ki67 and HER-2 as well as the pro-inflammatory cytokines IL- 1β, IL-6, TNFα, and INFγ.
An important relationship between inflammation and cancer development has been suggested for colorectal cancer. Inflammation is activated by soluble factors, cytokines and chemokines, which can be produced by tumor cells themselves or, more often, by cells recruited to the tumor microenvironment. Inflammatory cytokines and chemokines promote the survival of cancer cells as well as they mediate their resistance and perturb their differentiation [3]. Indeed, the tumor microenvironment represents an attractive target for preventive and therapeutic strategies since toxicity; resistance and treatment failure limit the clinical use of the first-line therapy in various types of cancer. The importance of understanding the molecular mechanisms that underlie cancer progression and drug resistance allows for the identification of crucial targets, some of which can play an important role in promoting inflammation events that may eventually lead to metastasis as well.
Metformin has been reported as an anticancer therapy [27-29]; yet little is known about its anti-inflammatory activity. It has been suggested to alter inflammation independently of its effects on glycemic control, although its mechanism remains to be fully elucidated, its anti-inflammatory effects have been reported to inhibited the production of pro-inflammatory cytokines in endothelial cells [30].
On the other hand, some other reports have shown that doxorubicin treatment induces inflammation in various cancer cells -such as urothelial cell lines- which after been exposed to doxorubicin display high levels of prostaglandin E2 and IL- 1β [31]. Our results in this regard, showed the opposite (i.e. IL-1β downregulation) most likely owing to different cellular origin of the samples. These results constitute novel findings for overcoming inflammation-related colorectal cancer.
Pro-inflammatory cytokines, such as TNFα, IL-6 and IL-1β, or transcription factors that are required for signaling by these cytokines, including NF-κB and STATs, are emerging as potential targets for anticancer therapy. Therefore, development of drugs that restore the tumor microenvironment or interrupt the crosstalk between it and the tumor itself constitute an important approach to the management of cancer. As seen in Figure 3, inflammatory-related cytokine profile with the triple-drug treatment was comparable and even better than the profile of healthy tissue.
This result further reinforces the relevance of this novel proposed triple therapy in overcoming the colitis-related colorectal cancer, as it restores the pro-inflammatory cytokine profile in addition to the aforementioned decrease in the number of tumors; therefore, this therapy might also be a perspective for controlling some other inflammation-related diseases such as IBD. It is noticeable that among components of inflammatory process and malignant transformation are influenced by the pro-inflammatory and anti-inflammatory cytokines such as the ones we observed were modified with the triple therapy (IL-1β, IL-6, TNFα, INFγ and IL-10 respectively). It has been reported previously the main role of IL-1β in the regulation of the inflammation process and its clear relation with the COX-2 enzyme levels [32], moreover, this pro-inflammatory cytokine takes part in the development of metastasis by the stimulation of metalloproteinases [33]. Therefore, it is feasible to speculate that this proposed triple-therapy also is useful for reducing the metastasis process.
Regarding induction of cleaved PARP-1 and caspase 3, our findings suggest the activation of apoptotic cell death, since PARP-1 is specifically proteolysed by caspases to 89 kDa catalytic fragment during the apoptotic program; moreover, its overactivation causes the energy depletion that triggers necrosis in response to high levels of DNA damage [34].
Similar to that of apoptosis, malfunction or deregulation of autophagy may also contribute to cancer disease because of its critical role for a pro-survival response during starvation, as well as for removing damaged and dysfunctional organelles and foreign particles, including microorganisms, and for maintaining tissue development, differentiation and homoeostasis [35]. An over-activation of this process may result in cell death and can be regulated through signaling pathways that promote cell proliferation and cellular metabolism such as the mTOR sensor, whose activity is directly proportional to the amount of available nutrients and thus, may or may not promote the activation and initiation of autophagy [36]. In addition, metformin, commonly used as an oral anti-hyperglycemic drug for the treatment of type 2 diabetes mellitus, can provoke starvation conditions, the consequent activation of autophagy [37], and a synergistic effect with the triple therapy through inhibition of mTOR. Autophagy was dramatically induced by LC3 conversion (LC3-I to LC3-II) and mTOR inhibition (detected as p-mTOR depletion) as noted in figure 6; this result can be supported in previous reports where the LC3 conversion is directly proportional to the number of autophagosomes showing the autophagy begins [38].
Concerning the glycolysis pathway -which is related to the deregulating cellular energetics, we observed a complete inhibition of the LDHA (Figure 6), the most important glycolysis-related enzyme and a potential prognostic marker in solid cancer types [39]. This is consistent with previous reports where knock-down of LDHA mediated tumor growth and metastatic effects in human hepatocellular carcinoma [40], and shows the potential of the triple therapy as a wide-spectrum cancer inhibitor.
Together, our studies showed that treatment of colitis-related colorectal cancer with the triple therapy of Metformin - Sodium Oxamate - Doxorubicin resulted in significant inhibition of tumor cell growth in vivo by affecting mTOR and glycolysis pathways as well as pro- and anti-inflammatory cytokines. Moreover, we were able to show an important tissue structure restoring. Although the anti-tumor effect of the Metformin - Sodium Oxamate - Doxorubicin combination is clearly demonstrated in this work, our experiments assess its response to two hallmarks of cancer: 'cellular energetics and 'tumor-promoting inflammation'. The response to 'genome instability and mutation', most likely related to the DNA-intercalating characteristics of doxorubicin, remains to be described in detail in a further work. In conclusion, we propose this triple therapy as a novel and potential candidate strategy for overcoming inflammatory-related colorectal cancer.
Supplementary Material
Supplementary figure.
Acknowledgements
G. F-G. Is grateful to Dirección General de Asuntos del Personal Académico for a postdoctoral scholarship.
Competing Interests
The authors declare that they have no competing interests.
References
1. Ferlay J SI, Ervik M, Dikshit R, Eser S, Mathers C, Rebelo M, Parkin DM, Forman D, Bray F. GLOBOCAN 2012 v1.0, Cancer Incidence and Mortality Worldwide: IARC CancerBase No. 11 [Internet]. Lyon, France: International Agency for Research on Cancer. 2013 http://globocan.iarc.fr
2. Colotta F, Allavena P, Sica A, Garlanda C, Mantovani A. Cancer-related inflammation, the seventh hallmark of cancer: links to genetic instability. Carcinogenesis. 2009;30(7):1073-81
3. Vyas D, Laput G, Vyas AK. Chemotherapy-enhanced inflammation may lead to the failure of therapy and metastasis. Onco Targets Ther. 2014;7:1015-23
4. Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144(5):646-74
5. Yang F, Teves SS, Kemp CJ, Henikoff S. Doxorubicin, DNA torsion, and chromatin dynamics. Biochim Biophys Acta. 2014;1845(1):84-9
6. Kisfalvi K, Eibl G, Sinnett-Smith J, Rozengurt E. Metformin disrupts crosstalk between G protein-coupled receptor and insulin receptor signaling systems and inhibits pancreatic cancer growth. Cancer Res. 2009;69(16):6539-45
7. Leclerc GM, Leclerc GJ, Kuznetsov JN, DeSalvo J, Barredo JC. Metformin induces apoptosis through AMPK-dependent inhibition of UPR signaling in ALL lymphoblasts. PLoS One. 2013;8(8):e74420
8. Fu LL, Wen X, Bao JK, Liu B. MicroRNA-modulated autophagic signaling networks in cancer. Int J Biochem Cell Biol. 2012;44(5):733-6
9. Hirsch HA, Iliopoulos D, Struhl K. Metformin inhibits the inflammatory response associated with cellular transformation and cancer stem cell growth. Proc Natl Acad Sci U S A. vol. 110. 2013:972-7
10. Oermann EK, Wu J, Guan KL, Xiong Y. Alterations of metabolic genes and metabolites in cancer. Semin Cell Dev Biol. 2012;23(4):370-80
11. Le A, Cooper CR, Gouw AM, Dinavahi R, Maitra A, Deck LM, Royer RE, Vander Jagt DL, Semenza GL, Dang CV. Inhibition of lactate dehydrogenase A induces oxidative stress and inhibits tumor progression. Proc Natl Acad Sci U S A. 2010;107(5):2037-42
12. Zhai X, Yang Y, Wan J, Zhu R, Wu Y. Inhibition of LDH-A by oxamate induces G2/M arrest, apoptosis and increases radiosensitivity in nasopharyngeal carcinoma cells. Oncol Rep. 2013;30(6):2983-91
13. Walenta S, Mueller-Klieser WF. Lactate: mirror and motor of tumor malignancy. Semin Radiat Oncol. 2004;14(3):267-74
14. Miskimins WK, Ahn HJ, Kim JY, Ryu S, Jung YS, Choi JY. Synergistic anti-cancer effect of phenformin and oxamate. PLoS One. 2014;9(1):e85576
15. Miao P, Sheng S, Sun X, Liu J, Huang G. Lactate dehydrogenase A in cancer: a promising target for diagnosis and therapy. IUBMB Life. 2013;65(11):904-10
16. Fantin VR, St-Pierre J, Leder P. Attenuation of LDH-A expression uncovers a link between glycolysis, mitochondrial physiology, and tumor maintenance. Cancer Cell. 2006;9(6):425-34
17. Academies NRCotN. Guide for care and use of laboratory animals. In Eighth Edition edn. Washington, D.C: The National Academy Press. 2011
18. Boivin GP, Washington K, Yang K, Ward JM, Pretlow TP, Russell R, Besselsen DG, Godfrey VL, Doetschman T, Dove WF. et al. Pathology of mouse models of intestinal cancer: consensus report and recommendations. Gastroenterology. 2003;124(3):762-77
19. Hofmann M, Stoss O, Shi D, Buttner R, van de Vijver M, Kim W, Ochiai A, Ruschoff J, Henkel T. Assessment of a HER2 scoring system for gastric cancer: results from a validation study. Histopathology. 2008;52(7):797-805
20. Freshney I. Primary Culture. Culture of Animal Cells: A Manual of Basic Technique and Specialized Applications; Fifth Edition edn. John Wiley & Sons, Inc. 2005:580
21. Tanaka T. A novel inflammation-related mouse colon carcinogenesis model induced by azoxymethane and dextran sodium sulfate. Cancer Sci. 2003 94(11)
22. Doublier S, Riganti C, Voena C, Costamagna C, Aldieri E, Pescarmona G, Ghigo D, Bosia A. RhoA silencing reverts the resistance to doxorubicin in human colon cancer cells. Mol Cancer Res. 2008;6(10):1607-20
23. Ai S, Jia T, Ai W, Duan J, Liu Y, Chen J, Liu X, Yang F, Tian Y, Huang Z. Targeted delivery of doxorubicin through conjugation with EGF receptor-binding peptide overcomes drug resistance in human colon cancer cells. Br J Pharmacol. 2013;168(7):1719-35
24. Gottesman MM. Mechanisms of cancer drug resistance. Annu Rev Med. 2002;53:615-27
25. Tu J, Yu Y, Liu W, Chen S. Significance of human epidermal growth factor receptor 2 expression in colorectal cancer. Exp Ther Med. 2015;9(1):17-24
26. Li Q, Tang Q, Zhang P, Wang Z, Zhao T, Zhou J, Li H, Ding Q, Li W, Hu F. et al. Human epidermal growth factor receptor-2 antibodies enhance the specificity and anticancer activity of light-sensitive doxorubicin-labeled liposomes. Biomaterials. 2015;57:1-11
27. Bo S, Ciccone G, Rosato R, Villois P, Appendino G, Ghigo E, Grassi G. Cancer mortality reduction and metformin: a retrospective cohort study in type 2 diabetic patients. Diabetes Obes Metab. 2012;14(1):23-9
28. Queiroz EA, Puukila S, Eichler R, Sampaio SC, Forsyth HL, Lees SJ, Barbosa AM, Dekker RF, Fortes ZB, Khaper N. Metformin induces apoptosis and cell cycle arrest mediated by oxidative stress, AMPK and FOXO3a in MCF-7 breast cancer cells. PLoS One. 2014;9(5):e98207
29. Nangia-Makker P, Yu Y, Vasudevan A, Farhana L, Rajendra SG, Levi E, Majumdar AP. Metformin: a potential therapeutic agent for recurrent colon cancer. PLoS One. 2014;9(1):e84369
30. Huang NL, Chiang SH, Hsueh CH, Liang YJ, Chen YJ, Lai LP. Metformin inhibits TNF-alpha-induced IkappaB kinase phosphorylation, IkappaB-alpha degradation and IL-6 production in endothelial cells through PI3K-dependent AMPK phosphorylation. Int J Cardiol. 2009;134(2):169-75
31. Kang SH, Chess-Williams R, Anoopkumar-Dukie S, McDermott C. Induction of inflammatory cytokines and alteration of urothelial ATP, acetylcholine and prostaglandin E2 release by doxorubicin. Eur J Pharmacol. 2013;700(1-3):102-9
32. Marszalek A, Szylberg L, Wisniewska E, Janiczek M. Impact of COX-2, IL-1beta, TNF-alpha, IL-4 and IL-10 on the process of carcinogenesis in the large bowel. Pol J Pathol. 2012;63(4):221-7
33. Shimizu S, Nishikawa Y, Kuroda K, Takagi S, Kozaki K, Hyuga S, Saga S, Matsuyama M. Involvement of transforming growth factor beta1 in autocrine enhancement of gelatinase B secretion by murine metastatic colon carcinoma cells. Cancer Res. 1996;56(14):3366-70
34. D'Amours D, Sallmann FR, Dixit VM, Poirier GG. Gain-of-function of poly(ADP-ribose) polymerase-1 upon cleavage by apoptotic proteases: implications for apoptosis. J Cell Sci. 2001;114(Pt 20):3771-8
35. Jain MV, Paczulla AM, Klonisch T, Dimgba FN, Rao SB, Roberg K, Schweizer F, Lengerke C, Davoodpour P, Palicharla VR. et al. Interconnections between apoptotic, autophagic and necrotic pathways: implications for cancer therapy development. J Cell Mol Med. 2013;17(1):12-29
36. Kim J, Kundu M, Viollet B, Guan KL. AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat Cell Biol. 2011;13(2):132-41
37. Gronich N, Rennert G. Beyond aspirin-cancer prevention with statins, metformin and bisphosphonates. Nat Rev Clin Oncol. 2013;10(11):625-42
38. Mizushima N, Yoshimori T. How to interpret LC3 immunoblotting. Autophagy. 2007;3(6):542-5
39. Girgis H, Masui O, White NM, Scorilas A, Rotondo F, Seivwright A, Gabril M, Filter ER, Girgis AH, Bjarnason GA. et al. Lactate dehydrogenase A is a potential prognostic marker in clear cell renal cell carcinoma. Mol Cancer. 2014;13:101
40. Sheng SL, Liu JJ, Dai YH, Sun XG, Xiong XP, Huang G. Knockdown of lactate dehydrogenase A suppresses tumor growth and metastasis of human hepatocellular carcinoma. FEBS J. 2012;279(20):3898-910
Author contact
![]() Corresponding authors: Carlos Pérez-Plasencia: carlos.pplascom; Tel.: +52-55-5623-1333 (ext. 39807). Nadia J Jacobo-Herrera: nadia.jacobocom; Tel.: +52-55-5487-0900 (ext. 2608).
Corresponding authors: Carlos Pérez-Plasencia: carlos.pplascom; Tel.: +52-55-5623-1333 (ext. 39807). Nadia J Jacobo-Herrera: nadia.jacobocom; Tel.: +52-55-5487-0900 (ext. 2608).