Impact Factor ISSN: 1837-9664

 Global reach, higher impact
Global reach, higher impactJ Cancer 2016; 7(15):2165-2172. doi:10.7150/jca.16925 This issue Cite
Research Paper
Massive parallel sequencing and digital gene expression analysis reveals potential mechanisms to overcome therapy resistance in pulmonary neuroendocrine tumors
Robert Fred Henry Walter1,2, Claudia Vollbrecht3, Daniel Christoph4, Robert Werner5, Jan Schmeller2, Elena Flom1,2, Georgia Trakada6, Aggeliki Rapti7, Vasilis Adamidis8, Wolfgang Hohenforst-Schmidt9, Jens Kollmeier5, Thomas Mairinger5, Jeremias Wohlschlaeger2, Paul Zarogoulidis8 ![]() , Konstantinos Porpodis8, Kurt Werner Schmid2, Fabian Dominik Mairinger2
, Konstantinos Porpodis8, Kurt Werner Schmid2, Fabian Dominik Mairinger2
1. Ruhrlandklinik, West German Lung Center, University Hospital Essen, University of Duisburg-Essen.
2. Institute of Pathology, University Hospital Essen, University of Duisburg-Essen.
3. Institute of Pathology, Charité Universitaetsmedizin, Berlin.
4. Department of Oncology, University Hospital Essen, University of Duisburg-Essen.
5. Institute of Pathology, Helios Klinikum Emil von Behring, Berlin.
6. Division of Pulmonology, Department of Clinical Therapeutics, National and Kapodistrian University of Athens School of Medicine, Alexandra Hospital, Athens, Greece.
7. 2nd Department of Pulmonary Medicine, "Sotiria" Hospital of Chest Diseases, Athens, Greece.
8. Pulmonary Department-Oncology Unit, "G. Papanikolaou" General Hospital, Aristotle University of Thessaloniki, Thessaloniki, Greece.
9. Medical Clinic I,''Fuerth'' Hospital, University of Erlangen, Fuerth, Germany.
Received 2016-7-21; Accepted 2016-9-18; Published 2016-10-25
Abstract
Background: Lung cancer is the leading cause of cancer-related deaths worldwide. 25% show neuroendocrine differentiation (typical/atypical carcinoids, large-/small-cell neuroendocrine carcinomas). Carcinoids present with long survival rates, but metastatic carcinoids correlate with decreased survival and are commonly insensitive to standard chemotherapy or radiation. Therefore, novel therapeutic strategies are urgently needed.
Material and methods: 70 representative tumor specimens were used for next-generation sequencing analysis of 14 genes related to therapy response. Additionally, mRNA-expression profiles of 60 matching samples were determined for 13 selected drug targets by using the NanoString nCounter technology.
Results: A number of features known to sensitize tumors for different targeted therapies could be identified, which hopefully improve the clinical management of this subgroup of lung neoplasias. In particular, EGFR expression was observed in the investigated tumors in a noteworthy manner. Additionally, MDM2 was strongly expressed in the majority of all samples whereas the expression of its physiological inhibitor, CDKN2A, was nearly absent in all low-grade tumors. TP53 showed a high frequency of variants in high-grade tumors but mutations were rare in carcinoids.
Conclusion: Based on our results, therapeutic approaches with MDM2-inhibitors and monoclonal anti-EGFR antibodies may be promising in pulmonary carcinoid tumors.
Keywords: NanoString, next-generation sequencing, biomarkers, personalized therapy, lung cancer.
INTRODUCTION
Lung cancer is the leading cause of cancer-related deaths worldwide [1-3], twenty-five percent of these belong to the group of neuroendocrine tumors [4]. These tumors encompass small-cell lung carcinoma (SCLC), large-cell neuroendocrine carcinoma (LCNEC), as well as typical (TC) and atypical carcinoids (AC) [5]. Patients suffering from TC have excellent survival rates with 87-100%, but some present with lymph-node metastasis (4-15%) [6-15]. Atypical carcinoids are more aggressive (survival rate 61-88%) and show higher frequency of nodal metastasis [8-14, 16]. According to the WHO classification from 1999, LCNEC was assigned to the non-small cell lung cancers (NSCLC) but shows similar biological behavior as SCLC with a five-year survival rate of 15%-57% [4, 8]. Up to 20% of all lung cancer incidences are SCLC showing the poorest five-year survival rates with less than 5% [4, 5, 8, 15]. High-grade neuroendocrine carcinomas of the lung occur almost exclusively in older patients (median 61 years) with a history of smoking, whereas lung carcinoids occur frequently in never smokers and younger patients (mean 45-55 years) [7, 15, 17, 18].
Carcinoid tumors are predominantly treated surgically, metastatic tumors are commonly not sensitive to chemotherapeutic regimes or radiation [4, 19]. Adjuvant chemo- or radiotherapy should be considered in completely resected AC with mediastinal lymph node involvement [6, 20, 21]. Combinations of chemotherapeutics are usually platinum- or streptozoticin-based [20].
For SCLC, chemotherapy with cisplatin plus etoposide is a well-established treatment since these tumors are notably sensitive to chemo- and radiotherapy [4]. An optimal treatment for LCNEC is still under investigation due to the relative rarity of this entity [4]. As defined by the WHO classification guidelines, LCNEC belongs to NSCLC, leaving it to the physician whether to treat it similarly to SCLC or NSCLC [8]. In fact, patients receiving SCLC-based regimens showed a significantly better outcome [20, 22].
The present study was conducted to identify markers for personalized therapeutic concepts in pulmonary neuroendocrine tumors, potentially leading to an improved clinical management.
MATERIAL AND METHODS
Demographic Data and Study Design
The study is based on a collective of seventy representative formalin-fixed, paraffin-embedded pulmonary neuroendocrine tumors (17 TC, 17 AC, 19 LCNEC, and 17 SCLC) used for sequencing. For expression analysis, 60 samples were investigated (16 TC and 13 AC, 16 LCNEC and 15 SCLC). The initial diagnosis was re-evaluated by two experienced pathologists (JWO, TMA). Specimens were taken from the Institute of Pathology at the University Hospital Essen (Germany) from 2005 till 2012. TNM-staging was based on the WHO Classification of Tumours guidelines (2004) [18]. The mean age at date of diagnosis was 58.6 years (median age: 59.0 years; 95% CI: 50.8-66.9 months). Survival data were available for 34 patients with twenty-two reported deaths at the time of data collection. Patients receiving chemotherapy before resection were excluded. The study was approved by the ethical committee of the University Hospital Essen (ID: 13-5382-BO). The investigations conform to the principles outlined in the declaration of Helsinki.
Sample Preparation
Genomic DNA was isolated on a Maxwell® 16 Research (Promega Corporation, Madison, USA) as recommended in the manufacturer's protocol. RNA extraction was performed using the RNeasy FFPE kit (Qiagen, Hilden, Germany) according to the manufacturer's recommendations. Nucleic acid quantification was performed using Qubit (Life Technologies, Carlsbad, USA) and Nanodrop 1000 instrument (Thermo Fisher Scientific, Waltham, USA). RNA integrity was assessed using an Agilent 2100 Bioanalyzer (Agilent Technologies, Santa Clara, USA) at the NanoString nCounter Core Facility at the University of Heidelberg (Germany) by smear analysis.
NanoString CodeSet Design and Expression Quantification
Important genes for different tumor-associated signaling pathways were included in the Custom CodeSet using the Standard Chemistry. The CodeSet contained a total of 91 genes and 13 of these genes were considered as potential pharmaceutical targets (ALK, CDKN2A, EGFR, FAS, FGFR1, FIGF, FLT4, IGF1, IGF2, KDR, MDM2, MET, MTOR).
Probe-sets for each gene were designed and synthesized at NanoString Technologies (Seattle, USA). Total RNA (100 ng) from FFPE material was measured at the NanoString nCounter Core Facility at the University of Heidelberg, Germany.
Next-Generation Sequencing
Sample preparation was done using the TruSeq Amplicon - Cancer Panel (Illumina Inc., San Diego, CA, USA) followed by paired-end sequencing on the MiSeq Personal Sequencer (Illumina) according the protocols provided by the manufacturer. The Cancer Panel covered the following 48 tumor-relevant genes (table 1) including 221 mutation hotspots. Library construction followed the TrueSeq Custom Amplicon - Library Preparation guide.
Overview of all genes covered by the NGS cancer hotspot panel. The Panel consists of two-times 221 probes for paired-end sequencing covering 221 mutational hotspots in 48 genes.
| ABL1 | AKT1 | ALK | APC | ATM | BRAF | CDH1 | CDKN2A |
|---|---|---|---|---|---|---|---|
| CSF1R | CTNNB1 | EGFR | ERBB2 | ERBB4 | FBXW7 | FGFR1 | FGFR2 |
| FGFR3 | FLT3 | GNA11 | GNAQ | GNAS | HNF1A | HRAS | IDH1 |
| JAK2 | JAK3 | KDR | KIT | KRAS | MET | MLH1 | MPL |
| NOTCH1 | NPM1 | NRAS | PDGFRA | PIK3CA | PTEN | PTPN11 | RB1 |
| RET | SMAD4 | SMARCB1 | SMO | SRC | STK11 | TP53 | VHL |
FastQ-files were aligned against the Hg19 build. For analysis of the aligned reads including variant calling, the software Avadis NGS (Strand Scientific Intelligence, California, USA) was used. Reads with a quality score <30 were discarded. After removal of SNPs, data filtering was done by excluding variants with <25 effective variant reads or below 10% variation frequency. Synonymous variants were removed.
Finally, variants were analyzed for their functional impact on the protein-activity by using MutationAssessor (release 2) [23] and implementation of the ANNOVAR algorithm [24], combining the tools SIFT [25], PolyPhen2 [26] and MutationTaster [27].
Statistical Analysis
All statistical analyses were performed using the R statistical programming environment (v3.1.3). For dichotomous factors and linear vectors the Wilcoxon Mann-Whitney rank sum test was applied. For variance analysis of variables with more than two categories the Kruskal-Wallis test was performed. Correlations between linear vectors were analyzed by Spearman's rank correlation test. Double dichotomous contingency tables were analyzed using Fisher's Exact test. To test dependency of ranked parameters with more than two groups the Pearson's Chi-squared test was used. COXPH-model was used to analyze overall survival (OS) and progression-free survival (PFS). PFS was calculated from the first day of chemotherapy until progression, death from any cause, or the last time of follow up. OS was defined as the time from diagnosis until the date of death or last follow-up. Surveillance of PFS and OS was stopped on August 31, 2014. Significant differences in PFS or OS were analyzed by the Wald-test, likelihood-ratio test and Score (logrank) test.
The level of statistical significance was defined as p≤0.05.
RESULTS
Gene Expression in Pulmonary Neuroendocrine Tumors
Gene expression analysis showed strong differences between tumors of the four different entities, but also within tumors of each subgroup. Most strikingly, EGFR gene expression is present in 87.5% of TC, 100% of AC, 65.3% of LCNEC and in 60% of SCLC. Likewise, FGFR1 is expressed in 97.3% of all carcinoids in a noteworthy manner. Besides, 62.5% of LCNEC and 80% of SCLC show strong expression of FGFR1. MDM2 was strongly expressed in the majority of all samples. 81.3% of TC, 76.9% of AC, 50% of LCNEC and 93.4% of SCLC present gene expression of MDM2. CDKN2A expression was rare in all low-grade neuroendocrine lung tumors, but present in some carcinomas showing high gene expression.
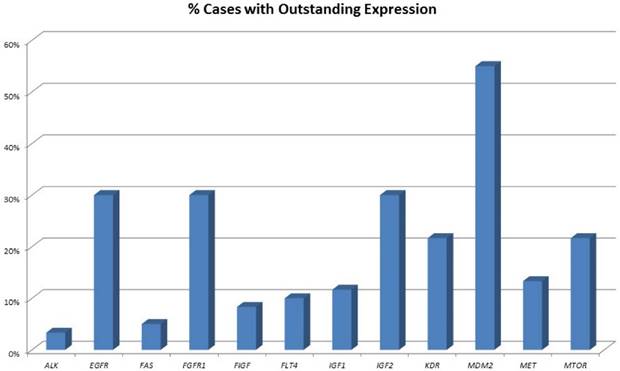
An overview of cases with increased expression levels is given in figure 1, an overview of all expression pattern is shown in table 2.
Percentage of tumours showing highly increased gene expression. One-third of all pulmonary neuroendocrine tumours show strong overexpression of EGFR and FGFR1. Of note, also one-third of tumours show high IGF2 expression and 10% present with high IGF1 expression. More than 50% of all tumours show highly elevated MDM2 expression.
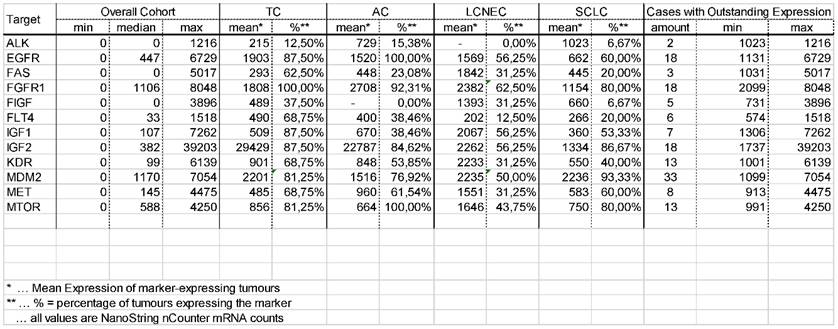
Results of the gene expression analysis. Minimum, maximum and median for all 13 therapy relevant markers in the overall cohort of pulmonary neuroendocrine tumours is shown. Additionally, the mean value of tumours expressing the mentioned markers as well as the percentage of tumours expressing these markers is shown for each entity. Besides, the number of cases showing an outstanding expression level, including minimum and maximum value, is listed.
Occurrence of Mutations in Pulmonary Neuroendocrine Tumors
86 functionally deleterious variants were determined within 13 therapy-relevant genes in 49 out of 70 samples. Four of the variants could be detected in TC, 14 in AC, 30 in LCNEC and 38 in SCLC.
In seven samples (10%; one TC, two AC, one LCNEC and three SCLC), variants in the EGFR gene were found, but none of them is known or predicted to activate the receptor. For ERBB2, a receptor tyrosine kinase (RTK), closely related to EGFR, three different variants could be observed in eleven samples.
Activating KRAS variants downstream of these receptors were found in three tumors (two LCNEC and one AC). Additionally, one sample showed activating mutations in the NRAS gene locus. BRAF variants occurred in three samples (4%), two LCNEC and one SCLC. Five different variants of PIK3CA were found in six tumor samples.
Interestingly, TP53 showed the highest frequency of variants with 31 alterations in 23 samples (33%), whereas the percentage was varying in the different neuroendocrine subtypes. 64.7% of all SCLC and 63.2% LCNEC showed a functional inactivation of P53 via TP53 mutations. No TP53 alterations were found in TC or in AC.
A summary of all mutations is given in Supplementary table 1.
Expression and mutation frequency are summarized in table 3 for each tumor entity.
Overview of results of the biomarker screening. Data for the percentage of tumours expressing each marker as well as the percentage of tumours showing inactivating genetic variants are shown.
| BRAF | 0% | 0% | 11% | 6% | ||||
|---|---|---|---|---|---|---|---|---|
| EGFR | 88% | 6% | 100% | 12% | 56% | 6% | 60% | 18% |
| ERBB2 | 12% | 12% | 16% | 24% | ||||
| FAS | 63% | 23% | 31% | 20% | ||||
| FGFR1 | 100% | 0% | 92% | 6% | 63% | 5% | 80% | 0% |
| FIGF | 38% | 0% | 31% | 7% | ||||
| FLT4 | 69% | 38% | 13% | 20% | ||||
| HRAS | 0% | 0% | 0% | 0% | ||||
| IGF1 | 88% | 38% | 56% | 53% | ||||
| IGF2 | 88% | 85% | 56% | 87% | ||||
| KDR | 69% | 0% | 54% | 6% | 31% | 0% | 40% | 0% |
| KIT | 0% | 12% | 0% | 0% | ||||
| KRAS | 0% | 6% | 11% | 0% | ||||
| MDM2 | 81% | 77% | 50% | 93% | ||||
| MET | 69% | 0% | 62% | 12% | 31% | 5% | 60% | 0% |
| MTOR | 81% | 100% | 44% | 80% | ||||
| NOTCH1 | 0% | 0% | 0% | 0% | ||||
| NRAS | 0% | 0% | 0% | 6% | ||||
| PIK3CA | 0% | 6% | 0% | 24% | ||||
| RET | 6% | 6% | 5% | 18% | ||||
| TP53 | 0% | 0% | 63% | 65% |
Association with Clinicopathological Parameters
EGFR, FLT4 and FIGF show highly significant differences in their expression levels between the four investigated tumor entities (p=0.0044, p=0.0054 and p=0.0096, respectively). Elevated EGFR gene expression (p=0.0002) is more prominent in carcinoid tumors compared to carcinomas. Also FGFR1 gene expression presents with significant differences (p=0.0301). Furthermore, upregulated EGFR gene expression associated significantly with lower IASLC-Grade (p=0.0005). FGFR1 as well as FLT4 show highly significant associations with tumor grade (p=0.0025 and p=0.0061, respectively). High FIGF (p=0.0210, HR: 1.40) and FAS (p=0.0233, HR: 1.27) gene expression levels seem to be associated with shortened PFS. In high-grade tumors, high FIGF mRNA levels identified a group of poor responders to therapy (COXPH: p=0.0347).
The appearance of TP53 variants was significantly associated with tumor type (p<0.0001) and is associated with carcinomas. PIK3CA shows variants in 5.9% of AC and 23.5% of SCLC, whereas TC and LCNEC present none of these variants (p=0.0116). Tumors infiltrating lymph nodes present with higher frequency of TP53 variants (p=0.0007), as well as ERBB2 variants (p=0.0051) or BRAF mutations (p=0.0468).
PIK3CA variants are associated with shortened OS (p=0.0019, HR: 2.54). Activating mutations in the KRAS gene (p=0.0003, HR: >10) are associated with a higher risk of progression.
DISCUSSION
Until now, studies investigating novel therapeutic approaches for pulmonary carcinoid tumors are lacking due to the rather long overall survival rates of patients with carcinoids. Nevertheless, tumors associated with a syndrome are resistant to chemo- and radiotherapy in most cases. This study is one of the first approaches to investigate a broad spectrum of biomarkers potentially predicting response to different chemotherapeutic agents. For mRNA expression analysis, the NanoString nCounter technology was used and additionally, 221 mutation hotspots were screened via massive parallel sequencing by synthesis.
Erlotinib, gefitinib or icotinib, in second generation also afatinib, are epidermal growth factor receptor tyrosine-kinase inhibitors (EGFR-TKI) used for the treatment of NSCLC harboring activating EGFR mutations [28]. Pathway activation downstream of EGFR, e.g. by KRAS mutations, is known to be a resistance mechanism for EGFR-TKI therapeutic approaches, but also the appearance of resistance mutations within the EGFR gene (e.g. p.T790M) has been described [29]. Moreover, continued activation of PI3K signaling via mutated PIK3CA was identified to abrogate gefitinib-induced apoptosis [29]. Further studies confirmed that the PI3K pathway is involved in different resistance mechanisms with respect to EGFR therapy approaches [30]. Consequently, different trials using PI-103, a PI3K/mTOR double inhibitor, in combination with EGFR-TKI therapy were initiated.
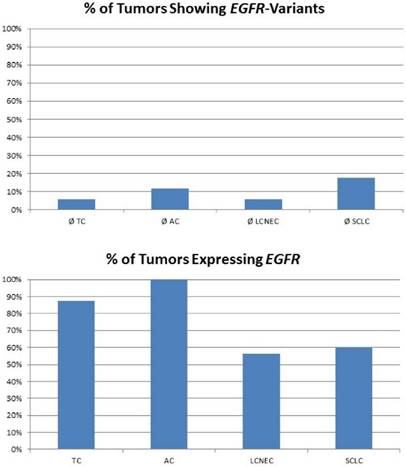
In our study, none of the tumors showed classical activating mutations in the EGFR gene. Of note, eight mutations impairing protein function were found in pulmonary neuroendocrine tumors regardless of the subtype (figure 2). A present study revealed that also cases with other non-classical mutations and complex mutational events had similar end-point outcomes to TKI therapy compared with classical activating mutations [31]. Besides, anti-EGFR antibody therapy with cetuximab may be a possible alternative. Cetuximab activity in NSCLC was found in tumors that expressed high levels of EGFR only [32], but also other monoclonal anti-EGFR antibodies were evaluated. These include matuzumab, panitumumab and most promising necitumumab, leading to an increased survival compared to cetuximab [33]. In our study, the majority of carcinoid tumors and more than half of all high-grade tumors show strongly upregulated expression levels of EGFR; one third of all NELC present with a highly increased EGFR gene expression level (figure 2). Furthermore, it has been reported that more than half of all carcinoids show increased EGFR copy numbers [34]. Thus, making them potential targets for monoclonal anti-EGFR antibodies.
The plot in the upper row shows the percentage of tumours with EGFR variants predicted to influence protein function. Nevertheless, classical EGFR mutations were not found in the collective investigated. The plot at the bottom shows the percentage of tumours expressing EGFR. Carcinoids present with elevated EGFR expression compared to carcinomas.
Lapatinib is currently approved for patients with ERBB2 (HER2)-positive metastatic breast cancer in combination with capecitabine after prior therapy with anthracycline, taxane, and trastuzumab [35, 36]. It is also approved in combination with letrozole in postmenopausal women with ERBB2-positive advanced breast cancer where endocrine therapy is indicated [36, 37]. Mutations in the ERBB2 (HER2) gene locus were reported in 1.5%-2% of all NSCLC, but this aberration occurred exclusively in adenocarcinomas [38, 39]. We found nine patients (13%) harboring the activating p.L785R and the p.R868W. It has been shown that patients with mutations and without amplification of the ERBB2 gene locus can respond to lapatinib [36, 39].
Activating BRAF mutations were found in two LCNEC and one SCLC. Patients harboring activating BRAF mutations are known to be sensitive for therapy with vemurafenib [40]. Activation downstream of BRAF, primary through RAS mutations, is reported to act as a potential resistance mechanism [41]. RAS mutations were observed in all tumor entities except TC, but never appeared in combination with BRAF mutations. The response of pulmonary neuroendocrine tumors harboring BRAF mutations to vemurafenib has to be proven in further studies.
A large part of human cancers shows TP53 inactivation, mostly by mutations or loss of heterozygosity (LOH) of the whole gene locus. Even in TP53 wild type tumors the functional protein is inactivated resulting from some putative mechanisms of amplification and overexpression of MDM2, the physiological inhibitor of P53 [42-46]. MDM2 itself is controlled by the tumor suppressor P14/ARF (encoded by the gene locus of CDKN2A). Numerous environmental influences and genetic alterations induced by platin compounds are able to activate P53 by post-translational modifications, which results in cell cycle arrest, cellular senescence, or apoptosis [47]. Nutlin-3A, a cis-imidazoline analogue, is a potent and selective MDM2 inhibitor [48, 49] that prevents MDM2-TP53-interaction by binding to the hydrophobic binding pocket of MDM2 leading to an immediate reactivation of P53 [48, 50, 51]. It is currently tested in a phase I clinical trial (NCT01143519, NCT00623870) [51]. Second-generation MDM2 inhibitors (RG7112, RO5045337, Idasanutlin, RG-7388, DS-3032b, SAR405838, CGM-097, MK8242, HDM201 etc.) are currently tested in phase I-III trials for various diseases (NCT01877382, NCT02319369, NCT00559533, NCT01985191, NCT02016729, NCT02343172 etc.)
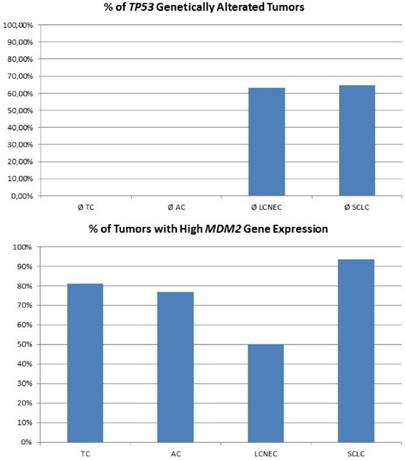
For high-grade neuroendocrine lung tumors, TP53 mutations were found in the majority of tumors (figure 3A). As described, the main alternative mechanism seems to be LOH, so MDM2 inhibition may not be successful. In contrast, carcinoids lack genetic alterations of TP53, but about 80% of samples show a dramatically high MDM2 gene expression level (figure 3B). Additionally, CDKN2A inhibiting MDM2 is absent in most pulmonary carcinoids. Therefore, pulmonary carcinoids seem to be an ideal target for Nutlin-3A therapy. A major issue with carcinoid tumors is the rarity of the disease and therefore it is difficult to conduct such studies. Also, there is an issue if drugs such as; everolimus could be administered as adjuvant therapy and for how long after surgery [52]. Everolimus could be used as treatment in the case that surgery is not possible, or could be used as adjuvant therapy in the case that surgery is not possible and only endoscopic debulking procedures are possible. The length of the therapy administration depends on multiple factors such as local disease progression and drug side effects. There are still many matters to clarify as disease diagnosis and management. [53-55]
The plot at the top is showing the percentage of tumours with inactivating mutations in the TP53 gene. The plot at the bottom shows the percentage of tumours with high MDM2 expression. MDM2 overexpression is one possible mechanism to avoid TP53-dependet apoptosis and cell senescence.
CONCLUSION
Carcinoids of the lung show long survival rates, but sometimes progress to a systemic disease. Carcinoids with a syndrome are resistant to chemo- and radiotherapy and new therapeutic approaches are needed. Based on our results, therapeutic approaches with MDM2 inhibitors and monoclonal anti-EGFR antibodies may be a promising novel therapeutic approach and need to be confirmed in further in vitro and in vivo studies.
SUPPLEMENTARY MATERIAL
Supplementary table 1.
Acknowledgements
We thank the nCounter Core Facility Heidelberg for providing the nCounter system and related services.
CONFLICT OF INTEREST
All authors disclose any affiliations that are considered to be relevant and important with any organization that to our knowledge has any direct interest in the subject matter discussed.
References
1. Eramo A, Lotti F, Sette G, Pilozzi E, Biffoni M, Di Virgilio A. et al. Identification and expansion of the tumorigenic lung cancer stem cell population. Cell death and differentiation. 2008;15:504-14 doi:10.1038/sj.cdd.4402283
2. Jett JR, Midthun DE. Screening for lung cancer: current status and future directions: Thomas A. Neff lecture. Chest. 2004;125:158S-62S
3. Mallick R, Patnaik SK, Yendamuri S. MicroRNAs and lung cancer: Biology and applications in diagnosis and prognosis. Journal of carcinogenesis. 2010:9 doi:10.4103/1477-3163.67074
4. Rekhtman N. Neuroendocrine tumors of the lung: an update. Archives of pathology & laboratory medicine. 2010;134:1628-38 doi:10.1043/2009-0583-RAR.1
5. Takei H, Asamura H, Maeshima A, Suzuki K, Kondo H, Niki T. et al. Large cell neuroendocrine carcinoma of the lung: a clinicopathologic study of eighty-seven cases. The Journal of thoracic and cardiovascular surgery. 2002;124:285-92
6. Thomas CF Jr, Tazelaar HD, Jett JR. Typical and atypical pulmonary carcinoids: outcome in patients presenting with regional lymph node involvement. Chest. 2001;119:1143-50
7. Travis WD, Rush W, Flieder DB, Falk R, Fleming MV, Gal AA. et al. Survival analysis of 200 pulmonary neuroendocrine tumors with clarification of criteria for atypical carcinoid and its separation from typical carcinoid. The American journal of surgical pathology. 1998;22:934-44
8. Asamura H, Kameya T, Matsuno Y, Noguchi M, Tada H, Ishikawa Y. et al. Neuroendocrine neoplasms of the lung: a prognostic spectrum. Journal of clinical oncology: official journal of the American Society of Clinical Oncology. 2006;24:70-6 doi:10.1200/JCO.2005.04.1202
9. Cardillo G, Sera F, Di Martino M, Graziano P, Giunti R, Carbone L. et al. Bronchial carcinoid tumors: nodal status and long-term survival after resection. The Annals of thoracic surgery. 2004;77:1781-5 doi:10.1016/j.athoracsur.2003.10.089
10. Beasley MB, Thunnissen FB, Brambilla E, Hasleton P, Steele R, Hammar SP. et al. Pulmonary atypical carcinoid: predictors of survival in 106 cases. Human pathology. 2000;31:1255-65 doi:10.1053/hupa.2000.19294
11. Fink G, Krelbaum T, Yellin A, Bendayan D, Saute M, Glazer M. et al. Pulmonary carcinoid: presentation, diagnosis, and outcome in 142 cases in Israel and review of 640 cases from the literature. Chest. 2001;119:1647-51
12. Garcia-Yuste M, Matilla JM, Cueto A, Paniagua JM, Ramos G, Canizares MA. et al. Typical and atypical carcinoid tumours: analysis of the experience of the Spanish Multi-centric Study of Neuroendocrine Tumours of the Lung. European journal of cardio-thoracic surgery: official journal of the European Association for Cardio-thoracic Surgery. 2007;31:192-7 doi:10.1016/j.ejcts.2006.11.031
13. Pelosi G, Scarpa A, Puppa G, Veronesi G, Spaggiari L, Pasini F. et al. Alteration of the E-cadherin/beta-catenin cell adhesion system is common in pulmonary neuroendocrine tumors and is an independent predictor of lymph node metastasis in atypical carcinoids. Cancer. 2005;103:1154-64 doi:10.1002/cncr.20901
14. Rea F, Rizzardi G, Zuin A, Marulli G, Nicotra S, Bulf R. et al. Outcome and surgical strategy in bronchial carcinoid tumors: single institution experience with 252 patients. European journal of cardio-thoracic surgery: official journal of the European Association for Cardio-thoracic Surgery. 2007;31:186-91 doi:10.1016/j.ejcts.2006.10.040
15. Travis WD. Lung tumours with neuroendocrine differentiation. Eur J Cancer. 2009;45(Suppl 1):251-66 doi:10.1016/S0959-8049(09)70040-1
16. Scott WJ. Surgical treatment of other bronchial tumors. Chest surgery clinics of North America. 2003;13:111-28
17. Swarts DR, Ramaekers FC, Speel EJ. Molecular and cellular biology of neuroendocrine lung tumors: evidence for separate biological entities. Biochimica et biophysica acta. 2012;1826:255-71 doi:10.1016/j.bbcan.2012.05.001
18. Travis WD, World Health Organization, International Agency for Research on Cancer, International Association for the Study of Lung Cancer, International Academy of Pathology. Pathology and genetics of tumours of the lung, pleura, thymus and heart. Oxford: IARC Press. 2004
19. Gridelli C, Rossi A, Airoma G, Bianco R, Costanzo R, Daniele B. et al. Treatment of pulmonary neuroendocrine tumours: State of the art and future developments. Cancer treatment reviews. 2012 doi:10.1016/j.ctrv.2012.06.012
20. Gridelli C, Rossi A, Airoma G, Bianco R, Costanzo R, Daniele B. et al. Treatment of pulmonary neuroendocrine tumours: State of the art and future developments. Cancer treatment reviews. 2013;39:466-72 doi:10.1016/j.ctrv.2012.06.012
21. Mackley HB, Videtic GM. Primary carcinoid tumors of the lung: a role for radiotherapy. Oncology (Williston Park). 2006;20:1537-43 discussion 44-5, 49
22. Rossi G, Cavazza A, Marchioni A, Longo L, Migaldi M, Sartori G. et al. Role of chemotherapy and the receptor tyrosine kinases KIT, PDGFRalpha, PDGFRbeta, and Met in large-cell neuroendocrine carcinoma of the lung. Journal of clinical oncology: official journal of the American Society of Clinical Oncology. 2005;23:8774-85 doi:10.1200/JCO.2005.02.8233
23. Reva B, Antipin Y, Sander C. Predicting the functional impact of protein mutations: application to cancer genomics. Nucleic acids research. 2011;39:e118. doi:10.1093/nar/gkr407
24. Wang K, Li M, Hakonarson H. ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic acids research. 2010;38:e164. doi:10.1093/nar/gkq603
25. Kumar P, Henikoff S, Ng PC. Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm. Nature protocols. 2009;4:1073-81 doi:10.1038/nprot.2009.86
26. Adzhubei IA, Schmidt S, Peshkin L, Ramensky VE, Gerasimova A, Bork P. et al. A method and server for predicting damaging missense mutations. Nature methods. 2010;7:248-9 doi:10.1038/nmeth0410-248
27. Schwarz JM, Rodelsperger C, Schuelke M, Seelow D. MutationTaster evaluates disease-causing potential of sequence alterations. Nature methods. 2010;7:575-6 doi:10.1038/nmeth0810-575
28. Sarosi V, Losonczy G, Francovszky E, Tolnay E, Torok S, Galffy G. et al. Effectiveness of erlotinib treatment in advanced KRAS mutation-negative lung adenocarcinoma patients: Results of a multicenter observational cohort study (MOTIVATE). Lung Cancer. 2014 doi:10.1016/j.lungcan.2014.07.011
29. Engelman JA, Mukohara T, Zejnullahu K, Lifshits E, Borras AM, Gale CM. et al. Allelic dilution obscures detection of a biologically significant resistance mutation in EGFR-amplified lung cancer. The Journal of clinical investigation. 2006;116:2695-706 doi:10.1172/JCI28656
30. Sequist LV, Waltman BA, Dias-Santagata D, Digumarthy S, Turke AB, Fidias P. et al. Genotypic and histological evolution of lung cancers acquiring resistance to EGFR inhibitors. Science translational medicine. 2011;3:75ra26. doi:10.1126/scitranslmed.3002003
31. Peng L, Song ZG, Jiao SC. Efficacy analysis of tyrosine kinase inhibitors on rare non-small cell lung cancer patients harboring complex EGFR mutations. Scientific reports. 2014;4:6104. doi:10.1038/srep06104
32. Amendt C, Staub E, Friese-Hamim M, Storkel S, Stroh C. Association of EGFR expression level and cetuximab activity in patient-derived xenograft models of human non-small cell lung cancer. Clinical cancer research: an official journal of the American Association for Cancer Research. 2014 doi:10.1158/1078-0432.CCR-13-3385
33. Pirker R. EGFR-directed monoclonal antibodies in non-small cell lung cancer. Targeted oncology. 2013;8:47-53 doi:10.1007/s11523-012-0244-7
34. Gilbert JA, Adhikari LJ, Lloyd RV, Rubin J, Haluska P, Carboni JM. et al. Molecular markers for novel therapies in neuroendocrine (carcinoid) tumors. Endocrine-related cancer. 2010;17:623-36 doi:10.1677/ERC-09-0318
35. Geyer CE, Forster J, Lindquist D, Chan S, Romieu CG, Pienkowski T. et al. Lapatinib plus capecitabine for HER2-positive advanced breast cancer. The New England journal of medicine. 2006;355:2733-43 doi:10.1056/NEJMoa064320
36. Tan AR, Dowlati A, Stein MN, Jones SF, Infante JR, Bendell J. et al. Phase I study of weekly paclitaxel in combination with pazopanib and lapatinib in advanced solid malignancies. British journal of cancer. 2014;110:2647-54 doi:10.1038/bjc.2014.233
37. Johnston S, Pippen J Jr, Pivot X, Lichinitser M, Sadeghi S, Dieras V. et al. Lapatinib combined with letrozole versus letrozole and placebo as first-line therapy for postmenopausal hormone receptor-positive metastatic breast cancer. Journal of clinical oncology: official journal of the American Society of Clinical Oncology. 2009;27:5538-46 doi:10.1200/JCO.2009.23.3734
38. Mazieres J, Peters S, Lepage B, Cortot AB, Barlesi F, Beau-Faller M. et al. Lung cancer that harbors an HER2 mutation: epidemiologic characteristics and therapeutic perspectives. Journal of clinical oncology: official journal of the American Society of Clinical Oncology. 2013;31:1997-2003 doi:10.1200/JCO.2012.45.6095
39. Rolfo C, Giovannetti E, Hong DS, Bivona T, Raez LE, Bronte G. et al. Novel therapeutic strategies for patients with NSCLC that do not respond to treatment with EGFR inhibitors. Cancer treatment reviews. 2014;40:990-1004 doi:10.1016/j.ctrv.2014.05.009
40. Kim G, McKee AE, Ning YM, Hazarika M, Theoret M, Johnson JR. et al. FDA Approval Summary: Vemurafenib for Treatment of Unresectable or Metastatic Melanoma with the BRAF V600E Mutation. Clinical cancer research: an official journal of the American Association for Cancer Research. 2014 doi:10.1158/1078-0432.CCR-14-0776
41. Sos ML, Levin RS, Gordan JD, Oses-Prieto JA, Webber JT, Salt M. et al. Oncogene Mimicry as a Mechanism of Primary Resistance to BRAF Inhibitors. Cell reports. 2014 doi:10.1016/j.celrep.2014.07.010
42. Jones SN, Roe AE, Donehower LA, Bradley A. Rescue of embryonic lethality in Mdm2-deficient mice by absence of p53. Nature. 1995;378:206-8 doi:10.1038/378206a0
43. Marine JC, Francoz S, Maetens M, Wahl G, Toledo F, Lozano G. Keeping p53 in check: essential and synergistic functions of Mdm2 and Mdm4. Cell death and differentiation. 2006;13:927-34 doi:10.1038/sj.cdd.4401912
44. Montes de Oca Luna R, Wagner DS, Lozano G. Rescue of early embryonic lethality in mdm2-deficient mice by deletion of p53. Nature. 1995;378:203-6 doi:10.1038/378203a0
45. Parant J, Chavez-Reyes A, Little NA, Yan W, Reinke V, Jochemsen AG. et al. Rescue of embryonic lethality in Mdm4-null mice by loss of Trp53 suggests a nonoverlapping pathway with MDM2 to regulate p53. Nature genetics. 2001;29:92-5 doi:10.1038/ng714
46. Ringshausen I, O'Shea CC, Finch AJ, Swigart LB, Evan GI. Mdm2 is critically and continuously required to suppress lethal p53 activity in vivo. Cancer cell. 2006;10:501-14 doi:10.1016/j.ccr.2006.10.010
47. Harris SL, Levine AJ. The p53 pathway: positive and negative feedback loops. Oncogene. 2005;24:2899-908 doi:10.1038/sj.onc.1208615
48. Vassilev LT, Vu BT, Graves B, Carvajal D, Podlaski F, Filipovic Z. et al. In vivo activation of the p53 pathway by small-molecule antagonists of MDM2. Science. 2004;303:844-8 doi:10.1126/science.1092472
49. Shangary S, Wang S. Small-molecule inhibitors of the MDM2-p53 protein-protein interaction to reactivate p53 function: a novel approach for cancer therapy. Annual review of pharmacology and toxicology. 2009;49:223-41 doi:10.1146/annurev.pharmtox.48.113006.094723
50. Gamble LD, Kees UR, Tweddle DA, Lunec J. MYCN sensitizes neuroblastoma to the MDM2-p53 antagonists Nutlin-3 and MI-63. Oncogene. 2012;31:752-63 doi:10.1038/onc.2011.270
51. Voltan R, Secchiero P, Ruozi B, Forni F, Agostinis C, Caruso L. et al. Nanoparticles engineered with rituximab and loaded with Nutlin-3 show promising therapeutic activity in B-leukemic xenografts. Clinical cancer research: an official journal of the American Association for Cancer Research. 2013;19:3871-80 doi:10.1158/1078-0432.CCR-13-0015
52. Flaum N, Valle JW, Mansoor W, McNamara MG. Everolimus in the treatment of neuroendocrine tumors of the respiratory and gastroenteropancreatic systems. Future oncology. 2016 doi:10.2217/fon.16.23
53. Wolin EM. Advances in the Diagnosis and Management of Well-Differentiated and Intermediate-Differentiated Neuroendocrine Tumors of the Lung. Chest. 2016 doi:10.1016/j.chest.2016.06.018
54. Cives M, Soares HP, Strosberg J. Will clinical heterogeneity of neuroendocrine tumors impact their management in the future? Lessons from recent trials. Current opinion in oncology. 2016;28:359-66 doi:10.1097/CCO.0000000000000299
55. Rugo HS, Hortobagyi GN, Yao J, Pavel M, Ravaud A, Franz D. et al. Meta-analysis of stomatitis in clinical studies of everolimus: incidence and relationship with efficacy. Annals of oncology: official journal of the European Society for Medical Oncology / ESMO. 2016;27:519-25 doi:10.1093/annonc/mdv595
Author contact
![]() Corresponding author: Paul Zarogoulidis, M.D, Ph. D., Pulmonary Department-Oncology Unit, “G. Papanikolaou” General Hospital, Aristotle University of Thessaloniki, Thessaloniki, Greece. Fax: 00302310992424; Mobile: 00306977271974; E-mail: pzarogcom.
Corresponding author: Paul Zarogoulidis, M.D, Ph. D., Pulmonary Department-Oncology Unit, “G. Papanikolaou” General Hospital, Aristotle University of Thessaloniki, Thessaloniki, Greece. Fax: 00302310992424; Mobile: 00306977271974; E-mail: pzarogcom.