Impact Factor ISSN: 1837-9664

 Global reach, higher impact
Global reach, higher impactJ Cancer 2016; 7(15):2270-2279. doi:10.7150/jca.16010 This issue Cite
Research Paper
Nucleophosmin Mutations Induce Chemosensitivity in THP-1 Leukemia Cells by Suppressing NF-κB Activity and Regulating Bax/Bcl-2 Expression
Shuaishuai Zhang1, Fengxian Qin2, Liyuan Yang1, Jingrong Xian1, Qin Zou1, Hongjun Jin 1, Lu Wang1, Ling Zhang1 ![]()
1. Key Laboratory of Laboratory Medical Diagnostics Designated by the Ministry of Education, College of Laboratory Medicine, Chongqing Medical University, Chongqing, China.
2. Department of clinical laboratory, Liuzhou Worker's Hospital, Guangxi, China.
Received 2016-4-29; Accepted 2016-8-22; Published 2016-11-25
Abstract
Nucleophosmin (NPM1) - a gene that encodes for a nuclear protein with multiple functions. Mutations in NPM1 are seen in approximately one-third of acute myeloid leukemia (AML) and are generally associated with good response to induction chemotherapy. However, the mechanisms underlying this chemosensitivity are still unknown. Recent studies have established that nuclear factor-κB (NF-κB) activation is a key response of leukemia cell to chemotherapy. In this study, we transfected human monocytic leukemia THP-1 cells with the vector expressing NPM1 mutation variant (NPM1mA), and confirmed overexpression of NPM1mA at mRNA and protein levels by reverse transcription PCR (RT-PCR) and immunohistochemistry, respectively. The effects of NPM1 mutations on chemotherapeutical agents induced apoptosis, NF-κB activity and gene expression were examined using flow cytometry, luciferase reporter assays, quantitative real time PCR (qRT-PCR) and Western blot. We found that overexpression of NPM1mA in THP-1 cells sensitized these cells to apoptosis induced by chemotherapeutical agents such as daunorubicin (DNR) and cytarabine (Ara-C). Moreover, we demonstrated that expression of NPM1 mA reduced the NF-κB transcription activity of THP-1 cells upon drug treatment. In addition, restoration of NF-κB activity via TNF-α stimulation could attenuate the effect of NPM1mA overexpression on DNR-and Ara-C-induced apoptosis. Interestingly, expression of NPM1mA could upregulate Bax and downregulate Bcl-2 at mRNA and protein levels in THP-1 cells when treated with DNR or Ara-C. We also demonstrated that restoration of NF-κB activity via TNF-α pre-treatment reversed the effect of NPM1mA on the Bax/Bcl-2 expression. Furthermore, evaluation of gene expression data from The Cancer Genome Atlas (TCGA) dataset revealed that NPM1-mutated patients showed a higher expression of Bax and a lower expression of Bcl-2. These results suggest that the NPM1 gene mutations could confer increased sensitivity to chemotherapeutic agents, at least in part, by suppressing NF-κB activity and regulating Bax/Bcl-2 expression.
Keywords: nucleophosmin, gene mutation, chemosensitivity, NF-κB, Bax, Bcl-2, acute myeloid leukemia.
Introduction
Nucleophosmin (NPM1), also named B23, is a multifunctional nucleolar protein that mainly localized in nucleoli but able to shuttle between nucleus and cytoplasm [1]. NPM1 plays crucial roles in ribosome maturation and export, centrosome duplication, cell cycle progression, histone assembly and response to a variety of stress stimuli [2-3]. NPM1 gene heterozygous mutations are present in roughly a third of patients with acute myeloid leukemia (AML), making it one of the most frequent genomic alterations in these patients [4] Mutated NPM1 protein disrupts the C-terminal nucleolar localization signal of nucleophosmin and generates a new nuclear export signal, which alters the normal balance in nuclear-cytoplasmic NPM1 shuttling and causes the characteristic cytoplasmic localization [5, 6]. In 2005, Falini B et al. [7] first observed cytoplasmic nucleophosmin (NPMc+) in AML and reported that the type A NPM1 mutation (NPM1 mA, 4 base TCTG insertion) was the most frequent in adults (75-80% of cases). NPMc+ AML present with specific clinical, phenotypical, and genetic features [8, 9]. Strikingly, several clinical studies indicated a higher rate of complete remission after chemotherapy in patients carrying the NPM1 mutations when compared to wild type [10-13]. However, the molecular basis of this observation remains at present unclear.
Nuclear factor-κB (NF-κB) is a member of the Rel family proteins, which regulates protein expression mediating cell cycle/proliferation, anti-apoptosis, and cytokine secretion [14]. NF-κB transcription factors are involved in disparate processes such as inflammation, growth and development, and drug resistance [15, 16]. NF-κB has been demonstrated to be abnormally activated in blast cells from a significant number of AML patients [17]. Indeed, it has been demonstrated that many types of AMLs produce tumor necrosis factor (TNF)-α, and this TNF-α activates NF-κB transcription factors [18]. Additionally, recent studies have established that NF-κB activation is a key response of leukemia cells to chemotherapy [19, 20]. It is well known that inhibition of NF-κB increases sensitivity to many chemotherapeutical agents [21-23]. Activated NF-κB seems to trigger a series of molecular reactions [24, 25]. For instance, NF-κB can inhibit pro-apoptotic protein Bax and induce the expression of anti-apoptotic protein Bcl-2, which involved in chemotherapy induced apoptosis [26, 27].
In this study, we found that enforced expression of NPM1 mA increased THP-1 leukemia cells sensitivity to chemotherapeutic agents and reduced the NF-κB transcription activity of THP-1 cells upon drug treatment. In addition, expression of NPM1mA upregulated Bax expression and downregulated Bcl-2 levels. Notably, the potentiating effect of NPM1 mA on chemosensitivity was rescued by pre-treatment with TNF-α. Furthermore, NPM1-mutated patients showed a higher expression of Bax and a lower expression of Bcl-2. The results presented here demonstrate that the NPM1 gene mutations may confer increased sensitivity to chemotherapeutic agents, at least in part, by regulating NF-κB activity and the apoptosis related proteins expression.
Materials and methods
Cell line and reagents
Human monocytic leukemia THP-1 cells (purchased from Shanghai Institutes for Biological Sciences) were maintained in RPMI-1640 medium (Gibco, MD, USA), which was supplemented with 10% fetal bovine serum (FBS, Gibco, MD, USA) and 100 U of penicillin and streptomycin (Sangon Biotech,Shanghai, China) in a 5% CO2-humidified incubator at 37°C. Daunorubicin (DNR) and cytarabine (Ara-C) were purchased from Sigma. TNF-α was purchased from Beyotime.
Plasmids and cell infection
The pEGFPC1-NPM1mA and empty pEGFPC1 vectors were kindly provided by Dr. B Falini (Institute of Hematology, University of Perugia, Perugia, Italy). THP-1 cells were transfected with either pEGFPC1-NPM1 mA or empty pEGFPC1 vector using xfectTM reagent (Clontech, BD, USA), they were named as the “NPM1mA” or “pEGFPC1” group respectively. The “Blank” group represented THP-1 cells without transfection. Stable cell lines were selected and maintained in G418 (Invitrogen, MD, USA). Numerous independent single colonies were subcloned and analyzed for mRNA expression with RT-PCR and for protein expression with immunocytochemistry staining.
Reverse transcription PCR (RT-PCR) and quantitative real time PCR (qRT-PCR)
Reverse transcription PCR (RT-PCR) was operated as follows: the total RNA of cells was extracted with TRIzol reagent (TaKaRa, Tokyo, Japan). Then, 2 micrograms of total RNA that was treated with DNA-free DNase were reverse-transcribed for cDNA synthesis. The amplification conditions were: 5 min at 94 °C (one cycle) and 30 sec at 94 °C; 30 sec at the annealing temperature of 55 °C; and 50 sec at 70 °C (35 cycles) and 72 °C for 10 min (one cycle). The amplification productions for NPM1mA and β-actin were confirmed by 1.5% agarose gel electrophoresis. Quantitative real-Time PCR (qRT-PCR) analysis was performed on a MJ Mini™ Gradient Thermal Cycler Real-Time PCR machine (Bio-Rad, California, USA) with the SYBR Green reaction (TaKaRa, Tokyo, Japan) kit in triplicate. PCR amplification mixtures (25 μl) contained 12.5 μl of SYBR Green reaction, 2 μl of a mixture of 2.5 mM reverse and forward primers, 8.5 μl of nuclease free water and 2 μl of cDNA template. The Bax and Bcl-2 gene expression levels were analyzed relative to the β-actin gene transcript levels. The primer sequences are listed in Table 1.
The sequence of PCR primers for each gene.
| Genes | Forward primer | Reverse primer | PCR fragment size (bp) |
|---|---|---|---|
| NPM1mA | TGGAGGTGGTAGCAAGGTTC | CTTCCTCCACTGCCAGACAGA | 446 |
| Bcl-2 | TTCTTTGAGTTCGGTGGGGTC | TGCATATTTGTTTGGGGCAGG | 300 |
| Bax | GGATGCGTCCACCAAGAA | GCACTCCCGCCACAAAGA | 386 |
| β-actin | GCTGTCCCTGTATGCCTCT | GATGTCACGCACGATTTCC | 220 |
Immunocytochemistry staining
Immunostaining was performed according to the alkaline phosphatase monoclonal anti-alkaline phosphatase (APAAP) technique. Approximately 1×105 cells were cytocentrifuged onto a clean glass slide, then fixed with 4% paraformaldehyde for 30 min, permeabilized with 0.25% cold triton for 15 min and incubated with 1% bovine serum albumin (Sigma, MO, USA) in PBS for 1 h at room temperature for blocking. After washing with 0.01 M PBS three times,
anti-NPM1 monoclonal antibody (Inc. Santa Cruz, CA, USA) was added at a concentration of 1:100. The slides were incubated overnight at 4°C. The detection steps were performed according to the manufacturer's (Zhong Shan Golden bridge Biotechnology, China) instructions. The presence of the NPM1 mutant protein was assessed by examining the cytoplasmic distribution of red precipitate particles.
Flow cytometry
Apoptosis was evaluated by flow cytometry for the detection of annexin V-positive cells. Cells were incubated separately with 0.25 μM DNR or 8.5 μM Ara-C for 24 h, respectivly. At the indicated time points, apoptosis induction was measured by double-staining the cells with Annexin-PE/7-AAD (Keygen, Nanjing, China) using a FACS Calibur flow cytometer (BD). Briefly, cells were washed once in phosphate buffered saline and once in 1x-binding buffer, then 5 μl of annexin V-FITC was added to the cells. Cells were incubated at room temperature for 15 min, after which 300ml 1x-binding buffer was added and cells were analyzed by flow cytometry. Apoptotic cells were defined as annexin V positive and propidium iodide negative. Each experiment was performed in triplicate and the final numerical value is expressed as mean adsorbance value ± SD.
Luciferase reporter assays
Nuclear factor-kB basal activity was determined by the ELISA method. A total of 106 cells in each group were cultrued in a 6-well plate and transfected with a firefly luciferase-reporter construct (pNF-κB-TA-luc, Beyotime, Shanghai, China) plus the control vector containing Renilla luciferase, pRL-TK Promega, Madison, WI) in a ratio 10:1. After 24 h, cells were incubated with 0.25 μM of daunorubicin and 8.5 μM Ara-C. Firefly and renilla luciferase activities were measured 24 h after treatment with the reagents using the Dual-Luciferase Reporter Assay System (Promega, Madison, WI) and the the ratio was set as the relative activities of NF-κB. All the experiments were performed in triplicate and the final numerical value is expressed as mean adsorbance value ± s.d.
Western blot
The cultured cells were washed and lysed in cell extraction buffer; a total of 50 μg of proteins was loaded into a sodium dodecyl sulfate polyacrylamide gel for electrophoresis and transferred onto PVDF membranes. PVDF membranes were blocked with 5% non-fat dry milk for 2 h at room temperature and then incubated with primary antibody overnight at 4°C. Membranes were washed with TBST (20 mM Tris-HCl, pH 7.6, 137 mM NaCl, 0.1% Tween 20) three times for 10 min each and then incubated with alkaline horseradish peroxidase (HRP)-conjugated anti-rabbit or anti-mouse secondary antibody (Zhong Shan Golden bridge Biotechnology, China) for 1 h at room temperature. Protein bands were visualized by incubating the membranes in chemiluminescence HRP substrate (Millipore, USA). Bax and Bcl-2 monoclonal antibodies (Santa Cruz Biotechnology, CA, USA) were used at 1:500 dilutions. β-actin monoclonal antibody (Zhong Shan Golden bridge Biotechnology, China) was added at a concentration of 1:1,000.
Analysis of gene expression data for AML patients in TCGA dataset
Gene expression data for AML patients from The Cancer Genome Atlas (TCGA) were downloaded from the TCGA website (https://tcga-data.nci.nih.gov/tcga/). A total of 165 AML cases stratified by leukemia French American British (FAB) morphology classification were included and 48 AMLs with NPM1 mutation were obtained from TCGA 2013 human AML dataset. RNA sequencing (RNA-Seq) and clinical data for AML samples were analyzed using the cBioPortal for Cancer Genomics [28, 29]. For statistical analysis, Bax/Bcl-2 mRNA expression was compared between the AMLs with NPM1 mutation and those with no NPM1 mutation.
Statistical analysis
SPSS17.0 software was used for statistical analysis. Results represent the mean ± SD of three independent experiments. A Student's t-test was performed to assess to compare averaged values. Wilcoxon rank sum test analysis was used to compare clinical data. *P<0.05 were considered statistically significant.
Results
Establishing THP-1 cells stably expressing NPM1 mA
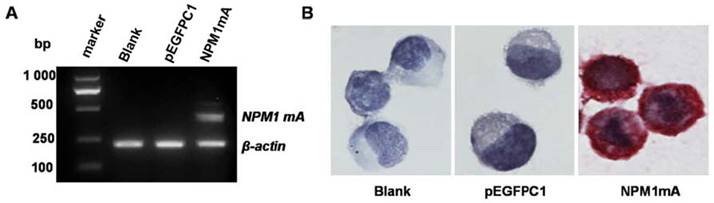
The eukaryotic expression vector pEGFPC1 and pEGFPC1-NPM1 mA were transfected into THP-1 cells. The expression of NPM1 mA mRNA was assayed by RT-PCR amplification. The results showed that the cells in NPM1 mA group could express NPM1 mA mRNA (Figure 1A), while the cells belonging to the two control groups could not. The cytoplasmic localization of the NPM1 mutant protein was observed by immunostaining. As shown in Figure 1B, the NPM1 mutant protein remained restricted to the cytoplasm in the NPM1 mA group, as evidenced by the presence of red precipitate particles.
The expression of NPM1mA mRNA and protein in transfected THP-1cells. (A)The expression of NPM1 mA mRNA was examined by RT-PCR. There was NPM1 mA mRNA expression in the NPM1 mA group, but not in the two control groups. (B) NPM1 protein detected by immunocytochemistry (APAAP, ×200). The cytoplasmic localization of NPM1 protein was observed in NPM1 mA group. An example of this localization is shown.
Effect of NPM1 mA on the chemosensitivity of THP-1 cells
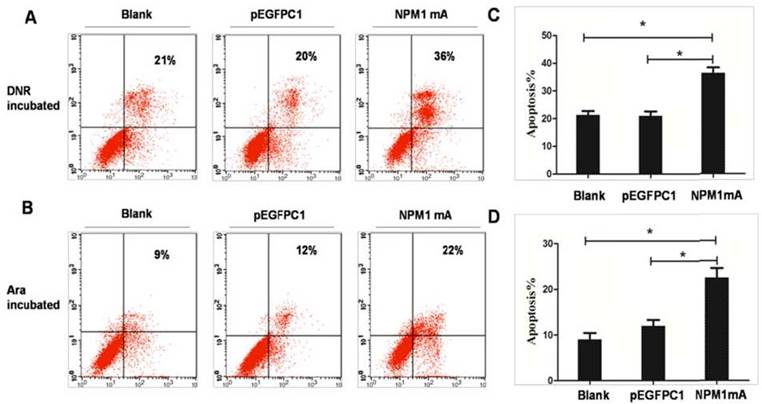
To observe the role of NPM1 mutations in inducing selective drug sensitivity, THP-1 cells expressing NPM1mA were incubated with DNR (0.25 μM) or Ara-C (8.5 μM) for 24h. Flow cytometric was performed to detect cell apoptosis (Figure 2A and 2B). As shown in Figure 2C, after DNR treatment, the percentage of apoptotic cells was significantly higher in NPM1mA group as compared to Blank and pEGFPC1 group (P<0.05). The mean value of annexin V-positive cells was 21.35±2.44% in Blank group and 20.96±1.64% in pEGFPC1 group after DNR incubation. While in NPM1mA cells, the mean value increased to 36.53±1.95% after DNR incubation. No notable difference was observed between the two control groups. Similarly, Ara-C induced a significantly higher rate of apoptosis in NPM1 mA group (22.53±2%) as compared to Blank and pEGFPC1 group (9±1.44% and 12±1.32%, respectively) (P<0.05) (Figure 2D). These results indicated that expressing NPM1 mA increased THP-1 leukemia cells sensitivity to chemotherapeutic agents.
Sensitization of THP-1 cells to DRN or Ara-C induced apoptosis through expression of NPM1mA. (A, C) THP-1 cells expressing NPM1mA or pEGFPC1 were treated with DNR (0.25 μM, 24h). (B, D) THP-1 cells expressing NPM1mA or pEGFPC1 were treated with Ara-C (8.5 μM, 24h). After the incubation of DNR or Ara-C, the cells were washed and the apoptosis rate was assessed by flow cytometry using Annexin V- FITC. The results in (A, B) are a representative experiment. The results in (C, D) represent the mean ± SD. of the percentage of Annexin V-positive cells from three determinations. Asterisks (*) indicate significant differences in relation to Blank or pEGFPC1 group cells.
Effect of NPM1 mA on the NF-κB activity of THP-1 cells
Recent studies have established that NF-κB activation is a key response of leukemia cell to chemotherapy [19, 20]. To define whether NF-κB activity is related to the chemosensitivity mediated by NPM1 mA, we used luciferase reporter assays to detect the NF-κB transcription activity of THP-1 cells in the presence and absence of chemotherapeutical agents. As shown in Figure 3A, NF-κB activity was significantly lower in NPM1mA group cells compared to the two control groups when treated with DNR (P<0.05). The mean basal level of NF-κB activity was 4.64±0.26% in NPM1mA group cells in the presence of DNR for 24 h. By contrast, in Blank and pEGFPC1 group, the mean basal level was 11.14±0.22%, 10.21±0.26%, respectively.
Effect of NPM1 mA on the NF-κB activity in THP-1 cells after drug treatment. (A) THP-1 cells expressing NPM1mA or pEGFPC1 were treated with DNR (0.25 μM, 24h). (B) THP-1 cells expressing NPM1mA or pEGFPC1 were treated with Ara-C (8.5 μM, 24h). NF-κB transcription activity was detected by luciferase reporter assays in the presence and absence of drug. Asterisks (*) indicate significant differences in relation to Blank or pEGFPC1 group.
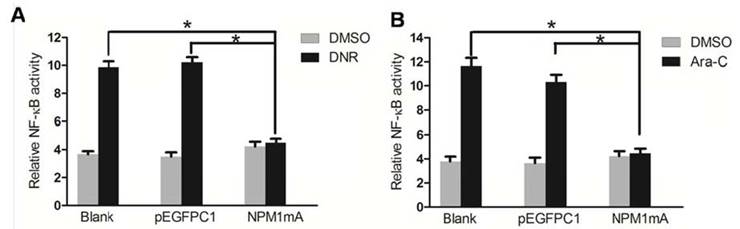
Similarly results were also observed in cells treatment with Ara-C (Figure 3B). NF-κB activity was significantly lower in NPM1mA group cells (4.56±0.37%) compared to the two control groups (11.45±0.26%, 10.78±0.47%, respectively) (P<0.05).
Restoration of NF-κB activity attenuated the effect of NPM1mA on chemosensitivity
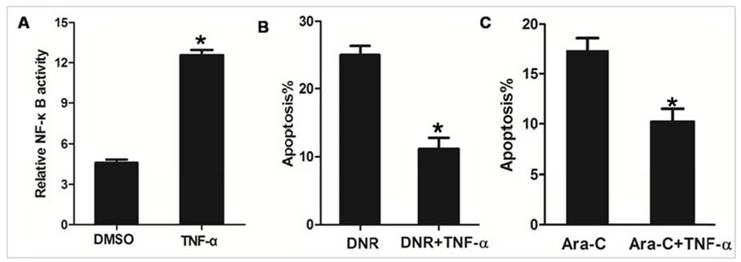
TNF-α is known to stimulate the activity of NF-κB [30]. As shown in Figure 4A, we observed that NF-κB transcription activity was significantly increased in NPM1mA group cells treated with TNF-α, as compared to cells treated with DMSO. To further confirm that NPM1mA expression mediates sensitization of AML cells to apoptosis through an NF-κB dependent mechanism, NPM1mA group cells were pre-treatment with TNF-α. As shown in Figure 4B, pre-treatment with TNF-α attenuated DNR or Ara-C induced apoptosis. Taken together, these data indicated that NF-κB involved in the chemosensitivity mediated by NPM1 mA.
TNF-α stimulation attenuated the effect of NPM1mA on THP-1 cells sensitive to drug. (A) THP-1 cells expressing NPM1 mA cells were treated with TNF-α (100 μM, 12h) and NF-κB transcription activity was detected by luciferase reporter assays. (B) THP-1 cells expressing NPM1 mA cells were treated with or without TNF-α (100 μM, 12h) following DNR (0.25 μM, 24h) incubation. (C) THP-1 cells expressing NPM1 mA cells were treated with or without TNF-α (100 μM, 12h) following Ara-C (8.5 μM, 24h) incubation. The apoptosis rate was assessed by flow cytometry using Annexin V-FITC. Asterisks (*) indicate significant differences in relation to control untreated cells.
Effect of NPM1 mA on the expression of Bax and Bcl-2
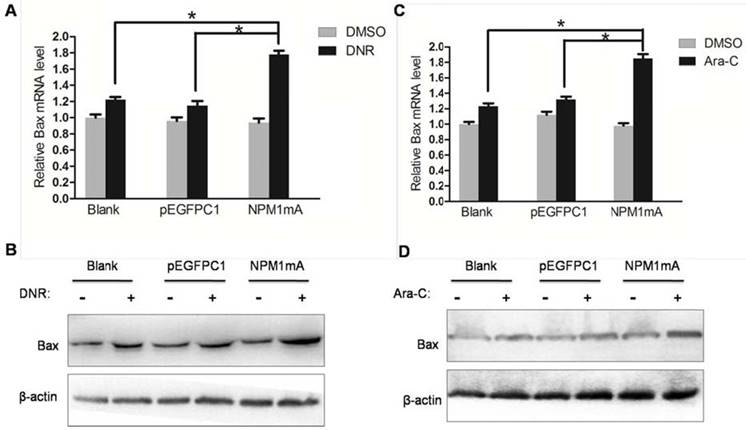
To further clarify the molecular mechanism of NPM1 mA contributing to chemosensitivity in leukemia cells. Number of Bcl-2 family proteins had been implicated in the regulation drug resistance of AML cell [31, 32]. And NF-κB has been demonstrated to regulate multiple Bcl-2 family members expression. Therefore, we examined the expression of pro-apoptotic proteins Bax as well as anti-apoptotic proteins Bcl-2 in THP-1 cells expressing NPM1 mA treated with DNR or Ara-C. As shown in Figure 5A and 5B, the mRNA and protein levels of Bax were significantly increased in NPM1 mA group cells after treatment with DNR compared with Blank group and pEGFPC1 group. Similarly results were also observed in cells treatment with Ara-C (Figure 5C and 5D).
Effect of NPM1mA on the expression of Bax in THP-1 cells after drug treatment. (A, B) THP-1 cells expressing NPM1mA or pEGFPC1 were treated with DNR (0.25 μM, 24h). (C, D) THP-1 cells expressing NPM1mA or pEGFPC1 were treated with Ara-C (8.5 μM, 24h). Quantitative RT-PCR and Western blot were used to assay the expression of Bax. Asterisks (*) indicate significant differences in relation to Blank or pEGFPC1 group cells.
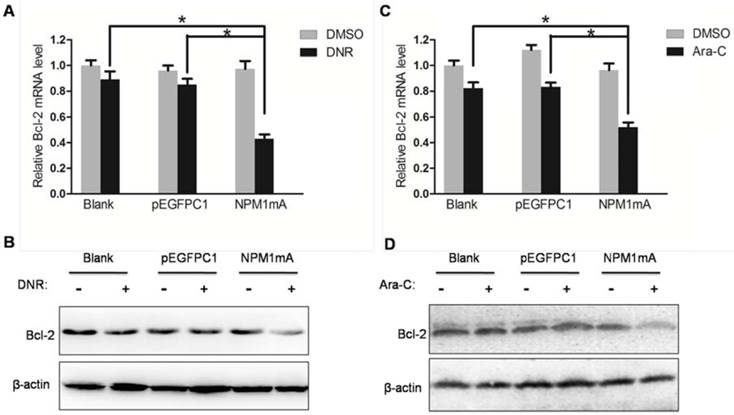
In addition, we asked whether anti-apoptotic Bcl-2 expression changed in THP-1cells expressing NPM1 mA. As shown in Figure 6, after treatment with DNR or Ara-C, the mRNA and protein levels of Bcl-2 were significantly decreased in NPM1 mA group compared with Blank group and pEGFPC1 group.
Effect of NPM1mA on the expression of Bcl-2 in THP-1 cells after drug treatment. (A, B) THP-1 cells expressing NPM1mA or pEGFPC1 were treated with DNR (0.25 μM, 24h). (C, D) THP-1 cells expressing NPM1mA or pEGFPC1 were treated with Ara-C (8.5 μM, 24h). Quantitative RT-PCR and Western blot were used to assay the expression of Bcl-2. Asterisks (*) indicate significant differences in relation to Blank or pEGFPC1 group cells.
Restoration of NF-κB activity attenuated the effect of NPM1mA on Bax/Bcl-2 expression
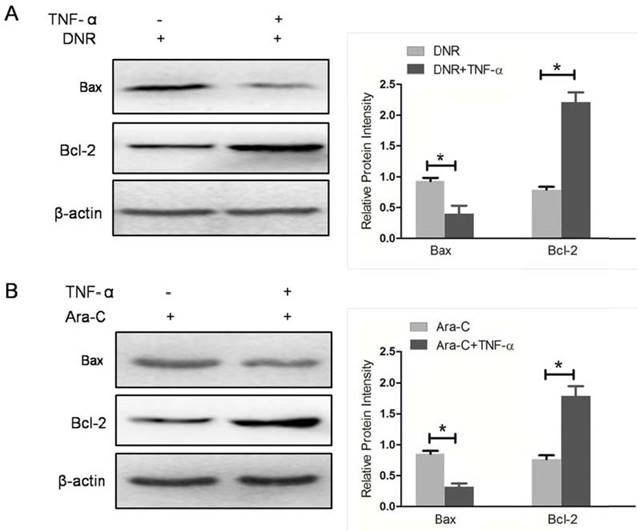
To furthermore confirm NF-κB activity involved in Bcl-2 family proteins expression mediated by NPM1 mA, the NF-κB activator, TNF-α, was used to pre-treat THP-1 cells expressing NPM1 mA when cells incubated with DNR or Ara-C. We detected the levels of Bax and Bcl-2 expression in THP-1 cells with NPM1 mA. As shown in Figure 7, TNF-α pre-treatment resulted in a significantly downregulated in Bax expression, while an upregulated in Bcl-2 expression (Figure 7).
TNF-α stimulation attenuated the effect of NPM1mA on Bax/Bcl-2 expression. (A) THP-1 cells expressing NPM1 mA cells were treated with or without TNF-α (100 μM, 12h) following DNR (0.25 μM, 24h) incubation. (B) THP-1 cells expressing NPM1 mA cells were treated with or without TNF-α (100 μM, 12h) following Ara-C (8.5 μM, 24h) incubation. Western blot was used to assay the expression of Bax and Bcl-2. All the proteins were quantified using image software and normalized against β-actin. Asterisks (*) indicate significant differences in relation to control untreated cells.
High Bax and low Bcl-2 mRNA expression in primary human AMLs with NPM1 mutation
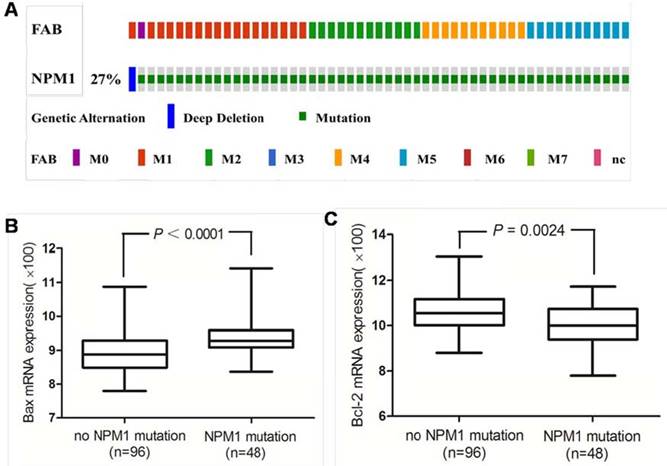
Next we investigated the Bax and Bcl-2 mRNA expression in primary human AMLs with NPM1 mutation. The 165 fully annotated human AML samples from TCGA were queried for NPM1 and NPM1 mutation was seen in M0, M1, M2, M4 and M5 FAB categories excluded M3, M6 and M7 (Figure 8A). Then we accessed the RNA-Seq data for 48 TCGA AMLs with NPM1 mutation through cBioPortal. The Bax mRNA expression levels were significantly elevated in the AMLs with NPM1 mutation compared with those with no NPM1 mutation (P<0.0001, Figure 8B), while Bcl-2 expression levels were significantly reduced in the AMLs with NPM1 mutation compared with those with no NPM1 mutation (P= 0.0024, Figure 8C).
High Bax and low Bcl-2 mRNA expression in primary human AMLs with NPM1 mutation. (A) NPM1 mutation was analyzed using cBioPortal on 165 fully annotated patients from the 2013 TCGA human AML dataset. The percentage of total mutated cases in the dataset was listed, and cases without NPM1mutation were not fully shown. Cases were sorted by the French American British (FAB) classification. (B) Bax mRNA expression data for AML cases from TCGA were assessed. Brackets indicate the 95% confidence intervals, and black lines indicate the median value of Bax mRNA expression. The Bax mRNA expression levels were significantly elevated in the AMLs with NPM1 mutation compared with those with no NPM1 mutation (P<0.0001). (C) Bcl-2 expression levels were significantly reduced in the AMLs with NPM1 mutation compared with those with no NPM1 mutation (P= 0.0024).
Discussions
AML has a poor prognosis in older patients, and intensive induction carries significant mortality [33]. Mounting evidence indicates that the difference between success and failure of treatment of AML is largely determined by genotypic leukemia-specific differences among patients [34]. AML patients harboring NPM1 mutations without an FLT3-ITD mutation showed improved survival when treated with intensive chemotherapy and better prognosis [10-13], but the mechanisms still have to be elucidated. Study the molecular basis of this observation will help us better treat chemoresistance AML. Here, our data show that NPM1 gene mutations may confer increased sensitivity to chemotherapeutic agents via regulating NF-κB activity and Bcl-2 family proteins expression.
Leukemic cells bearing NPM1 mutations frequently show myelomonocytic or monocytic features with dysplasia of two or more cell lineages [35]. In this study, THP-1 cells derived from human monocytic leukemia without NPM1 mutation and FLT3-ITD were selected as a cellular model, and the cell lines stably expressing NPM1 mutant A variant (NPM1 mA) were established. We observed that enhanced expression of NPM1 mA increased drug sensitivity of THP-1 cells. Mitchell BS et al. [36] found that introduction NPM1 mA into K562 cells sensitized these cells to apoptosis induced by drugs such as bortezomib and arsenic trioxide (ATO). Importantly, These results are consistent with previous clinical trial studies that AML patients harboring NPM1 mutations are associated with responsiveness to chemotherapy and better prognosis [10-13].
Recent studies have established that NF-κB activation is a key response of leukemia cell to chemotherapy [19]. There was report that many types of AMLs produce TNF-α, and this TNF-α acts in an autocrine fashion to promote AML cells survival and self-renewal through activation of NF-κB [18]. In addition, inhibition of NF-κB increased sensitivity of AML to many chemotherapeutical agents [22]. In our experiments, we found that expression of NPM1 mA reduced the NF-κB transcription activity of THP-1 cells upon drug treatment. Furthermore, restoration of NF-κB by TNF-α pre-treatment eliminated the effect of NPM1 mA on apoptosis induced by DNR or Ara-C. Sanjit K. Dhar et al. [37] demonstrated that wild type NPM protein could interact with NF-κB proteins, and influence NF-κB transcription activity. NPM1 mutation protein was demonstrated to retain functional interactions with both their nuclear partners and wild-type NPM [38]. Thus, NPM1 mutation protein may interact with NF-κB proteins and influence NF-κB-induced gene expression. These results indicated that NF-κB activity was involved in NPM1mA regulated chemosensitivity of leukemia cells.
The accumulating evidence indicates that Bcl-2 family proteins are important in AML survival and resistance to chemotherapy [31, 32]. The balance of pro- and anti-apoptotic proteins is an important determinant of cell sensitivity to apoptosis [39]. The expression of these pro- and anti-apoptotic proteins is in turn regulated by NF-κB [40]. Therefore, we extended our investigation to study possible modifications of NF-κB downstream molecules, specifically the Bcl-2 family of apoptosis regulatory proteins. We found that expression of NPM1 mA upregulated Bax and downregulated Bcl-2 at both mRNA and protein levels in cells treated with DNR or Ara-C. In addition, restoration of NF-κB attenuated the effect of NPM1mA on Bax/Bcl-2 expression. Previous studies demonstrated that overexpression of Bcl-2 was associated with drug resistance in hematologic malignancies [41], whereas a high Bax level was found to be a good prognostic indicator in AML [42]. Based on Bax and Bcl-2 were involved in the effect of NPM1mA on chemosensitivity. We further confirmed the Bax and Bcl-2 mRNA expression in primary human AMLs with NPM1 mutation. Evaluation of mRNA expression data from TCGA dataset indicated that the AMLs with NPM1 mutation had significantly higher Bax mRNA expression levels and lower Bcl-2 mRNA expression levels. The results were consistent with a former study that AML patients carrying the NPM1 mutations showed a high Bax/Bcl-2 ratio [43]. This finding suggests that NPM1 mutation protein may regulate Bax/Bcl-2 expression and increase chemosensitivity of leukemia cells. In a recent study, Jochen Greiner et al. [44] reported that NPM1 mutant-triggered CD8+ and CD4+ T-cell immune response may result in a T-cell orchestration that could be responsible for the favorable prognosis of AML patients with NPM1 mutations. Thus, in addition to NF-κB pathway, further mechanistic studies are still needed to determine in NPM1 mutation-induced leukemia chemosensitization.
In conclusion, our studies demonstrated that NPM1 gene mutations may confer increased sensitivity to chemotherapeutic agents by inhibiting NF-κB activity and regulating Bax/Bcl-2 expression in vitro. This new information casts a light on the favorable prognosis of patients carrying the NPM1 mutation, and further studies should explore the role of NPM1 mutants in leukemia chemosensitization in a proper animal model and clinical study.
Abbreviations
NPM1: nucleophosmin; AML: acute myeloid leukemia; NPM1mA: type A NPM1 mutation; NPMc+: cytoplasmic nucleophosmin; DNR: daunorubicin; Ara-C: cytarabine; NF-κB: nuclear factor-κB; TNF-α: tumor necrosis factor-α;
RT-PCR: reverse transcription PCR; qRT-PCR: quantitative real time PCR; TCGA: The Cancer Genome Atlas; FAB: French American British.
Acknowledgements
This work was supported by the National Natural Science Foundation of China (No. 81271913) and the Technology Foundation for Selected Overseas Chinese Scholar, Ministry of Personnel of China (No. 2013009). This study was also partly supported by a Graduate Fellowship in research innovation from the Chongqing Municipal Education Commission (No. CYS16140).
Competing Interests
The authors have declared that no competing interest exists.
References
1. Okuwaki M. The structure and functions of NPM1/Nucleophsmin/B23, a multifunctional nucleolar acidic protein. Journal of biochemistry. 2008;143(4):441-448
2. Lindstrom MS. NPM1/B23: A Multifunctional Chaperone in Ribosome Biogenesis and Chromatin Remodeling. Biochem Res Int. 2011;2011:195209
3. Falini B, Mecucci C, Saglio G. et al. NPM1 mutations and cytoplasmic nucleophosmin are mutually exclusive of recurrent genetic abnormalities: a comparative analysis of 2562 patients with acute myeloid leukemia. Haematologica. 2008;93(3):439-442
4. Di Matteo A, Franceschini M, Chiarella S. et al. Molecules that target nucleophosmin for cancer treatment: an update. Oncotarget. 2016;7(28):44821-44840
5. Falini B, Bolli N, Liso A. et al. Altered nucleophosmin transport in acute myeloid leukaemia with mutated NPM1: molecular basis and clinical implications. Leukemia. 2009;23(10):1731-1743
6. Falini B, Bolli N, Shan J. et al. Both carboxy-terminus NES motif and mutated tryptophan(s) are crucial for aberrant nuclear export of nucleophosmin leukemic mutants in NPMc+ AML. Blood. 2006;107(11):4514-4523
7. Falini B, Mecucci C, Tiacci E. et al. Cytoplasmic nucleophosmin in acute myelogenous leukemia with a normal karyotype. N Engl J Med. 2005;352(3):254-266
8. Falini B. Acute myeloid leukemia with mutated nucleophosmin (NPM1): molecular, pathological, and clinical features. Cancer treatment and research. 2010;145:149-168
9. Haferlach C, Mecucci C, Schnittger S. et al. AML with mutated NPM1 carrying a normal or aberrant karyotype show overlapping biologic, pathologic, immunophenotypic, and prognostic features. Blood. 2009;114(14):3024-3032
10. Dohner K, Schlenk RF, Habdank M. et al. Mutant nucleophosmin (NPM1) predicts favorable prognosis in younger adults with acute myeloid leukemia and normal cytogenetics: interaction with other gene mutations. Blood. 2005;106(12):3740-3746
11. Schnittger S, Schoch C, Kern W. et al. Nucleophosmin gene mutations are predictors of favorable prognosis in acute myelogenous leukemia with a normal karyotype. Blood. 2005;106(12):3733-3739
12. Shahab S, Shamsi TS, Ahmed N. Prognostic Involvement of Nucleophosmin Mutations in Acute Myeloid Leaukemia. Asian Pacific Journal of Cancer Prevention. 2013;14(10):5615-5620
13. Becker H, Marcucci G, Maharry K. et al. Favorable prognostic impact of NPM1 mutations in older patients with cytogenetically normal de novo acute myeloid leukemia and associated gene- and microRNA-expression signatures: a Cancer and Leukemia Group B study. Journal of clinical oncology. 2010;28(4):596-604
14. Razani B, Cheng G. NF-kappaB: much learned, much to learn. Science signaling. 2010;3(138):pe29
15. Klein U, Ghosh S. The Two Faces of NF-kappaB Signaling in Cancer Development and Therapy. Cancer cell. 2011;20(5):556-558
16. Tornatore L, Thotakura AK, Bennett J. et al. The nuclear factor kappa B signaling pathway: integrating metabolism with inflammation. Trends in cell biology. 2012;22(11):557-566
17. Guzman ML, Neering SJ, Upchurch D. et al. Nuclear factor-kappaB is constitutively activated in primitive human acute myelogenous leukemia cells. Blood. 2001;98(8):2301-2307
18. Volk A, Li J, Xin J. et al. AML Cells Utilize TNF-Driven JNK Signaling As a Critical NF-κB-Independent Survival Signal. Blood. 2013;122:2890
19. Jacamo R, Chen Y, Wang Z. et al. Reciprocal leukemia-stroma VCAM-1/VLA-4-dependent activation of NF-kappaB mediates chemoresistance. Blood. 2014;123(17):2691-2702
20. Turco MC, Romano MF, Petrella A. et al. NF-kappaB/Rel-mediated regulation of apoptosis in hematologic malignancies and normal hematopoietic progenitors. Leukemia. 2004;18(1):11-17
21. Gupta SC, Sundaram C, Reuter S. et al. Inhibiting NF-kappaB activation by small molecules as a therapeutic strategy. Biochimica et biophysica acta. 2010;1799(10-12):775-787
22. Haruki K, Shiba H, Fujiwara Y. et al. Inhibition of nuclear factor-kappaB enhances the antitumor effect of tumor necrosis factor-alpha gene therapy for hepatocellular carcinoma in mice. Surgery. 2013;154(3):468-478
23. Braun T, Carvalho G, Fabre C. et al. Targeting NF-kappaB in hematologic malignancies. Cell death and differentiation. 2006;13(5):748-758
24. Stark LA, Dunlop MG. Nucleolar sequestration of RelA (p65) regulates NF-kappaB-driven transcription and apoptosis. Molecular and cellular biology. 2005;25(14):5985-6004
25. Yang YI, Ahn JH, Lee KT. et al. RSF1 is a positive regulator of NF-kappaB-induced gene expression required for ovarian cancer chemoresistance. Cancer research. 2014;74(8):2258-2269
26. Qu Y, Qu B, Wang X. et al. Knockdown of NF-kappaB p65 subunit expression suppresses growth of nude mouse lung tumour cell xenografts by inhibition of Bcl-2 apoptotic pathway. Cell biochemistry and function. 2015;33(5):320-325
27. Ortiz-Lazareno PC, Bravo-Cuellar A, Lerma-Diaz JM. et al. Sensitization of U937 leukemia cells to doxorubicin by the MG132 proteasome inhibitor induces an increase in apoptosis by suppressing NF-kappa B and mitochondrial membrane potential loss. Cancer cell international. 2014;14(1):13
28. Cerami E, Gao J, Dogrusoz U. et al. The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer discovery. 2012;2(5):401-404
29. Gao J, Aksoy Bü A, Dogrusoz U. et al. Integrative Analysis of Complex Cancer Genomics and Clinical Profiles Using the cBioPortal. Science signaling. 2013;6(269):pl1
30. Lee RE, Walker SR, Savery K. et al. Fold change of nuclear NF-kappaB determines TNF-induced transcription in single cells. Molecular cell. 2014;53(6):867-879
31. Reed JC. Bcl-2-family proteins and hematologic malignancies: history and future prospects. Blood. 2008;111(7):3322-3330
32. Brinkmann K, Kashkar H. Targeting the mitochondrial apoptotic pathway: a preferred approach in hematologic malignancies? Cell death & disease. 2014;5(3):e1098
33. Burnett A, Wetzler M, Lowenberg B. Therapeutic advances in acute myeloid leukemia. J Clin Oncol. 2011;29(5):487-494
34. Lowenberg B. Acute myeloid leukemia: the challenge of capturing disease variety. Hematology Am Soc Hematol Educ Program. 2008:1-11
35. Falini B, Sportoletti P, Martelli MP. Acute myeloid leukemia with mutated NPM1: diagnosis, prognosis and therapeutic perspectives. Current opinion in oncology. 2009;21(6):573-581
36. Huang M, Thomas D, Li MX. et al. Role of cysteine 288 in nucleophosmin cytoplasmic mutations: sensitization to toxicity induced by arsenic trioxide and bortezomib. Leukemia. 2013;27(10):1970-1980
37. Dhar SK, Lynn BC, Daosukho C. et al. Identification of nucleophosmin as an NF-kappaB co-activator for the induction of the human SOD2 gene. The Journal of biological chemistry. 2004;279(27):28209-28219
38. Leong SM, Tan BX, Bte Ahmad B. et al. Mutant nucleophosmin deregulates cell death and myeloid differentiation through excessive caspase-6 and -8 inhibition. Blood. 2010;116(17):3286-3296
39. Sarosiek KA, Ni Chonghaile T, Letai A. Mitochondria: gatekeepers of response to chemotherapy. Trends in cell biology. 2013;23(12):612-619
40. Yinjun L, Jie J, Yungui W. Triptolide inhibits transcription factor NF-kappaB and induces apoptosis of multiple myeloma cells. Leukemia research. 2005;29(1):99-105
41. Bradbury DA, Zhu YM, Russell NH. et al. Bcl-2 expression in acute myeloblastic leukaemia: relationship with autonomous growth and CD34 antigen expression. Leukemia Lymphoma. 1997;24(3-4):221-228
42. Ong YL, McMullin MF, Bailie KE. et al. High bax expression is a good prognostic indicator in acute myeloid leukaemia. British journal of haematology. 2000;111(1):182-189
43. Del Poeta G, Ammatuna E, Lavorgna S. et al. The genotype nucleophosmin mutated and FLT3-ITD negative is characterized by high bax/bcl-2 ratio and favourable outcome in acute myeloid leukaemia. British journal of haematology. 2010;149(3):383-387
44. Greiner J, Ono Y, Hofmann S. et al. Mutated regions of nucleophosmin 1 elicit both CD4(+) and CD8(+) T-cell responses in patients with acute myeloid leukemia. Blood. 2012;120(6):1282-1289
Author contact
![]() Corresponding author: Ling Zhang, College of Laboratory Medicine, Chongqing Medical University, No. 1, Yixueyuan Road, Chongqing, 400016, China. Tel: +86 023-68485240, Fax: +86 0 23-68485239 Email: lingzhangedu.cn.
Corresponding author: Ling Zhang, College of Laboratory Medicine, Chongqing Medical University, No. 1, Yixueyuan Road, Chongqing, 400016, China. Tel: +86 023-68485240, Fax: +86 0 23-68485239 Email: lingzhangedu.cn.