Impact Factor ISSN: 1837-9664

 Global reach, higher impact
Global reach, higher impactJ Cancer 2017; 8(1):140-145. doi:10.7150/jca.15838 This issue Cite
Short Research Communication
The Novel PIM1 Inhibitor NMS-P645 Reverses PIM1-Dependent Effects on TMPRSS2/ERG Positive Prostate Cancer Cells And Shows Anti-Proliferative Activity in Combination with PI3K Inhibition
Luca Mologni1 ![]() *, Vera Magistroni1
*, Vera Magistroni1 ![]() *, Francesco Casuscelli2, Marisa Montemartini2, Carlo Gambacorti-Passerini1,3
*, Francesco Casuscelli2, Marisa Montemartini2, Carlo Gambacorti-Passerini1,3
1. School of Medicine and Surgery, University of Milan-Bicocca, Monza, Italy;
2. Nerviano Medical Sciences srl, Nerviano, Milan, Italy;
3. Hematology and Clinical Research Unit, San Gerardo Hospital, Monza, Italy.
* These authors contributed equally to the work.
Received 2016-4-13; Accepted 2016-9-18; Published 2017-1-1
Abstract
PIM1 is over-expressed in multiple tumors, including prostate cancer (PCa). PIM1 upregulation is mediated by direct binding of the ERG transcription factor to its promoter. About 50% of PCa cases are characterized by the presence of the TMPRSS2/ERG fusion, leading to ERG over-expression and thus to PIM1 transcriptional activation. PIM kinases are considered as weak oncogenes, but when combined with additional genetic alterations can induce strong transforming effects. Here we show anti-proliferative activity of the newly described PIM1 inhibitor NMS-P645 in combination with the PI3K inhibitor GDC-0941 in TMPRSS2/ERG positive and negative PCa cells. Treatment with NMS-P645 alone can reverse PIM1-mediated pro-survival signals in prostate cells, such as activation of STAT3 through Tyr705 phosphorylation and resistance to taxane-based treatments, but does not exert a strong anti-tumoral effect. However, the simultaneous treatment with NMS-P645 and GDC-0941 induces a significant anti-proliferative response in PCa cells. These results support the use of combination strategies with PIM and PI3K inhibitors as effective treatment for PCa cases.
Keywords: Prostate cancer, PIM1, PI3K, kinase, inhibitor.
Introduction
Prostate cancer (PCa) is still a significant medical burden in developed countries. The fusion between the prostate-specific TMPRSS2 gene (21q22.2) and the coding region of the ERG transcription factor (21q22.3) leads to ERG over-expression and characterizes about 50% of PCa cases [1]. Despite the high frequency of this genetic rearrangement, expression of TMPRSS2/ERG fusion alone is not sufficient to generate a fully transformed phenotype in prostate cells, both in vitro and in vivo [2-5]. One of the consequences of aberrant ERG expression is the upregulation of PIM1 transcription, through direct binding of ERG to PIM1 promoter [5, 6]. The PIM (Proviral Integration site of mouse Moloney leukemia virus) serine/threonine kinase family comprises three distinct kinases (PIM1, PIM2 and PIM3) highly conserved throughout evolution [7]. All three enzymes lack a regulatory domain, suggesting a constitutive activity which is directly correlated with their expression levels [8]. PIM1 is known to be upregulated in multiple tumors through different pathways, including ERG, JAK/STAT, hypoxia [9] and NF-kB [10-12]. In particular, STAT3 and STAT5 are known to bind to PIM1 promoter, upregulating its transcription [13, 14]. It is widely accepted that PIM kinases are weak oncogenes, since their over-expression in prostate, liver and lymphoid tissues induces tumoral transformation after a long latency and with a low penetrance [15]. We and others have shown that, in PCa, PIM1 over-expression is associated with increased genomic instability after taxane-based treatments [5, 16, 17]. The simultaneous presence, in prostate cells, of additional genetic alterations such as MYC upregulation, induces a strong transforming effect in PIM1 overexpressing cells [18], suggesting a cooperative oncogenic property. The synergistic activity with other oncogenes is further supported by the increased tumorigenic potential of advanced-stage PCa cell lines after induction of PIM1 over-expression [19]. These evidences, together with recent pre-clinical data on drugs blocking PIM kinase activity, suggest that PIM1 inhibition could be most effective when used in combination with additional targeted therapies or chemotherapies [11]. Several studies indicate the presence of a cross-talk between PIM and AKT kinases [11, 20]. The PI3K/AKT pathway is commonly hyper-activated in prostate cancer, but the therapeutic efficacy of its inhibition is limited by the presence of compensatory pathways mediated, at least in part, by PIM1 activity [20]. Here we show the biological activity of the newly described PIM1 inhibitor NMS-P645 [21] in prostate cancer cell lines, demonstrating its anti-proliferative role when used in combination with the PI3K inhibitor GDC-0941 [22].
Materials and Methods
Cell lines
The human prostate cell lines RWPE-1, 22Rv1 and VCaP were obtained from the American Type Culture Collection (ATCC). Immortalized nonmalignant RWPE-1 cells were maintained in keratinocyte-serum free medium supplemented with epidermal growth factor and bovine pituitary extract. 22Rv1 and VCaP cells were maintained in RPMI and DMEM medium, respectively, supplemented with 10% FBS.
Plasmids and transfections
pHCMV2-empty and phCMV2_HA_tERG plasmids have been described elsewhere together with the creation of RWPE-1_Empty and RWPE-1_tERG stable transfectants [5]. To obtain the phCMV2_PIM1 vector, we amplified the PIM1 cDNA from RWPE-1 cells with PIM1_Fw 5'tagaattctgatgctcttgtccaaaatc3' and PIM1_Rw 5'TACGCGGCCGCCTATTTGCTGGGCCCCG3' primers. The amplified fragment was inserted into phCMV2 through an EcoRI/NotI digestion followed by ligation with T4 DNA ligase (New England Biolabs, Ipswich, MA, USA). RWPE-1 cells were transfected with Fugene 6 (Roche, Applied Science, Germany) according to manufacturer's instruction and selected for geneticin (Invitrogen, Carlsbad, CA, USA) resistance at 0.25 mg/ml to obtain the RWPE-1_PIM1 cell line. Short interfering RNA-mediated knockdown of PIM1 and ERG was performed with siRNA from Dharmacon (GE Healthcare, UK) as described [5].
Compounds
NMS-P645 was synthesized by Nerviano Medical Sciences as described [21]. GDC-0941 and vandetanib were purchased from Selleck Chemicals. Taxol was from Sigma Aldrich (Merck group, Germany). All compounds were dissolved at the desired concentration following manufacturer's guidelines.
Western Blot analysis
Western Blot was performed as previously described [5] using the following antibodies: αPIM1 (12H8) (Santa Cruz Biotechnology, CA, USA), αHA (Covance, Princeton, NJ, USA), αActin (Sigma-Aldrich, Merck group, Germany), αSTAT3 (Calbiochem, Merk group, Germany); phospho-specific antibodies were purchased from Cell Signalling Technology (Denvers, MA, USA): αp-STAT3Y705 (clone 3E2, cat. #9138) , αp-p70S6KT389 (#9234) and αp-RPS6S240/244 (#5364), anti-MIG6 (#2440), α-RPS6 (#2217), α-p70S6K(2708), pSRC (#2101).
Proliferation Assay
Exponentially growing cells were plated in at least three replicates in 96-well plates at a density of 3000 cells/well. Cells were treated with drugs or vehicle alone for the indicated time. One µCi of [3H]thymidine was added to each well 8 hours before harvesting onto glass fiber filters by a Tomtec automated cell harvester. Incorporation of [3H]thymidine was measured using a filter scintillation counter (1450 MicroBeta Wallac Trilux, Perkin Elmer, MA, USA).
Results
NMS-P645 reverses PIM1-induced aneuploidy and STAT3 activation in prostate cells
The immortalized prostate cell line RWPE-1 was stably transfected with the TMPRSS2/ERG-coding sequence (RWPE-1_tERG) or the empty vector (RWPE-1_Empty) as described in [5] (Figure 1A). tERG over-expression induces PIM1 upregulation, as demonstrated in [5] and shown in Figure 1B. It has previously been shown that PIM1 over-expression is able to induce aneuploidy in RWPE-1 cells especially after treatment with taxane agents [5, 17]. In RWPE-1_tERG cells, the PIM1 inhibitor NMS-P645 reversed taxol-induced aneuploidy, reducing the >4n population after 36 hours of taxol treatment to basal content, from 5.5% to 1.7% (Figure 1C). Furthermore, NMS-P645 re-sensitized RWPE-1_tERG cells to taxane cytotoxicity, to the levels observed in RWPE-1_Empty cells (subG0 Empty_Taxol: 48.5%; subG0 tERG_Taxol: 35.9%; subG0 tERG_Taxol_NMS-P645: 49%).
The tERG-PIM1 axis favors resistance to taxane-based agents and induces STAT3 activation through PIM1 upregulation. (A) Anti-HA immunoblot of whole cell lysates of RWPE-1 transfected with empty vector or HA-tagged tERG. Actin was used as a loading control. (B) Anti- PIM1, activated STAT3 (pSTAT3Y705) and total STAT3 immunoblot of RWPE-1_Empty, RWPE-1_tERG and RWPE-1_PIM1 cells. The PIM1 band in RWPE-1_PIM1 cells corresponds to the ectopic HA-tagged PIM1 and appears at a slightly higher MW compared to the endogenous PIM1 in RWPE-1_tERG lane. A graph representing the fold change of p-STAT3 signal relative to total STAT3 in RWPE-1 transfectants is shown. The values are normalized on RWPE-1_Empty signal. (C) PropIdium Iodide (PI) staining of RWPE-1 transfectants in the presence (+) or in the absence (-) of Taxol and NMS-P645 after 36 hours treatment. The fraction (%) of polyploid and apoptotic cells is indicated in the right-hand side and in the top-left corner of each plot, respectively. (D) Silencing of ERG (siERG) or PIM1 (siPIM1) reduces STAT3 tyrosine phosphorylation in RWPE-1_tERG transfectants. RWPE-1_Empty cells are shown as a control; siNT, scrambled control RNAi. The fold change of the p-STAT3 signal relative to total STAT3 in RWPE-1 transfectants is shown with the values normalized on RWPE-1_tERG_siNT. (E-F) Inhibition of STAT3Y705 phosphorylation in RWPE-1_tERG (E) and VCaP (F) cells by NMS-P645. The p-STAT3/total STAT3 signal ratio is shown in the graph for VCaP cells. Values are normalized on non-treated cells (-). (G) Anti-MIG6 immunoblot of whole cells lysates from RWPE-1 transfectants. (H) Anti- pSTAT3Y705 , total STAT3, pSRCy416 of RWPE-1 transfectants treated for 6 hours with vandetanib at the indicated concentrations. Actin was used as loading control. (I) Schematic representation of the proposed model for PIM1-mediated STAT3 activation.
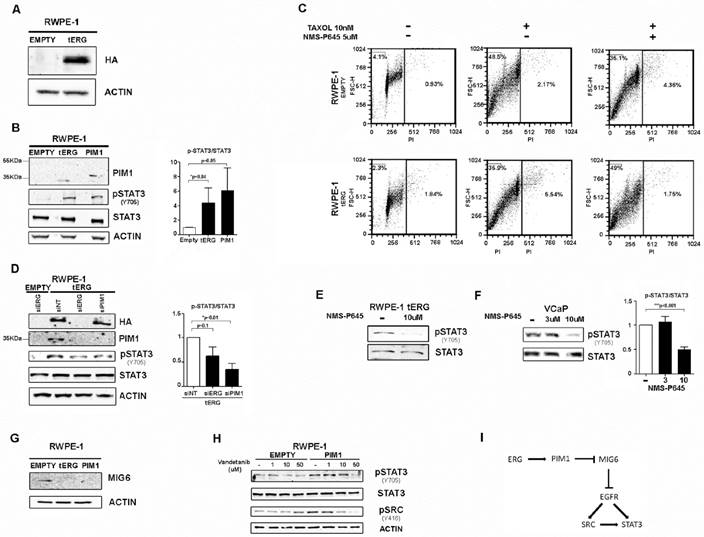
The expression of PIM1 gene is regulated by active STAT3 [14]. Here we demonstrate that PIM1 expression can work backwards and activate STAT3 inducing its phosphorylation at Tyr705 in RWPE-1 cells, both in the presence and in the absence of tERG, as shown in RWPE-1 cells stably transfected with PIM1 (Figure 1B). Specific silencing of PIM1 in RWPE1_tERG cells reduces STAT3 phosphorylation, indicating a direct association between PIM1 expression and STAT3 activation (Figure 1D). Similarly, tERG silencing decreases STAT3 phosphorylation through inhibition of PIM1 expression (Figure 1D). Accordingly, PIM1 inhibition with NMS-P645 reverses STAT3 activation in the tERG-positive RWPE-1_tERG and VCaP cells (Figure 1E-F).
As PIM1 is a Ser/Thr kinase, it cannot directly phosphorylate STAT3 on Tyr 705, suggesting the presence of additional effectors mediating STAT3 tyrosine phosphorylation, that are directly altered by PIM1. We found that in RWPE-1 transfectants PIM1 over-expression induces MIG6 down-regulation, a known EGFR inhibitor [23] (Figure 1G). In addition, treatment with the EGFR inhibitor vandetanib reduces STAT3 activation in RWPE1-PIM1 cells, but not in control (EMPTY) cells (Figure 1H). The pharmacological inhibition of EGFR also affects activation of SRC, a known EGFR effector and STAT3 activator (Figure 1H) [24]. We thus suggest a role for MIG6/EGFR pathway in the PIM1-mediated STAT3 activation (Figure 1I).
Anti-proliferative activity of NMS-P645 in combination with the PI3K inhibitor GDC-0941 in prostate cancer cells
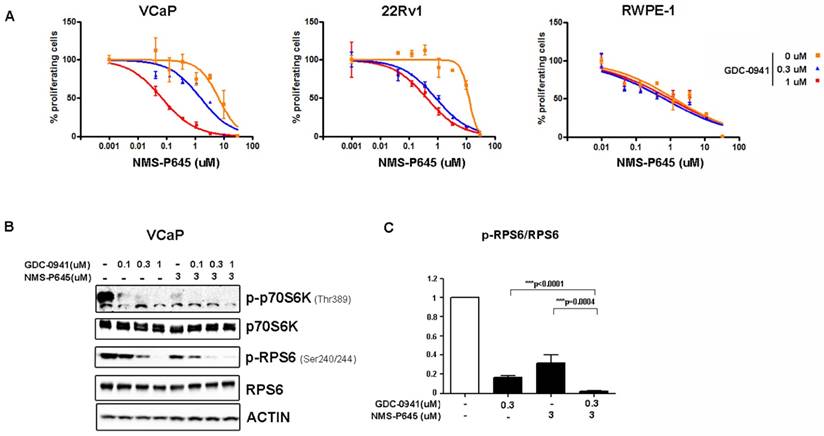
To test the anti-proliferative activity of NMS-P645 in tERG-positive prostate cells, we evaluated the proliferation rate of cell lines with a tritiated-thymidine incorporation assay after 72 hours of treatment. NMS-P645 alone did not show specific activity for PCa cell lines compared to non-tumoral cells (RWPE-1, BPH1) both in the presence (VCaP, RWPE-1-tERG) or in the absence of tERG fusion (22Rv1, DU-145, LNCaP) (Table 1). The same result was observed for PIM1 over-expressing cells (RWPE-1-PIM1). Since PIM1 inhibitors show stronger effect when used in combination with additional drugs, we treated tERG-positive (VCaP) and negative (22Rv1) PCa cell lines with NMS-P645 in combination with different compounds. NMS-P645 exhibited synergistic activity with the PI3K inhibitor GDC-0941 both in the presence and in the absence of tERG. As shown in Figure 2A, dose-response curves of NMS-P645 shifted towards the left by 1 to 2 logs in the presence of increasing concentrations of GDC-0941 in PCa cells (VCaP and 22Rv1) but not in normal prostate cells (RWPE-1), indicating specific cooperative cell growth inhibition in tumoral prostate tissues. Statistical analysis of the dose-median effect by Chou-Talalay method [25] indicated synergism in the two PCa cell lines and antagonism in normal RWPE-1 cells (Table 2). To dissect the biological mechanism at the basis of the synergistic activity between NMS-P645 and GDC-0941, we analyzed whole cell lysates of VCaP cells after treatment with the two drugs: both compounds inhibited the PI3K/AKT/mTOR pathway, as shown by inhibition of p70S6K phosphorylation. However, the combined treatment showed a superior effect on phosphorylation of the ribosomal protein S6 (RPS6), a major downstream effector of the PI3K/AKT/mTOR pathway (Figure 2B).
Sensitivity of prostate-derived cell lines to NMS-P645, analyzed by tritiated thymidine-incorporation assay. IC50 values (mean ± SD) are reported.
| Cell line | Cell type (known genetic alterations) | IC50 [μM] |
|---|---|---|
| VCaP | PCa (TMPRSS2/ERG) | 2.7 ± 1.1 |
| 22Rv1 | PCa | 5.2 ± 1.1 |
| DU-145 | PCa (UBE2L3/KRAS) | 9.7± 1.0 |
| LNCaP | PCa (MIPOL1/ETV1) | 5.4 ± 1.1 |
| BPH1 | Benign Prostate Hyperplasia | 4.9 ± 1.1 |
| RWPE1 | Normal Prostate Epithelium | 4.2 ± 1.2 |
| RWPE1-Empty | Stable Transfectant (empty vector) | 4.5 ± 1.2 |
| RWPE1-tERG | Stable Transfectant (TMPRSS2/ERG) | 4.0 ± 1.1 |
| RWPE1-PIM1 | Stable Transfectant (PIM1) | 3.5 ± 1.2 |
Treatment efficacy of NMS-P645 in combination with the PI3K inhibitor GDC-0941 in prostate cancer cells. (A) Dose-response curves of normal prostate cells (RWPE-1), tERG-positive (VCaP) and tERG-negative (22Rv1) PCa cells treated with NMS-P645 as single agent or in the presence of fixed concentrations of GDC-0941. (B) Immunoblot analysis for activated p-p70S6KT389 and p-RPS6S240/244 in VCaP cells treated with vehicle (-) or the indicated concentrations of GDC-0941 and NMS-P645. Total p70S6K and RPS6 immunoblots from the same lysates were run to show that the total protein levels are not affected. Actin is shown as a loading control. (C) The pRPS6/total RPS6 signal is shown in the graph as average of four different experiments. Values are normalized on non-treated cells.
Combination Indexes obtained in prostate cancer (VCaP and 22Rv1) and non-cancerous (RWPE-1) cell lines by NMS-P645/GDC-0941 simultaneous treatment. Graded symbols represent the average CI and are derived from the original publication [25].
| Cell line | Ratio | Combination Index (CI) at | Grade Synergism | ||
|---|---|---|---|---|---|
| ED50 | ED75 | ED90 | |||
| VCaP | 1:10 | 0.846 | 0.726 | 0.661 | ++ |
| 22Rv1 | 1:10 | 0.484 | 0.543 | 0.610 | +++ |
| RWPE-1 | 1:10 | 1.282 | 1.454 | 1.668 | - - - |
Discussion
The serine/threonine kinase PIM1 is overexpressed in several tumors, including prostate cancer, with an established role in facilitating tumorigenesis. It thus represents an encouraging target for anticancer drug discovery. The novel PIM1 inhibitor NMS-P645 has been recently described and shows strong specificity for PIM family members against a panel of kinases representing all the human protein kinase families [21]. We here demonstrate the activity of this compound in PCa cells, showing its growth inhibition effects in combination with the PI3K inhibitor GDC-0941. The use of NMS-P645 alone does not exert specific anti-tumoral activity in PCa cell lines, but it restores sensitivity to Taxol-based treatment in PIM1-overexpressing prostate cells (Figure 1C). In addition, we showed that NMS-P645 treatment can prevent PIM1-mediated activation of STAT3 (Figure 1B, 1D). Active STAT3 is known to bind to PIM1 promoter, thus increasing its expression level [14]. In our cell line models, PIM1 over-expression induces STAT3 activation, here shown by increased phosphorylation at Tyr705, revealing the existence of a positive feed-back loop between PIM1 and STAT3 in prostate cells. As PIM1 is a Ser/Thr kinase, the induction of STAT3 tyrosine phosphorylation must be indirect. We did not observe any phosphorylation on Ser727 (data not shown). We suggest that PIM1 could affect STAT3 activation in RWPE-1 cells through modulation of EGFR activity mediated by inhibition of MIG6 expression (Figure 1G-I).
The combination of NMS-P645 with GDC-0941 shows a promising synergistic effect that is selective for prostate cancer cells versus normal prostate epithelium, thus suggesting a rational for the concomitant use of NMS-P645 and PI3K pathway inhibitors for prostate cancer treatment (Figure 2). Since we recently showed that PIM1 over-expression can be driven by the presence of the TMPRSS2/ERG fusion [5], we tried to evaluate whether this combination therapy could be specific for TMPRSS2/ERG-positive PCa cell lines. We found that the treatment with NMS-P645 and GDC-0941 is active in both TMPRSS2/ERG-positive and -negative PCa cells. This is not entirely unexpected, given that PIM1 expression can be driven by several mechanisms, including activation of the JAK/STAT pathway [26]. PIM1 expression in the TMPRSS2/ERG-negative 22Rv1 cells is lower compared to VCaP cells (TMPRSS2/ERG-positive) [5], but is probably sufficient to exert a role in the tumorigenesis process.
In conclusion, we here describe the biological activity of the novel PIM inhibitor NMS-P645 in prostate cancer cell lines. The description of the synergism between NMS-P645 and GDC-0941 in PCa cells confirms the efficacy and specificity of combination strategies in prostate cancer through the concomitant inhibition of PIM1 and PI3K pathways. Further experiments are ongoing to elucidate the efficacy of this therapeutic approach in vivo.
Acknowledgements
This work was supported by Associazione Italiana Ricerca sul Cancro 2013 (IG-14249 to C.G.P.), and by European Union's Horizon 2020 Marie Skłodowska-Curie Innovative Training Networks (ITN-ETN) under grant agreement No.: 675712CGP.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Tomlins SA, Laxman B, Varambally S, Cao X, Yu J, Helgeson BE. et al. Role of the TMPRSS2-ERG gene fusion in prostate cancer. Neoplasia. 2008;10:177-88
2. Zong Y, Xin L, Goldstein AS, Lawson DA, Teitell MA, Witte ON. ETS family transcription factors collaborate with alternative signaling pathways to induce carcinoma from adult murine prostate cells. Proc Natl Acad Sci U S A. 2009;106:12465-70
3. Klezovitch O, Risk M, Coleman I, Lucas JM, Null M, True LD. et al. A causal role for ERG in neoplastic transformation of prostate epithelium. Proc Natl Acad Sci U S A. 2008;105:2105-10
4. Carver BS, Tran J, Chen Z, Carracedo-Perez A, Alimonti A, Nardella C. et al. ETS rearrangements and prostate cancer initiation. Nature. 2009;457:E1 discussion E2-3
5. Magistroni V, Mologni L, Sanselicio S, Reid JF, Redaelli S, Piazza R. et al. ERG deregulation induces PIM1 over-expression and aneuploidy in prostate epithelial cells. PloS one. 2011;6:e28162
6. Goldberg L, Tijssen MR, Birger Y, Hannah RL, Kinston SJ, Schutte J. et al. Genome-scale expression and transcription factor binding profiles reveal therapeutic targets in transgenic ERG myeloid leukemia. Blood. 2013;122:2694-703
7. Nawijn MC, Alendar A, Berns A. For better or for worse: the role of Pim oncogenes in tumorigenesis. Nature reviews Cancer. 2011;11:23-34
8. Qian KC, Wang L, Hickey ER, Studts J, Barringer K, Peng C. et al. Structural basis of constitutive activity and a unique nucleotide binding mode of human Pim-1 kinase. The Journal of biological chemistry. 2005;280:6130-7
9. Chen J, Kobayashi M, Darmanin S, Qiao Y, Gully C, Zhao R. et al. Pim-1 plays a pivotal role in hypoxia-induced chemoresistance. Oncogene. 2009;28:2581-92
10. Iwakura T, Mohri T, Hamatani T, Obana M, Yamashita T, Maeda M. et al. STAT3/Pim-1 signaling pathway plays a crucial role in endothelial differentiation of cardiac resident Sca-1+ cells both in vitro and in vivo. Journal of molecular and cellular cardiology. 2011;51:207-14
11. Warfel NA, Kraft AS. PIM kinase (and Akt) biology and signaling in tumors. Pharmacology & therapeutics. 2015;151:41-9
12. Zhu N, Ramirez LM, Lee RL, Magnuson NS, Bishop GA, Gold MR. CD40 signaling in B cells regulates the expression of the Pim-1 kinase via the NF-kappa B pathway. J Immunol. 2002;168:744-54
13. Yip-Schneider MT, Horie M, Broxmeyer HE. Transcriptional induction of pim-1 protein kinase gene expression by interferon gamma and posttranscriptional effects on costimulation with steel factor. Blood. 1995;85:3494-502
14. Matikainen S, Sareneva T, Ronni T, Lehtonen A, Koskinen PJ, Julkunen I. Interferon-alpha activates multiple STAT proteins and upregulates proliferation-associated IL-2Ralpha, c-myc, and pim-1 genes in human T cells. Blood. 1999;93:1980-91
15. Aguirre E, Renner O, Narlik-Grassow M, Blanco-Aparicio C. Genetic Modeling of PIM Proteins in Cancer: Proviral Tagging and Cooperation with Oncogenes, Tumor Suppressor Genes, and Carcinogens. Frontiers in oncology. 2014;4:109
16. Roh M, Gary B, Song C, Said-Al-Naief N, Tousson A, Kraft A. et al. Overexpression of the oncogenic kinase Pim-1 leads to genomic instability. Cancer Res. 2003;63:8079-84
17. Roh M, Song C, Kim J, Abdulkadir SA. Chromosomal instability induced by Pim-1 is passage-dependent and associated with dysregulation of cyclin B1. J Biol Chem. 2005;280:40568-77
18. Wang J, Kim J, Roh M, Franco OE, Hayward SW, Wills ML. et al. Pim1 kinase synergizes with c-MYC to induce advanced prostate carcinoma. Oncogene. 2010;29:2477-87
19. Chen WW, Chan DC, Donald C, Lilly MB, Kraft AS. Pim family kinases enhance tumor growth of prostate cancer cells. Mol Cancer Res. 2005;3:443-51
20. Cen B, Mahajan S, Wang W, Kraft AS. Elevation of receptor tyrosine kinases by small molecule AKT inhibitors in prostate cancer is mediated by Pim-1. Cancer research. 2013;73:3402-11
21. Casuscelli F, Ardini E, Avanzi N, Casale E, Cervi G, D'Anello M. et al. Discovery and optimization of pyrrolo[1,2-a]pyrazinones leads to novel and selective inhibitors of PIM kinases. Bioorganic & medicinal chemistry. 2013;21:7364-80
22. Folkes AJ, Ahmadi K, Alderton WK, Alix S, Baker SJ, Box G. et al. The identification of 2-(1H-indazol-4-yl)-6-(4-methanesulfonyl-piperazin-1-ylmethyl)-4-morpholin-4-yl-t hieno[3,2-d]pyrimidine (GDC-0941) as a potent, selective, orally bioavailable inhibitor of class I PI3 kinase for the treatment of cancer. Journal of medicinal chemistry. 2008;51:5522-32
23. Siu A, Virtanen C, Jongstra J. PIM kinase isoform specific regulation of MIG6 expression and EGFR signaling in prostate cancer cells. Oncotarget. 2011;2:1134-44
24. Oda K, Matsuoka Y, Funahashi A, Kitano H. A comprehensive pathway map of epidermal growth factor receptor signaling. Molecular systems biology. 2005;1:2005 0010
25. Chou TC. Theoretical basis, experimental design, and computerized simulation of synergism and antagonism in drug combination studies. Pharmacol Rev. 2006;58:621-81
26. Shirogane T, Fukada T, Muller JM, Shima DT, Hibi M, Hirano T. Synergistic roles for Pim-1 and c-Myc in STAT3-mediated cell cycle progression and antiapoptosis. Immunity. 1999;11:709-19
Author contact
![]() Corresponding author: Luca Mologni, PhD. School of Medicine and Surgery, University of Milan-Bicocca, via Cadore 48, Monza, Italy. Email: luca.mologniit. Tel. +39 0264488148 Or Vera Magistroni, PhD. School of Medicine and Surgery, University of Milan-Bicocca, via Cadore 48, Monza, Italy. Email: vera.magistroniit. Tel. +39 0264488362.
Corresponding author: Luca Mologni, PhD. School of Medicine and Surgery, University of Milan-Bicocca, via Cadore 48, Monza, Italy. Email: luca.mologniit. Tel. +39 0264488148 Or Vera Magistroni, PhD. School of Medicine and Surgery, University of Milan-Bicocca, via Cadore 48, Monza, Italy. Email: vera.magistroniit. Tel. +39 0264488362.