Impact Factor ISSN: 1837-9664

 Global reach, higher impact
Global reach, higher impactJ Cancer 2017; 8(2):178-189. doi:10.7150/jca.16387 This issue Cite
Research Paper
Targeting Metabolic Remodeling in Triple Negative Breast Cancer in a Murine Model
Verónica García-Castillo1,2, Eduardo López-Urrutia1, Octavio Villanueva-Sánchez3, Miguel Á. Ávila-Rodríguez4, Alejandro Zentella-Dehesa3, Carlo Cortés-González6, César López-Camarillo5, Nadia J Jacobo-Herrera3 ![]() , Carlos Pérez-Plasencia2,6
, Carlos Pérez-Plasencia2,6 ![]()
1. Posgrade in Experimental Biology, Metropolitan University of Mexico;
2. FES-Iztacala, UBIMED, National Autonomous University of Mexico, UNAM, Tlalnepantla, Mexico;
3. Biochemistry Unit, National Nutrition Institute of Mexico “Salvador Zubirán”, Mexico;
4. PET/CT Unit, Faculty of Medicine, UNAM;
5. Posgrade in Genomic Sciences, Autonomous University of México City, Mexico;
6. Genomics Lab, National Cancer Institute of Mexico, Mexico.
Received 2016-6-5; Accepted 2016-10-12; Published 2017-1-13
Abstract
Background: Chemotherapy is the backbone of systemic treatment for triple negative breast cancer (TNBC), which is one of the most relevant breast cancers molecular types due to the ability of tumor cells to develop drug resistance, highlighting the urgent need to design newer and safer drug combinations for treatment. In this context, to overcome tumor cell drug resistance, we employed a novel combinatorial treatment including Doxorubicin, Metformin, and Sodium Oxamate (DoxMetOx). Such pharmacological combination targets indispensable hallmarks of cancer-related to aerobic glycolysis and DNA synthesis.
Materials and Methods: Thirty-five female nude mice were transplanted subcutaneously with MDA-MB-231 triple negative human cancer cell line. Once tumors were visible, mice were treated with doxorubicin, metformin, oxamate or all possible pharmacologic combinations. Treatments were administered daily for 15 days and tumors were measured by calipers every day. MicroPET images were taken in three different occasions, basal state, in the middle of the treatment, and at the end of treatment. Western blot analyses, qRT-PCR, flow cytometry, and cytotoxicity assays were performed to elucidate the mechanism of cell death promoted by the drugs in vitro.
Results: In this work we assessed the proof of concept of metabolic correction in solid tumors as an effective drug treatment; hence, mice bearing tumors treated with the DoxMetOx therapy showed a complete inhibition of the tumor mass growing in 15 days of treatment depicted by the micro PET images. In vitro studies displayed that the three drugs together act by inhibiting both, mTOR-phosphorylation and expression of LDH-A gene, promoting apoptosis via dependent on the caspase-3 pathway, accompanied by cleavage of PARP. Moreover, induction of autophagy process was observed by the accumulation of LC3-II, a primordial protein implicated in the conformation and elongation of the autophagolysosome.
Conclusions: The lack of effective drugs to inhibit TNBC growth is the main cause of therapy failure and tumor relapse. We have showed that targeting crucial molecular pathways in cancer by the combination of Doxorubicin, Metformin, and Oxamate resulted as an efficient and rapid tumor growth inhibitor in a triple negative xenograft model. Our findings are promising for patients diagnosed with TNBC tumors, for which unfortunately there are no reliable drug therapies.
Keywords: Triple Negative Breast Cancer (TNBC), metformin, oxamate, doxorubicin, mTOR, aerobic glycolysis, xenograft, micro PET.
Introduction
Breast cancer (BC) is a global health problem as a leading cause of cancer-related deaths in women (1). Only in 2012, nearly to 1.7 million new cases of BC were registered around the world, which represented close to 12% of all cancers in that year (2). About half the breast cancer cases and 60% of deaths are estimated to occur in economically developing countries. Currently, BC is classified into five major groups based on molecular profiling. Triple negative breast cancer (TNBC) is defined clinically as negative for estrogen and progesterone receptors (ER-negative, PR-negative) as well as negative for human epidermal growth factor receptor type 2 (HER2-negative) (3).
Worldwide, approximately 15% of breast tumors correspond to TNBC (4,5). At present, there is no specific therapy for these patients; however, the standard treatment consists of surgery, radiation and co-adjuvant treatments based on anthracyclines (doxorubicin, gemcitabine, capecitabine, vinorelbine, ixabepilone) and taxanes (6). The main consequences of current chemotherapy agents are cardiotoxicity, and both neuro- and hemato-toxicity in patients with prolonged treatments (6).
Solid tumors present a disturbed metabolism, i.e. glycolytic rate, and glucose absorption increments that in consequence produce high lactate concentrations, known as the Warburg effect or aerobic glycolysis (7). Thus, cancer cells require glucose as a primary source of energy to supply ATP to maintain their active metabolism and proliferation (8). A key enzyme regulating this process is the isoform A of the lactate dehydrogenase (LDH-A), which catalyzes the reversible conversion of lactate to pyruvate (9). Released lactate into the extracellular matrix increase cancer cell motility and invasion through the activation of metalloproteases (10). Besides can inhibit activation, differentiation and expression of antigens in dendritic cells (11). Hence, lactate has been proposed as an onco-metabolite (12). Tumor lactate content has been considered as a prognostic parameter in the clinic for cervical and head-and-neck cancers after observed that patients with high lactate concentration are likely to develop metastasis and to present lower survival probabilities (13,14). All these distinctive attributes of tumor cells promise therapeutic targets. Hence, it is feasible to use selective drugs to inhibit directly glycolysis and ATP generation, promoting apoptosis in tumor cells with reduced cytotoxicity in normal cells.
Several findings support the use of oxamate (Ox)- a competitive inhibitor of LDH-A and aspartate aminotransferase (AAT)- as a glycolysis and cellular growth inhibitor in vitro and in vivo studies with HeLa cells, cervical, breast and astrocytoma cancer (15,16,17,18,19,20). Moreover, since its chemical structure is very similar to pyruvate, it is likely to be safe for humans (20). It is known that once LDH-A is inhibited, levels of ATP diminish and mitochondrial reactive oxygen species (ROS) production increases, accelerating tumoral cell death through apoptosis (20,21,22,23). Therefore, it is rational to assume that LDH-A inhibition blocks glycolysis and potentially provokes deleterious changes in the tumor metabolism.
Recently, evidence of Metformin -the most commonly prescribed antidiabetic drug for type II diabetes- as an anticancer drug has been gathering (24). Metformin inhibits proliferation in different cancer cell lines including breast, glial, prostate, and gastric cancer (25,26,27,28) through the inhibition of the respiratory chain -complex 1-, decreasing ATP synthesis and thus producing an increment in the activity of the AMP-promoted kinase (AMPK) (29,30). AMPK is a crucial regulator of lipids and glucose in response to metabolic alterations and intracellular levels of ATP (31). When AMPK is active, Mammalian Target of Rapamycin (mTOR) is inhibited and so are ATP-dependent processes such as protein, fatty acid, and cholesterol biosynthesis, as well as gluconeogenesis (32). A very new investigation demonstrated that the use of phenformin (a biguanide antidiabetic drug) together with oxamate is a potent anticancer formulation that leads to cell death by apoptosis in vitro, and reduces tumor growth in vivo (20).
With the previous evidence in mind, we hypothesize that drug-targeting molecular and metabolic pathways that are aberrantly-activated in tumor cells, we could inhibit tumor growth cell by inhibiting Warburg effect, and nucleic acid synthesis; activating apoptosis and autophagy. For that purpose, we used a combination of Doxorubicin, Metformin, and Sodium Oxamate (DoxMetOx). As a result, the pharmacological combination was able to dramatically reduce the tumor growth since the first administration. Moreover, in vitro analyses showed that apoptosis was activated and caspase-3 and PARP were cleaved in cells treated with DoxMetOx. In addition, inhibition of mTOR phosphorylation occurred at the same time that LDH-A was inhibited, both at mRNA and protein levels. Such data suggest that both processes (mTOR phosphorylation and transcriptional activation of LDH-A) could be connected. Finally, treated cells with DoxMetOx also activated LC3-II, a molecular marker of autophagy. Taken together these results, strengthen the hypothesis that concurrent inhibition of mTOR and LDH-A leads tumor cells to activate apoptosis and autophagy. Our investigation sheds light to patients diagnosed with TNBC, a tumor with lower survival expectation than other breast cancer phenotypes.
Methods
Drugs
Doxorubicin (Doxolem) (Dox); Metformin (1-1dimethylbiguanidine hydrochloride, Santa Cruz® sc-202000A) (Met); Sodium Oxamate (Ox) (Santa Cruz sc-215880). Concentrations in vitro: as indicated. Dose in vivo: Met 200 mg/Kg, Ox 15 mg/Kg, and Dox 1 mg/Kg.
Cell culture
The MDA-MB-231 human mammary adenocarcinoma cells, with mutation in p53 and negative expression of epidermal growth factor receptor type 2 (HER2) receptor, they express low levels of androgen receptors, and are highly invasive, were purchased from the American Type Culture Collection (ATCC). Cells were cultured in GIBCO® RPMI-1640 media supplemented with 10% FBS (Biowest®), 5 mL/L penicillin/streptomycin (GIBCO®), and maintained at 37°C, 5% CO2 in air and 95% humidity.
Cytotoxicity Assay by Sulforhodamine B (SRB) assay
MDA-MB-231 cells were incubated in 96-well plates at a density of 7000 cells/well. The cells were treated with different concentrations of Dox, Met or Ox for 4, 24, 48 and 72 h. In a separate plate (day 0), 7000 cells/well were seeded, incubated and allowed to attach for 4 hours. At the end of every incubation time, either with or without treatment, cells were fixed with cold trichloroacetic acid 10% (TCA) at 4°C for one h. TCA was removed, and plates washed with tap water and air dried. Finally, 100 μL of SFR-B were added to each well and incubated at room temperature for 30 min, wells were washed with 1% acetic acid to remove unbound dye. The dye was dissolved in 10 mM Tris base solution (pH 10.5) and stirred over 5 min. Optical density (OD) was measured in an Epoch Microplate Spectrophotometer (Bio-Tek) at 510 nm. Results are expressed as the concentration that inhibits 50% of control growth after the incubation period (IC50). The values were estimated from a semi-log plot of the drug concentration (mM/mL and μM/mL) against the percentage of growth inhibition (33).
MDA-MB-231 subcutaneous xenograft model
All in vivo experiments were performed following animal care guidelines approved by the Committee for Animal Research and Procedures of the National Nutrition Institute of Mexico "Salvador Zubirán" (Project No. 768). Thirty-five female nude mice (nu/nu) were used when they were 4-week-old, food and water were provided ad libitum, and animals were housed in cages with microisolators in a 12-h light and dark cycle. MDA-MB-231 cells (10 x 106 in 100 μL of 0.9% saline solution) were implanted subcutaneously into the right flank of all mice and allowed to grow up to the subcutaneous xenografts tumors became visible (approximately 50 mm3 in size), mice were random assigned to be treated intraperitoneally for 15 days with all possible combination of drugs (as single treatment and combined drugs). Tumor sizes were measured by calipers every day and their volumes were calculated according to the formula of (width2 × length)/2 (34). Hence, seven different groups of five mice each were treated as follows: 1) positive control, Dox (1 mg/Kg); 2) Met (200 mg/Kg); 3) Ox (15 mg/Kg); 4) Dox/Met (1 mg/Kg/200 mg/kg); 5) Dox/Ox (1 mg/Kg/15 mg/Kg); 6) Met/Ox (200 mg/Kg/15 mg/Kg); and 7) Dox/Met/Ox (1/200/15 mg/Kg). All animals were humanely euthanized. At the end of the model development, a representative sample of three animals from group seven was kept for three months for disease recurrence monitoring.
(18F)-FDG micro PET
Micro PET imaging was performed at Faculty of Medicine, National Autonomous University of Mexico (UNAM), by using the Micro PET Focus 120 Concorde Microsystem, Siemens equipment and the OSEM 3D Siemens software. This technique was employed to follow disease progress in the animal model. Briefly, mice were anesthetized with an isoflurane/oxygen (0.5 L/min) induction chamber during 5 min; 200 μL of (18F)-FDG were injected via caudal vein, with a bio-distribution time of 45 min; static images were acquired during 30 min. Micro PET images were taken on three different occasions: 1) basal state before treatment started, 2) in the middle of the treatment, and 3) at the end of treatment.
Primary Culture
A primary culture was grown from tumors of the xenograft. Cultures were maintained in RPMI-1640 medium supplemented with SFB 10% under incubation conditions and treated with Dox, Ox or Met and their combinations during 4, 8, 12 and 24 h, keeping the same concentrations used in the others in vitro experiments. These cells were harvested for Western blotting and qRT-PCR assays.
qRT-PCR
Total RNA was purified from MDA-MB-231 cell line using RNeasy Mini Kit (Qiagen, Valencia, CA). The quality and concentration of RNA in samples was determined using gel electrophoresis and Epoch Microplate Spectrophotometer (Bio-Tek). 500 ng from the different treatments conditions were used for cDNA synthesis using iScript cDNA synthesis kit (Bio-Rad Laboratories, Hercules, CA), according to the manufacturer's instructions. The LDH-A primer set employed to analyze gene expression levels was designed by an experimentally verified computer algorithm, and tested in a quality control assay to guarantee a yield of a single band of 165 bp by agarose gel electrophoresis. The sequence of primers was as follows: forward 5´ TTGCAACCGCTTCC ATAACACGG 3´ and reverse 3´ TGGTCCAGCGTAACGTGAACATCT 5´. Real-time PCR was performed using FastStart SYBR Green master. In Light Cycler 480 instrument II (Roche, Mannheim, Germany) according to manufacturer´s protocol. Triplicate RT samples were utilized in each assay; data were normalized with ß-actin housekeeping gene and in a parallel way was used GAPDH. The comparative Ct method (ΔΔCt) was employed to quantify gene expression, and the relative quantification was calculated as 2-ΔΔCt.
Western blotting
After pharmacological treatments, total protein from primary cultured cells was extracted using RIPA buffer (Santa Cruz Biotechnology sc-24948). From these extracts, 50 μg of protein were separated by SDS-polyacrylamide gel electrophoresis and transferred to a polyvinylidene difluoride (PVDF) membrane (GE Healthcare, USA) in a semi-dry chamber Trans Blot Turbo (Bio-Rad) at 25 V 1 mA for 30 min. After blocking with 5% of non-fat milk for 2 h, the membrane was incubated with the specific antibody overnight at 4°C on a rocking platform, washed, and then incubated with the corresponding secondary antibody for 2 h at room temperature. The blot was visualized using the SuperSignal West Femto chemiluminescent substrate (Pierce) in a C-Digit scanner (LI-COR)™ employing the IMAGE STUDIO (LI-COR) software.
The primary antibodies for apoptosis were purchased from Santa Cruz Biotechnology (Santa Cruz, CA): anti-PARP-1 (1:3000, sc-8007) and anti-Caspase-3 (H2777) (1:3000, sc-7148). Antibodies for autophagy: anti-LC3 from Cell Signaling Technology (1:2000, #2775), mTOR (1:3000, Cat. 2983), Phosphorylated-mTOR from Invitrogen (1:3000, Cat. 2971); and, for glycolysis anti-LDH-A (1:1000, sc-27230) from Santa Cruz Biotechnology. All secondary antibodies were obtained from Santa Cruz Biotechnology: anti-mouse (1:5000, sc-23719), anti-rabbit (1:5000, sc-2370) and anti-goat (1:3000, sc-2020). The positive control for apoptosis was UV (10 min of exposure). β-actin (1:3000, Santa Cruz Biotechnology sc-4778) was used as an internal control.
Statistics
All experiments were carried out in triplicate unless mentioned. Differences among treatments were evaluated by ANOVA using the Dunnett method for multiple comparisons. For all statistical analyzes, we used the GraphPad PRISM version 5.0 software with an acceptable significance of p<0.05.
Results
Cytotoxic effect of the DoxMetOx therapy in the MDA-MB-231 cells
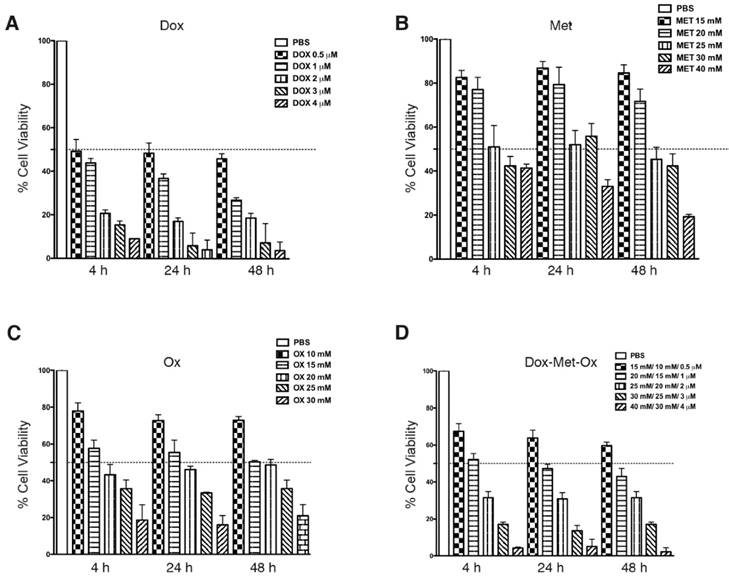
MDA-MB-231 cells were treated with Met, Ox, or Dox, or all possible combinations as described in Materials and Methods. The IC50's were obtained by the SRB assay. The IC50 values for single drugs were of rapid achievement (4h) and were also stable along time (maintaining the same effect from 4 to 48 h). Figure 1A-C displays IC50 values (Dox 0.5 μM, Met 25 mM, and Ox 15 mM). As depicted in Figure 1D, DoxMetOx keeps the same IC50 values as for monotherapy and respect rules of persistent-time-action and initiating point. Even more, the IC50 values when tested as duplets, the concentrations stayed the same (data not shown). The IC50 values used were the same in all in vitro studies.
Cytotoxic effect of Metformin, Oxamate or Doxorubicin, and their combinations against MDA-MB-231 cells in the SRB assay. MDA-MB-231 cells were treated with Met, Ox, or Dox alone, and all possible combinations for 4, 24, 48 and 72 hours at different concentrations. Estimated IC50 values are Dox 0.5 μM, Met 25 mM, and Ox 15 mM.
DoxMetOx reduced tumor growth in vivo tracked by (18F) FDG micro PET
Progression of disease and response to treatment was monitored by micro PET image acquisition employing (18F)-FDG as a metabolic radionuclide tracer as described in material and methods, following the timetable illustrated in Figure 2A. In Figure 2B-D, are shown the last images of the evolution of tumor growth after treatments. The 18F-deoxiglucose accumulates in the brain, heart and bladder, zones of the body that are in bright red in the image. Tumor cells, due to their high proliferation rate, have a greater intake of deoxyglucose, thus coloring the tumor in red as occur in the other organs. As it can be noticed in Figure 2D when tumors treated with DoxMetOx, there is no deoxyglucose intake in the area where the MDA-MB-231 cells were transplanted. All pictures were taken from a representative sample of each group.
Tumor growth of MDA-MB-231 xenotransplant in nude mice after different treatments. A) The schedule followed for drug administration and microPET monitoring. B-D) Results of the third micro PET imaging acquisition. The disease progression was monitored by the molecular imaging technique micro PET using the radionuclide (18F)FDG on three different occasions: basal state, day eight and day 15 (end of treatment). The acquisitions were performed in transverse and coronal cuts. Animals were euthanized at the end of the treatments. B) Monotherapies: Dox, Met, and Ox; C) Duplets: Met/Dox; Dox/Ox and Met/Ox; and D) DoxMetOx (*p<0.05). In all cases were used the same doses: Dox 1 mg/Kg, Met 200 mg/Kg, and Ox 15 mg/Kg. The (18F)FDG is accumulated in the brain, heart and bladder, colored in bright red in the picture. Tumors are also colored in red in the right flank of the animals. The DoxMetOx group showed a complete remission of the tumor mass at the end of treatment, no presence of the radionuclide accumulation or disease recurrence were detected. Assays were performed at least on three independent occasions with three animals in each experiment. E) Nude mice were inoculated with 10x106 MDA-MB-231 cells on the right flank. Once the tumor reached a volume of 50 mm3, treatments started. Drugs were administered daily via i.p. for 15 days and tumor masses were measured and graphed ±S.E. The response of the animals to monotherapies Doxorubicin (1 mg/Kg) (p=0.05), Metformin (200 mg/Kg) and Oxamate (15 mg/Kg). F) Effect of duplets (Dox/Met; Dox/Ox; Met/Ox) and DoxMetOx against tumors. DoxMetOx reduced tumor mass growth (*p<0.005) in comparison to the other possible drugs' combinations.
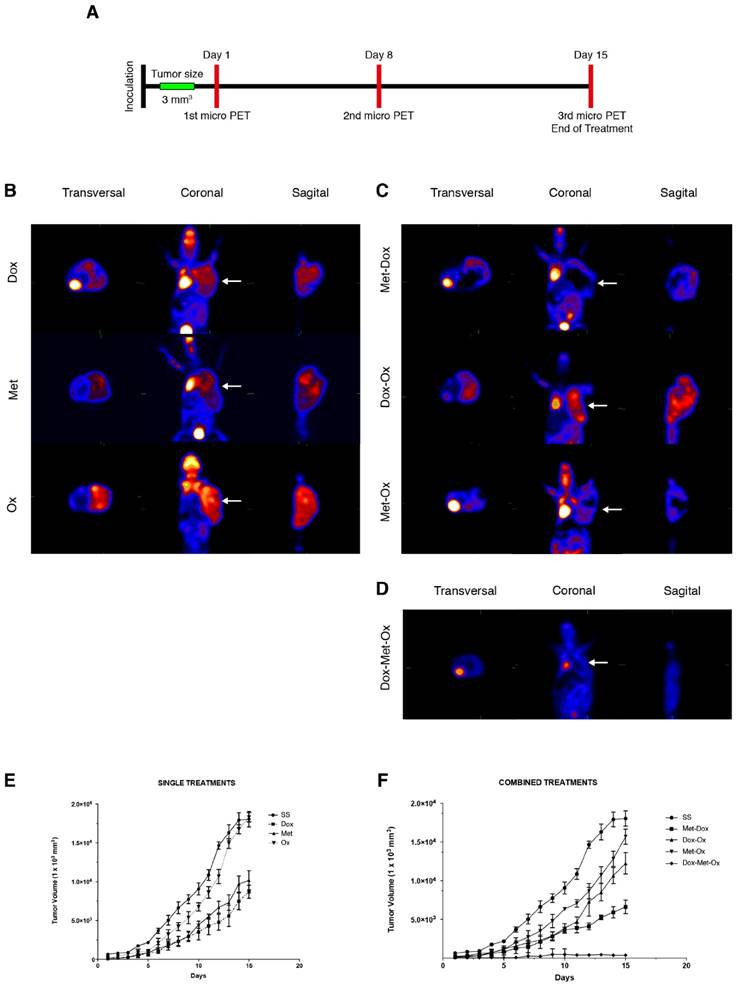
The tumors were measured daily to generate a graph of the tumor response to treatments. As depicted in Figure 2E and 2F, monotherapies had no significant reduction in tumor size except for Dox (*p=0.05). Also, none of the duplets tested (Met/Ox, Dox/Ox, or Met/Dox) enhanced the effect of Dox alone. On the contrary, when tumors treated with DoxMetOx they stop growing in 48 h, maintaining the same volume until day eight, to finally lead to an absence of measurable tumor mass (*p=0.05). Furthermore, in a gross external examination, the animals that belonged to DoxMetOx did not experiment toxic effects during the treatment, and moreover, three animals were kept in the vivarium for five months and no disease recurrence was observed.
Activation of Apoptosis by DoxMetOx treatment
To evaluate apoptosis cell death in MDA-MB-231 cells by the different treatments, we next carried out an assay of Western blot using PARP-1 and caspase 3 as apoptosis markers. When drugs were administrated as monotherapy (Figure 3A-C), both, Dox and Ox, slightly increased the expression of the full-length procaspase 3 or cleavage of PARP-1 in comparison to Met, who presented a major expression of both proteins and in shorter times. Single treatments had a time-dependent pro-apoptotic effect. Whereas the DoxMetOx therapy showed a substantial up-regulation of cleaved PARP-1 from the 4 h time point, which corresponds to the detection of cleaved effector caspase 3, maintaining the effect along tested times. The duplet Met/Ox had similar result since 12 h with cleaved PARP-1.
Apoptosis activated by DoxMetOx. A) Apoptosis induction identified by Western blot detection of PARP-1 and caspase 3. We searched for the effect of tested drugs or combinations at short times. As can be clearly observed (E) DoxMetOx could activate both, PARP-1 and caspase 3 at 4 h point time, as a continuous process along 24 hours. F) Finally, to corroborate our results we assayed apoptosis by means of flow cytometry after staining with Annexin V-FITC/propidium iodide. As can be observed in representative scatter plots of PI (y-axis) vs. Annexin V (x-axis), DoxMetOx therapy induced early apoptosis (lower right quadrant) since 48 h and at 72 h the induction of apoptosis was in 99% of tumoral cells.
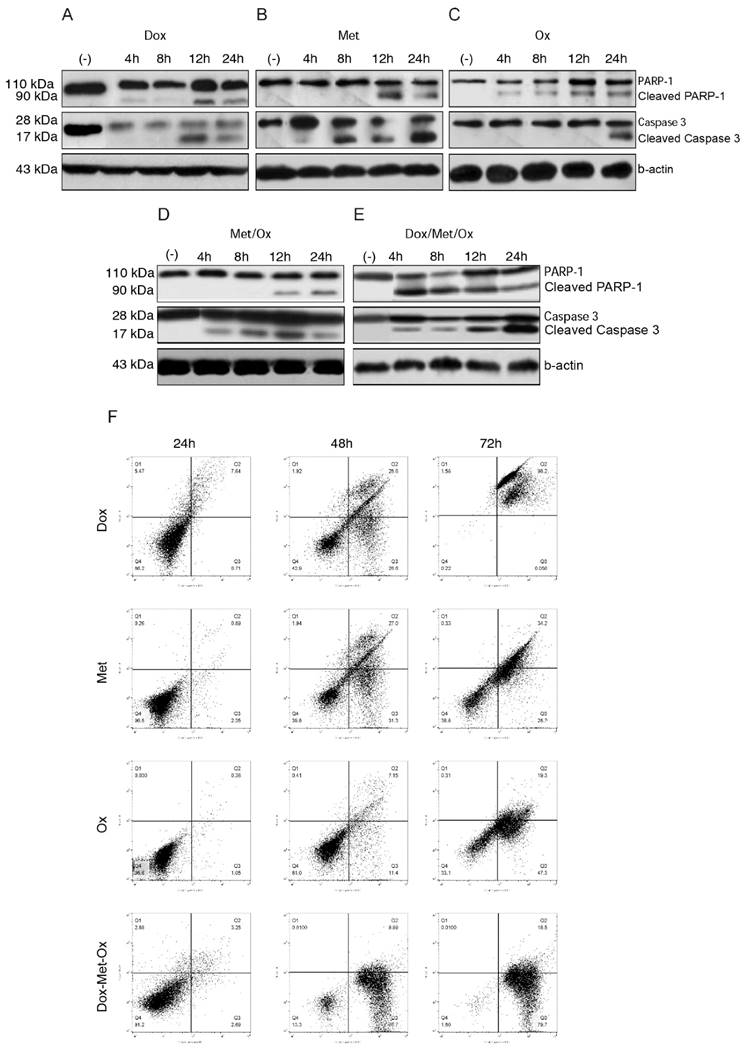
Western blot results, proved the induction of apoptosis in MDA-MB-231 breast cancer cell line by DoxMetOx, by both caspase-dependent and PARP-dependent apoptosis cell death, enhancing the Met proapoptotic activity
Moreover, the annexin V assays provided support for the pro-apoptotic effect of the combination DoxMetOx. As depicted in Figure 3F, when the three drugs are together, after 48 h of treatment 87% of the cells enter into apoptosis, effect that remains for 72 h. In comparison, when cells are treated with the drugs individually, doxorubicin is highly toxic after 72 h (Figure 3F). On the other side, metformin and oxamate are less aggressive but also time-dependent (Figure 3F). Metformin and Oxamate after 48 h induced early apoptosis in 31% and 11%, respectively. Interestingly, the proapoptotic effect of the oxamate increases in time to 47% (Figure 3F).
DoxMetOx is likely to induce autophagy through LC3 protein activation and mTOR inhibition
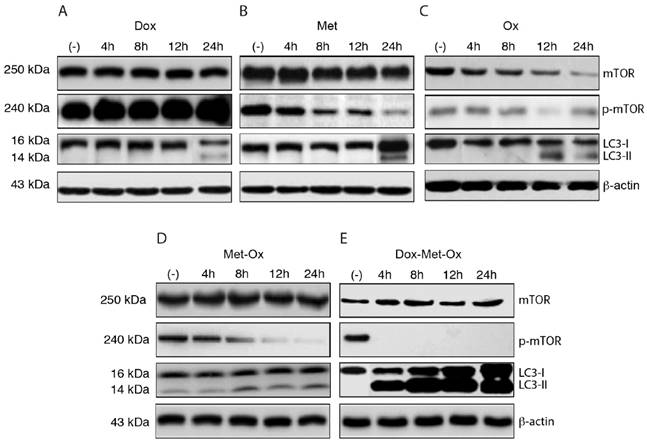
The induction of autophagy was evaluated through LC3 protein activation and inhibition of phosphorylated-mTOR (p-mTOR) by Western Blotting (Figure 4A-E). As single treatments, Met and Ox moderately favored the inhibition of p-mTOR and increased LC3-II accumulation (Figure 4B and C); on the contrary, such effect was not observed with Dox, which unexpectedly increased the p-mTOR signal (Figure 4A). The duplet Met-Ox inhibited p-mTOR after 12 h of treatment, and it does fairly increment the conversion of LC3-I to LC3-II (Figure 4D). In contrast, DoxMetOx promotes the inhibition of phosphorylated mTOR and a significant increment in the conversion of LC3-I to LC3-II at 4 h (Figure 4E). These data suggest that the DoxMetOx therapy could also induce tumor cell autophagy in early stages even more efficiently than Met alone, which in our results seem to require longer time to inhibit p-mTOR (Figure 4B).
Early autophagy activation and inhibition of p-mTOR. DoxMetOx was able to inhibit p-mTOR at 4 h, and the effect was maintained up to 24 h. The same occurred with LC3-II accumulation, in contrast to monotherapies or duplets.
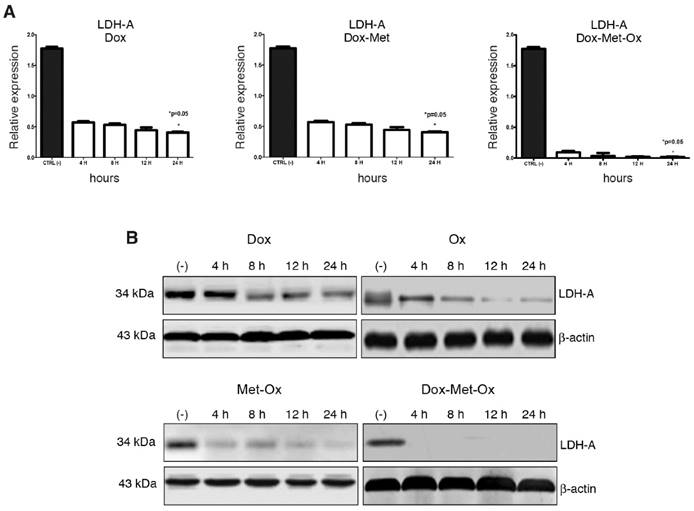
DoxMetOx inhibits LDH-A expression
RT-PCR and Western blot experiments were performed to assess expression levels of LDH-A as a marker of glycolysis. As depicted in Figure 5, Oxamate -the specific inhibitor of LDH-A- had a time-dependent inhibition response; it could require a longer treatment or a higher concentration since incomplete LDH-A inhibition was observed. Dox did not inhibit LDH-A expression. Nevertheless, when Dox was added to the Met-Ox duplet, the LDH-A was undetected by end-point RT-PCR or Western blot since four hours, and the effect persisted up to 24 h. No single treatment or other combinations showed a similar inhibition effect.
LDH-A inhibition by triple therapy. mRNA and protein levels were analyzed by qRT-PCR and western blot, respectively. A) DoxMetOx inhibited the protein levels of LDH-A. B) mRNA levels were undetectable when DoxMetOx was administered.
Discussion
Triple negative breast cancer therapy is still a challenge due the complexity of the disease. Standard treatments such as hormonal therapy or trastuzumab are not functional, because of the lack of the receptors Her2, ER y PGR (35,36) (Couch, et al., 2015; Bernier and Poortmans, 2016;). On the other side, the use of cisplatin and doxorubicin, derive in nephro and cardiotoxicity consequences, respectively. Currently, treatment options explored for this type of BC are focused on drugs controlling cell proliferation and induction of cell death. For instance, targeting the epidermal growth factor receptors (EGFR), which is overexpressed in TNBC using monoclonal antibodies such as cetuximab or inhibitors as gefitinib (37,38) [Reddy, 2011; Baselga 2002]. Inhibition of PARP1 has been very successful, especially because it has been demonstrated that the combination of such agents with cisplatin or doxorubicin, reduces their secondary toxic effects; and inhibitors of Ras, MEK/ERK, mTOR, heat shock proteins, or VEGF, proteins implicated in cell proliferation and overexpressed in tumor cells (Reddy, 2011).
Anticancer therapies have been developed through the years, but the deregulation of cellular energetics has been approached less frequently. Our work shows the striking effect of a novel combined therapy comprising Metformin, Oxamate, and Doxorubicin, which target key hallmarks of cancer: nucleic acid synthesis, mTOR and LDH-A inhibition leading to apoptosis and autophagy activation. The experimental group of mice that received DoxMetOx therapy exhibited complete tumor growth suppression and in vitro results showed evidence of enhanced apoptosis and autophagy, which is consistent with an energetic metabolism reprogramming. Metformin is not limited to its function as the preferred drug for diabetes, as it has been demonstrated that it has antineoplastic activity as well. In vitro, the Metformin inhibits cell proliferation (32), and induces caspase- and PARP-dependent apoptotic cell death in several breast cancer cell lines (26,39). In vivo, it has been a successful tumor growth inhibitor in colon, breast, pancreatic, and lung cancers (25,26,27,40,41,42,43). Nevertheless, it is known that its use as an anticancer monotherapy drug is not effective.
Oxamate has been associated with antimetabolic effect, the inhibition of TCA cycle mediator production, and reduction of the glycolytic flux and the electron chain transport activity (17). Moreover, in vivo it has shown tumor growth reduction in two hepatocellular cancer cell lines (44). Additionally, Oxamate significantly arrests lung cells in G0/G1 phase (45), and Metformin induces cell cycle arrest in G1 phase in TNBC cell lines (26). Finally, Doxorubicin is a well-known antineoplastic drug, which is a DNA intercalator that acts as a nucleic acid synthesis inhibitor (46).
The IC50 values obtained for the tested drugs are consistent with values reported somewhere else (45,47), and the doses used in vivo of the three drugs were in the lower end of the recommended clinical dosage range for humans in the case of metformin and doxorubicin (48), and for animals in the event of oxamate (21). Therefore, the doses employed in our work are in agreement with other findings both in vitro and in vivo related to the toxicity range of the drugs.
In the murine model, mice carrying tumors subjected to the DoxMetOx therapy stopped growing after 24 h of the first drug administration and tumor volume kept stable for six days. Nevertheless, on day 11 tumor mass was complete eradicated, analyzed by both FDG-microPET and tumor volume measure. Indeed, the action of the DoxMetOx therapy was surprisingly fast; in the murine model it took only 24 h to stop tumor growth, followed by a complete elimination of the xenograft. Furthermore, animals of the DoxMetOx group were free of disease five months after finishing therapy, which confirms tumor elimination; these animals did not present obvious toxicity signs during the 15 days of the DoxMetOx therapy.
Recently, Miskimins and colleagues (20) published that the combination of oxamate and phenformin, act in synergy inhibiting proliferation in the breast, melanoma, colon, lung and prostate cancer cells. Also, in vivo this drugs combination showed a considerable reduction of the tumor size after 21 days of treatment. They suggested that the phenformin-oxamate combination increases apoptosis through the inhibition of complex I in the mitochondria by phenformin and simultaneously targeting the LDH in the cytosol by oxamate. Even though phenformin is a chemical analog of metformin, we did not observe any significant inhibition of tumor growth when metformin and oxamate were co-administered in vivo in our study.
According to data presented here, none of the three drugs used as monotherapy favored apoptosis or autophagy (Figures 3A-B and 4A-C). However, when the combination DoxMetOx was tested in vitro, they had an enhanced proapoptotic effect and showed the initiation of autophagy. Our results demonstrated that DoxMetOx leads to apoptosis by two mechanisms, the caspase- and PARP-dependent pathways. Such findings are supported by the flow cytometry results, where DoxMetOx showed a rising effect of apoptosis since the first 48 h after treatment keeping the same activity until 72 h. It is notable that the combination of the three drugs diminished the cytotoxic effect of the doxorubicin and restored the apoptosis death capacity of cells in short times.
Respecting to LDH-A expression a surprising result was observed, DoxMetOx was capable to inhibit LDH-A completely both at mRNA and protein level; suggesting an inhibition of transcription of the gene; however, more experiments are needed to clarify this point. Such effect was unexpected since neither oxamate as monotherapy nor the duplets that included it, had substantial effects on LDH-A inhibition or apoptosis death, as the previously reported for lung cancer cells (45). The LDH-A enzyme is known to be overexpressed in breast cancer (49,50), and its inhibition blocks aerobic glycolysis in tumor cells without harming normal cells that do not require LDH-A in the presence of oxygen; this protective effect to non-malignant cells could explain the non-toxic effect of the DoxMetOx to the surrounding tissue in the animals or even toxic systemic visible symptoms in nude mice.
The role of autophagy in cancer is part of an important debate. It has been documented that autophagy has a pro-tumorigenic function in the invasion and metastasis processes (51,52). Whereas there is also evidence, that supports tumor suppressor function in early stages through the protein Beclin-1 (53). Our results provide insight into the crosstalk between apoptosis and autophagy generated by anticancer therapies. A key step involved in this event is the inhibition of mTOR, the target of new drugs, which currently are under clinical trials for lung, blood, breast, and kidney cancers (54).
Phosphorylated-mTOR was inhibited in vitro by the DoxMetOx therapy in four hours. At the same time point, the drugs lead to accumulation of LC3-II that is an essential protein in the formation of the phagophore, initial step of autophagy efflux that lately leads to its elongation and closure to form the autophagosome, that when fused with lysosomes initiate the degradation of the different cellular contents in the autophagolysosome (55). Thus, the detection of LC3-II is significant to identify the autophagy process or even autophagic death.
The evidence provided by our group so far suggest that the DoxMetOx therapy kills the tumor cell in a very effective way and in short times (4 h). However, it is important for further investigation to describe cellular effects within a shorter period to unravel the cellular mechanisms switched on implicated in cell death and tumor mass suppression. In this context, we propose that when drugs are in combination, tumor cells fall into an energetic crisis and, as a consequence, apoptotic death is reactivated, either initiated or followed by autophagy, suggesting that all events are possibly derived mainly from the inhibition of glycolysis via the LDH-A inhibition.
Simultaneously, our research group found that in an associated inflammatory model of colorectal cancer in Balb/c mice, the administration of the combined treatment at the same dose, accomplished a reduction in the number and size of tumors in comparison to Dox used as positive control. These results correspond to our findings in mice carrying tumors of the TNBC cell line MDA-MB-231. Interestingly, after 4 h of treatment with the DoxMetOx therapy, three biological events were recorded: apoptosis cell death, induction of autophagy, and glycolysis inhibition in both murine models.
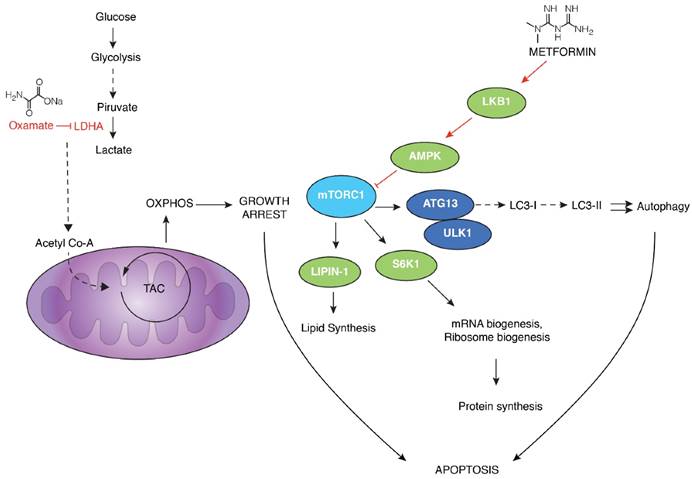
Proposed mechanism of action by the combination of metformin and oxamate treatment in triple negative breast cancer. Sodium oxamate targets lactate dehydrogenase A (LDHA) results in the increase of oxidative phosphorylation which in turn increase oxidative stress and subsequent growth arrest; ensuing inhibition of glycolysis. Metformin induces the activation of LKB1 and phosphorylation of AMPK, leading to inactivation of mTORC1 and subsequent biosynthetic pathways such as mRNA biogenesis, ribosome biogenesis, and lipid synthesis, among others. This is a crucial step conducting to concurrent activation of apoptosis, mainly due to activation of autophagy and energy depletion.
Conclusions
Currently, therapeutic explorations in cancer are focused on disease metabolism considering, on one hand, a specific action on cancer cells, and on the other, procuring no-side effects on normal cells. Understanding the biology and behavior of cancer cells is fundamental towards these aims.
Present work shows a new therapeutic strategy for triple negative breast cancer that dramatically reduced the tumor growth in mice in a short time, where the effectiveness was preserved for five months after finishing the therapy. The chosen drugs seem to act directly on tumor cells by inhibiting glycolysis and mTOR signaling, and activating mechanisms that lead eventually to apoptosis. Moreover, our therapy enriches with molecular evidence, the link between autophagy and apoptosis mechanisms of death. These findings are highly promising for future clinical trials on humans.
Acknowledgements
This manuscript complies in partial fulfillments author requirements for Ph.D. degree for V.G.-C. at Posgrado en Biología Experimental, Universidad Autónoma Metropolitana (UAM), Mexico. V.G.-C. is grateful to CONACyT for a graduate student scholarship (Grant 307249). We thank M. en C. Mallely C. Ávila-García and M. en C. Victor M. Lara-Camacho (PET Cyclotron Unit, Faculty of Medicine, National Autonomous University) for their help in micro PET assays. Authors are grateful to Dr. Alberto Huberman for his contribution to the improvement of the manuscript.
Authors' contributions
JHNJ and PPC conceived and designed the study. VSO performed and coordinated the mouse xenograft study. GCV and LUE performed the cytotoxicity evaluation, Western blots, and RT-PCR assays. GCV and ARMA performed the microPET imaging acquisition. LUE participated in the data analysis and figures editing. ZDA contributed to the discussion and data analysis. JHNJ and PPS carried out the literature search and wrote the manuscript. All authors read and approved the final manuscript.
Competing interests
The authors declare that they have no competing interests.
References
1. Jemal A, Bray F, Center MM, Ferlay J, Ward E, Forman D. Global Cancer Statistis. CA Cancer J Clin. 2011;61:69-90
2. Ferlay J, Soerjomataram I, Ervik M, Dikshit R, Eser S, Mathers C, Rebelo M, Parkin DM, Forman D, Bray F. GLOBOCAN 2012 v1.0, Cancer Incidence and Mortality Worldwide: IARC CancerBase No. 11 (Internet).
3. Irvin WJ, Carey LA. What is triple negative breast cancer? Eur J Cancer. 2008;44:2799-2805
4. Dent R, Trudeau M, Pritchard KI, Hanna WM, Kahn HK, Sawka CA, Lickley LA, Rawlinson E, Sun P, Narod SA. Triple negative breast cancer: clinical features and patterns of recurrence. Clin Cancer Res. 2007;13:4429-4434
5. Lara-Medina F, Pérez-Sánchez V Saavedra-Pérez D, Blake-Cerda M Arce C, Motola-Kuba D Villarreal-Garza C, González-Angulo AM Bargalló E, Aguilar JL Mohar A, Arrieta O. Triple-Negative Breast Cancer in Hispanic Patients. Cancer. 2011:3658-3669
6. Cárdenas-Sánchez J, Bargalló-Rocha E, Erazo-Valle A, Maafs-Molina E, Poitevin-Chacón A. Consenso Mexicano sobre diagnóstico y tratamiento del cáncer mamario. 5th Edition. Elsevier. 2013:125 http://consensocancermamario.com
7. Warburg O. On the origin of cancer cells. Science. 1956;123(3191):309-314
8. Pelicano H, Martin DS Xu R-H, Huang P. Glycolysis inhibition for anticancer treatment. Oncogene. 2006;25:4633-4646
9. Miao P, Sheng S, Sun X, Liu J, Huang G. Lactate dehydrogenase A in cancer: a promising target for diagnosis and therapy. IUBMB Life. 2013;65:904-910
10. Lee GH, Yan C, Shin SJ, Hong SC, Ahn T, Moon A, Park SJ, Lee YC, Yoo WH, Kim HT, Kim DS, Chae SW, Kim HR, Chae HJ. BAX inhibitor-1 enhances cancer metastasis by altering glucose metabolism and activating the sodium-hydrogen exchanger: the alteration of mitochondrial function. Oncogene. 2010;29(14):2130-41
11. Gottfried E, Kunz-Schughart LA, Ebner S, Mueller-Klieser W, Hoves S, Andreesen R, Mackensen A, Kreutz M. Tumor-derived lactic acid modulates dendritic cell activation and antigen expression. Blood. 2006;107(5):2013-2021
12. Avena P, Anselmo W, Wang Ch, Pestell RG, Lamb RS, Casaburi I, Andò S, Martinez-Outschoorn UE, Lisanti MP. Compartment-specific activation of PPARγ governs breast cancer tumor growth, via metabolic reprogramming and symbiosis. Cell Cycle. 2013;12(9):1360-70
13. Walenta S, Wetterling M, Lehrke M, Schwickert G, Sundfør K, Rofstad EK, Mueller-Klieser W. High lactate levels predict likelihood of metastases, tumor recurrence, and restricted patient survival in human cervical cancers. Cancer Res. 2000;60(4):916-921
14. Brizel DM, Schroeder T, Scher RL, Walenta S, Clough RW, Dewhirst MW, Mueller-Klieser W. Elevated tumor lactate concentrations predict for an increased risk of metastases in head-and-neck cancer. Int J Radiat Oncol Biol Phys. 2001;51(2):349-353
15. Beckner ME, Stracke ML, Liotta LA, Schiffmann E. Glycolysis as primary energy source in tumor cell chemotaxis. J Natl Cancer Inst. 1990;82(23):1836-1840
16. Liu H, Hu YP, Savaraj N, Priebe W, Lampidis TJ. Hypersensitization of tumor cells to glycolytic inhibitors. Biochemistry. 2001;40(18):5542-5547
17. Liu H, Savaraj N, Priebe W, Lampidis TJ. Hypoxia increases tumor cell sensitivity to glycolytic inhibitors: a strategy for solid tumor therapy (Model C). Biochem Pharmacol. 2002;64(12):1745-1751
18. Thornburg JM, Nelson KK, Clem BF, Lane AN, Arumugam S, Simmons A, Eaton JW, Telang S, Chesney J. Targeting aspartate aminotransferase in breast cancer. Breast Cancer Res. 2008;10(5):R84
19. Zhao YH, Zhou M, Liu H, Ding Y, Khong HT, Yu D, Fodstad O, Tan M. Upregulation of lactate dehydrogenase A by ErbB2 through heat shock factor 1 promotes breast cancer cell glycolysis and growth. Oncogene. 2009;28(42):3689-3701
20. Miskimins WK, Ahn HJ, Kim JY, Ryu S, Jung Y-S, Choi JY. Synergistic anti-cancer effect of phenformin and oxamate. Plos One. 2014;9(1):e85576
21. Papaconstantinou J, Colowick SP. The role of glycolysis in the growth of tumor cells. II. The effect of oxamic acid on the growth of HeLa cells in tissue culture. J Biol Chem. 1961;236:285-288
22. Fantin VR, St-Pierre J, Leder P. Attenuation of LDH-A expression uncovers a link between glycolysis, mitochondrial physiology, and tumor maintenance. Cancer Cell. 2006;9:425-434
23. Xie H, Valera VA, Merino MJ, Amato AM, Signoretti S, Linehan WM, Sukhatme VP, Seth P. LDH-A inhibition, a therapeutic strategy for treatment of hereditary leiomyomatosis and renal cell cancer. Mol Cancer Ther. 2009;8:626-635
24. Libby G, Donnelly LA, Donnan PT, Alessi DR, Morris AD, Evans JM. New users of metformin are at low risk of incident cancer: a cohort study among people with type 2 diabetes. Diabetes Care. 2009;32(9):1620-1625
25. Ben Sahra I, Laurent K, Loubat A, Giorgetti-Peraldi S, Colosetti P, Auberger P, Tanti JF, Le Marchand-Brustel Y, Bost F. Antidiabetic drug metformin exerts an antitumor effect in vitro and in vivo through a decrease of cyclin D1 level. Oncogene. 2008;27:3576-86
26. Liu B, Fan Z, Edgerton SM, Deng XS, Alimova IN, Lind SE, Thor AD. Metformin induces unique biological and molecular responses in triple negative breast cancer cells. Cell Cycle. 2009;8:2031-2040
27. Hirsch HA, Iliopoulos D, Tsichlis PN, Struhl K. Metformin selectively targets cancer stem cells, and acts together with chemotherapy to block tumor growth and prolong remission. Cancer Res. 2009;69:7507-7511
28. Kato K, Gong J, Iwama H, Kitanaka A, Tani J, Miyoshi H, Nomura K, Mimura S, Kobayashi M, Aritomo Y, Kobara H, Mori H, Himoto T, Okano K, Suzuki Y, Murao K, Masaki T. The antidiabetic drug metformin inhibits gastric cancer cell proliferation in vitro and in vivo. Mol Cancer Ther. 2012;11(3):549-560
29. Hardie DG. Role of AMP-activated protein kinase in the metabolic syndrome and in heart disease. FEBS Lett. 2008;582(1):81-89
30. Poels J, Spasic MR, Callaerts P, Norga KK. Expanding roles for AMP-activated protein kinase in neuronal survival and autophagy. Bioessays. 2009;31(9):944-952
31. Shackelford DB, Shaw RJ. The LKB1-AMPK pathway: metabolism and growth control in tumour suppression. Nat Rev Cancer. 2009;9(8):563-575
32. González-Angulo AM, Meric-Bernstam F. Metformin: a therapeutic opportunity in breast cancer. Clin Cancer Res. 2010;16(6):1695-1700
33. Vichai V, Kirtikara K. Sulforhodamine B colorimetric assay for cytotoxicity screening. Nat Protoc. 2006;1(3):1112-1116
34. Hedley BJ, Chu JE, Ormond DG, Beausoleil MS, Boasie A, Alan AL, Xeno Costas A. Recombinant human erythropoietin in combination with chemotherapy increases breast cancer metastasis in preclinical models. Clin Cancer Res. 2011;17(19):6151-6162
35. Couch FJ, Hart SN, Sharma P, Toland AE, Wang X, Miron P, Olson JE, Godwin AK, Pankratz VS, Olswold C, Slettedahl S, Halberg E, Guidugli L, Davila JI, Beckmann MW, Janni W, Rack B, Ekici AB, Slamon DJ, Konstantopoulou I, Fostira F, Vratimos A, Fountzilas G, Pelttari LM, Tapper WJ, Durcan L, Cross SS, Pilarski R, Shapiro CL, Kemp J, Yao S, Garber J, Cox A, Brauch H, Ambrosone C, Nevanlinna H, Yannoukakos D, Slager SL, Vachon CM, Eccles DM, Fasching PA. Inherited mutations in 17 breast cancer susceptibility genes among a large triple-negative breast cancer cohort unselected for family history of breast cancer. J Clin Oncol. 2015;33(4):304-311
36. Bernier J, Poortmans PMP. Surgery and radiation therapy of triple-negative breast cancers: from biology to clinics. Breast. 2016;28:148-155
37. Reddy KB. Triple-negative breast cancer: an updated review on treatment options. Current Oncology. 2011;18(4):e173-e179
38. Baselga J. Why the epidermal growth factor receptor? The rationale for cancer therapy. Oncologist. 2002;7(suppl 4):2-8
39. Anisimov VN, Berstein LM, Egormin PA, Piskunova TS, Popovich IG, Zabezhinski MA, Kovalenko IG, Poroshina TE, Semenchenko AV, Provinciali M, Re F, Franceschi C. Effect of metformin on life span and on the development of spontaneous mammary tumors in HER-2/neu transgenic mice. Exp Gerontol. 2005;40:685-693
40. Buzzai M, Jones RG, Amaravadi RK, Lum JJ, DeBerardinis RJ, Zhao F, Viollet B, Thompson CB. Systemic treatment with the antidiabetic drug metformin selectively impairs p53-deficient tumor cell growth. Cancer Res. 2007;67:6745-6752
41. Algire C, Zakikhani M, Blouin MJ, Shuai JH, Pollak M. Metformin attenuates the stimulatory effect of a high-energy diet on in vivo LLC1 carcinoma growth. Endocr Relat Cancer. 2008;15:833-839
42. Kisfalvi K, Eibl G, Sinnett-Smith J, Rozengurt E. Metformin disrupts crosstalk between G protein-coupled receptor and insulin receptor signaling systems and inhibits pancreatic cancer growth. Cancer Res. 2009;69:6539-6545
43. Ward RA, Brassington C, Breeze AL, Caputo A, Critchlow S, Davies G, Goodwin L, Hassall G, Greenwood R, Holdgate GA, Mrosek M, Norman RA, Pearson S, Tart J, Tucker JA, Vogtherr M, Whittaker D, Wingfield J, Winter J, Hudson K. Design and synthesis of novel lactate dehydrogenase A inhibitors by fragment-based lead generation. J Med Chem. 2012;55(7):3285-3306
44. Fiume L, Manerba M, Vettraino M, Di Stefano G. Impairment of aerobic glycolysis by inhibitors of lactic dehydrogenase hinders the growth of human hepatocellular carcinoma cell lines. Pharmacology. 2010;86(3):157-162
45. Yang Y, Su D, Zhao L, Zhang D, Xu J, Wan J, Fan S, Chen M. Different effects of LDH-A inhibition by oxamate in non-small cell lung cancer cells. Oncotarget. 2014;5(23):11886-11896
46. Escudero-Ortiz V, Ramón-López A, Duart MA, Pérez-Ruixo JJ, Valenzuela B. Populational pharmacokinetics of doxorubicin applied to personalised its dosing in cancer patients. Farm Hosp. 2012;36(4):282-291
47. Spallarossa P, Altieri P, Aloi C, Garibaldi S, Barisione C, Ghigliotti G, Fugazza G, Barsotti A, Brunelli C. Doxorubicin induces senescence or apoptosis in rat neonatal cardiomyocytes by regulating the expression levels of the telomere binding factors 1 and 2. Am J Physiol Heart Cir Physiol. 2009;297:H2169-H2181
48. Drug information database. http://www.drugs.com/pro/metformin.html. Accessed 25th of November. 2014
49. Koukourakis MI, Kontomanolis E, Giatromanolaki A, Sivridis E, Liberis V. Serum and tissue LDH levels in patients with breast/gynaecological cancer and benign diseases. Gynecol Obstet Invest. 2009;67:162-168
50. Arora R, Schmitt D, Karanam B, Tan M, Yates C, Dean-Colomb W. Inhibition of the Warburg effect with a natural compound reveals a novel measurement for determining the metastatic potential of breast cancers. Oncotarget. 2015;6(2):662-78
51. Hait WN, Jin S, Yang JM. A matter of life or death (or both): understanding autophagy in cancer. Clin Cancer Res. 2006;12(7):1961-1965
52. Gump JM, Thorburn A. Autophagy and apoptosis: what is the connection? Trends Cell Biol. 2011;21(7):387-392
53. Tschan MP, Simon HU. The role of autophagy in anticancer therapy: promises and uncertainties. J Intern Med. 2010;268(5):410-418
54. Macintosh RL, Ryan KM. Autophagy in tumour cell death. Semin Cancer Biol. 2013;23(5):344-351
55. Tanida I, Ueno T, Kominami E. LC3 and autophagy. Methods Mol Biol. 2008;445:77-88
Author contact
![]() Corresponding authors: Carlos Pérez-Plasencia, e-mail: carlos.pplas: +52-55-5623-1333 (ext. 39807). Or Nadia J Jacobo-Herrera, e-mail: nadia.jacobocom. Tel.: +52-55-54870900 (ext. 2608).
Corresponding authors: Carlos Pérez-Plasencia, e-mail: carlos.pplas: +52-55-5623-1333 (ext. 39807). Or Nadia J Jacobo-Herrera, e-mail: nadia.jacobocom. Tel.: +52-55-54870900 (ext. 2608).