Impact Factor ISSN: 1837-9664

 Global reach, higher impact
Global reach, higher impactJ Cancer 2017; 8(2):190-198. doi:10.7150/jca.15481 This issue Cite
Research Paper
Doxycycline-Regulated p16MTS1 Expression Suppresses the Anchorage-Independence and Tumorigenicity of Breast Cancer Cell Lines that Lack Endogenous p16
Maria C Todd1 ![]() , Thomas A Langan2*, Robert A Sclafani3
, Thomas A Langan2*, Robert A Sclafani3
1. Biology Department, Southwestern University, Georgetown, TX USA.
2. Department of Pharmacology, University of Colorado School of Medicine, Aurora, CO 80045 USA.
3. Department of Biochemistry and Molecular Genetics, University of Colorado School of Medicine, Aurora, CO 80045 USA.
* In memory of our dear friend and colleague, Dr. Thomas Langan.
Received 2016-3-8; Accepted 2016-9-17; Published 2017-1-13
Abstract
The RB pathway controls the critical transition from G1 into S phase of the mammalian cell cycle. Deregulation of the RB pathway by means of RB or p16 inactivation has been implicated in the development of virtually all human cancers. Such findings have led to the view that the loss of RB-mediated regulation at the G1/S checkpoint is a precondition for human malignancy. Our analysis of the RB-positive MCF-7 and ZR75.1 breast cancer cell lines revealed a lack of endogenous p16 protein expression as a result of the homozygous deletion and methylation of the p16 gene at the CDKN2A locus, respectively. We employed the TET-OFF inducible expression system to investigate the effects of non-growth inhibitory levels of functional p16 protein upon the in vitro and in vivo transformed properties of the MCF-7 and ZR75.1 cell lines. Stable transfectants of MCF-7 and ZR75.1 cells were isolated that expressed different levels of p16 protein in the absence of doxycycline (DOX) but continued to proliferate in culture. Transfectants that expressed modest levels of p16 (relative to SV40 T antigen-transformed HBL-100 breast epithelial cells) demonstrated a marked suppression of anchorage-independent growth in soft agar. Further, the induction of moderate and high levels of p16 (relative to HBL-100) resulted in the suppression of tumorigenicity of both MCF-7 and ZR75.1 cells as assayed by injection into nude mice. From these data, we concluded that RB pathway restoration by non-growth inhibitory levels of p16 protein was sufficient to revert breast cancer cells to a non-transformed and non-tumorigenic state.
Keywords: TET-inducible expression system, p16, breast cancer, methylation, homozygous deletion.
Introduction
Owing largely to improvements in early detection and treatment, the mortality rate of women with breast cancer has decreased dramatically in recent years. However, in the United States, breast cancer remains the second most frequently diagnosed cancer in women with an estimated 595,690 breast cancer-related deaths predicted for 2016 (1). It is essential, therefore, to identify and develop targeted therapies for this disease.
The majority of breast tumors and cell lines express the cell cycle regulatory protein RB but harbor defects of one or more of the other components of the 'RB pathway', which override RB function. The most frequent aberration is the loss of p16 protein expression which occurs in 60-70% of breast cancer cell lines (2-4) and approximately 50% of primary breast tumors (5,6) and is often found in association with overexpression of cyclin D1 protein (for a review see Hunter and Pines, 1994 (7)). Analyses of the underlying mechanisms responsible for the loss of p16 expression have revealed that the CDKN2A gene is subject to homozygous deletion, methylation and, less commonly, mutation (8-14).
The prevalence of p16 inactivation in breast cancer and indeed, in multiple other types of cancer (10,14) is consistent with its essential role within the 'RB pathway' as a negative regulator of the G1/S transition (15,16). During G1, p16 binds and inhibits the activity of the cyclin D-dependent kinases, CDK4 and CDK6. Further, the accompanying p16-mediated disruption of cyclin D/CDK4/6 complexes results in the redistribution of the cyclin-dependent kinase inhibitors, p21 and p27, from cyclin D/CDK4/6 complexes (where they are non-inhibitory) to cyclin E/CDK2 complexes where they inhibit CDK2 kinase activity (17-19). In the absence of CDK4/6 and CDK2-mediated RB phosphorylation, hypophosphorylated RB is able to bind both E2F family members and the histone deacetylase, DHAC1 (20), resulting in the active repression of transcription from E2F-responsive promoters (21).
In addition to its fundamental regulatory role at the G1/S cell cycle transition, the elevated levels of p16 detected in primary cultures of senescent fibroblast, uroepithelial and keratinocyte cells suggest that it is also involved in the control of cellular senescence (22-25). Indeed, Lu et al. (2012) (26) more recently observed p16-mediated induction of senescence in MDA-MB-231 breast cancer cells transduced with the AdRSVp16 recombinant virus. Consistent with the above findings, loss of p16 expression has been reported in association with escape of senescence in normal mammary epithelial, uroepithelial, fibroblast and mesothelial cells (25, 27-29).
The high frequency of p16 loss in breast cancer makes it a logical target for gene-replacement therapy or chemotherapy using p16-peptidomimetics. It is critical, however, that both the p16 and RB status of the tumors be known as we and others have shown that p16-/RB+ cancer cells are growth arrested in response to adenoviral-mediated overexpression of p16 but p16+/RB- and p16+/RB+ cancer cells are completely resistant to the growth suppressive effects of p16 (30-32,18). From these data, we suggest that p16-replacement therapies would benefit the specific (and largest) subgroup of breast cancer patients: those with p16-/RB+ tumors, more characteristic of the luminal B subtype (33,34) In contrast, p16 therapy would likely prove less efficacious for patients with basal-like and triple negative breast cancers, the majority of which are p16+/RB- (35-38).
Although the above adenoviral studies are important in that they revealed the sensitivity of p16-/RB+ cancer cell lines to p16 (18, 30-32), the level of protein expressed by the adenovirus was exceptionally high and arrested the growth of both cancer cells and normal bronchial epithelial (NHBE) and foreskin fibroblast cells (18). Clearly, an effective therapy should inhibit only the growth of cancer cells. In addition, cells that are growth-arrested in response to adenoviral-mediated overexpression of p16 cannot be assessed for key hallmarks of transformed cells such as anchorage independence and tumorigenicity in nude mice. Thus, the goal of the current study was to examine the effect of near-normal, non-growth inhibitory levels of p16 expression upon the transformed and tumorigenic properties of breast cancer cells. To this end, we have constructed two breast cancer cell lines with an inducible DOX-regulated p16 gene using the TET-OFF expression system (39). Stable transfectants of the MCF-7 and ZR75.1 cells (both of which lack endogenous p16 and express RB) were isolated that expressed different levels of p16 protein in the absence of DOX. Unlike cells infected with adenoviral p16 constructs, however, the levels of p16 expressed by the TET-transfectants were not sufficient to arrest the cells in G1 thus enabling us to study the study the effect of different levels of p16 on anchorage-dependence and tumorigenicity in proliferating breast cancer cells. Indeed, we found that non growth-inhibitory levels of p16 were sufficient both to restore the anchorage-dependent phenotype as assayed by plating in soft agar and suppress the in vivo tumorigenicity of the MCF-7 and ZR75.1 breast cancer cell lines.
Materials and Methods
Breast cancer cell lines
Two breast cancer cell lines, MCF-7 and ZR75.1, and one normal, SV40-transformed breast epithelial cell line, HBL-100, were obtained from the University of Colorado Cancer Center Shared Resource. The non-transformed, immortalized breast epithelial cell line, MCF-12A, was obtained from the American Type Culture Collection (Manassas, VA, USA). The MCF-7, ZR75.1 and HBL-100 cell lines were cultured in MEM media supplemented with 10% fetal calf serum, 0.2% sodium bicarbonate, 10 mM HEPES, 1% non-essential amino acids, 2 mM L-glutamine and 6 ng/ml insulin (Life Technologies, Grand Island, NY USA). The MCF-12A cell line was culture in Ham's F12/DME (1:1) supplemented with 10% fetal calf serum, 20 ng/ml EGF, 500 ng/ml hydrocortisone, 100 ng/ml cholera toxin and 10 μg/ml insulin (Life Technologies).
Antibodies
The anti-cyclin D1 and anti-p16 antibodies were obtained from EMD Millipore (Billerica, MA USA) and BD Biosciences (San Jose, CA USA), respectively. Anti-pRb was a gift from Dr. Wen-Hua Lee (University of California, Irvine, CA). The horseradish peroxidase-conjugated secondary antibodies were obtained from Bio-Rad (Hercules, CA USA).
TET-OFF plasmid constructs
The 'regulatory' plasmid, pUDH15-1 and 'response' plasmid, pTET-SPLICE, were kindly provided by Dr. Bujard (Heidelberg, Germany). The pUDH15-1 plasmid was modified by the cloning of the neomycin-resistance gene into the XhoI site to enable selection in G418. A 0.5 kb p16 cDNA fragment was subcloned from pUC19 (gift from Dr. A. Kamb, Myriad Genetics, Utah, USA) into the HindIII-SpeI sites of the pTET-SPLICE plasmid (to create the PTET-SPLICE-p16 plasmid), the latter of which carries a gene for zeocin resistance.
Oligonucleotides
The pUDH15-1 oligonucleotide primer sequences were as follows: 5' TAG ATG TGC TTT ACT AAG TC 3' (sense) and 5' ACT TGA TGC TCT TGA TCT TC 3' (antisense). The p16 exon 2 oligonucleotide primer sequences (c5.1) are from Hussussian et al. (1994) (40).
Protein extraction and western blot analysis
Cells were harvested, washed in PBS, and resuspended in Laemmli sample buffer (41). The extracts were then boiled for 4 minutes, sheared through a 26-gauge syringe needle, aliquoted, and stored at -80°C.
Approximately 100 μg of each protein extract was subjected to SDS/PAGE and transferred either to nitrocellulose membranes (ThermoFisher Scientific, Pittsburgh, PA USA) for p16 blots or Immobilon P membranes (Merck Millipore Ltd, Co. Cork, Ireland) for cyclin D1 and RB blots for 45 minutes at 0.45 A using the Genie Electrophoretic Blotter (Idea Scientific, Minneapolis, MN USA). Membranes were stained with Ponceau dye to control for equal loading and immunodetection performed using the enhanced chemiluminescence (ECL) kit (GE Healthcare, Buckinghamshire, UK) according to the manufacturer's instructions.
DNA isolation
DNA was prepared by incubating cells at 55°C in lysis buffer (10 mM Tris pH 8.0, 2.0 mM EDTA pH 8.0, 10 mM NaCl, 5% SDS) containing 1 mg/ml Proteinase K. The samples were then subjected to two phenol-chloroform extractions and one chloroform:isoamylalcohol (24:1) extraction, followed by ethanol precipitation.
Homozygous deletion analysis by PCR
A 20 μl reaction mixture contained a final concentration of 200 ng of genomic DNA, PCR buffer (10 mM tris-HCl, pH 8.3; 5 mM KCl; 0.1% gelatin) 200 μM dNTPs; 1.5 mM MgCl2; and 0.06 units ul-1 Taq Polymerase, all of which were supplied by Promega (Madison, WI USA). For amplification of a single locus, 0.2 μM of either the c5.1 (p16) primers (40) or D9S199 primers (Research Genetics, Huntsville, AL, USA) were used, and for amplification of both loci by multiplex PCR, 0.2 μM of the c5.1 (p16) and 0.25 μM of the D9S199 primers were used. Following an initial denaturation period of 7 minutes at 95°C, the DNA was subjected to 20 cycles of amplification consisting of denaturation for 1 minute at 94°C, annealing for 1 minute, with a starting temperature of 55°C and decreasing by 0.5°C per cycle for the first 20 cycles, and elongation for 1 minute at 72°C. The final 10 cycles consisted of denaturation for 1 minute at 94°C, annealing for 1 minute at 45°C and elongation for 1 minute at 72°C followed by a final elongation step of 10 minutes at 72°C. The PCR products were subjected to electrophoresis on a 3% NuSieve gel, stained with ethidium bromide and visualized with uv light.
DNA analysis by Methylation-specific PCR (MSP)
DNA samples were modified with sodium bisulfite according to the method of Herman et al. (1996) (11), and precipitated with ammonium acetate (3M final concentration) and two volumes of ethanol. The resulting templates were subjected to PCR using oligonucleotides designed from the promoter of the p16 gene specific for wildtype, methylated or unmethylated DNA (11). A 20 μl reaction mixture contained a final concentration of 20 ng of genomic DNA, 120 ng of each oligonucleotide, PCR buffer (10 mM tris-HCL, pH 8.3; 5 mM KCl; 0.1% gelatin); 200 μM dNTPs; 1.5 mM MgCl2; and 0.06 units μl-1 Taq polymerase (added once the reaction temperature reached 95°C) all of which were supplied by Promega (Madison, WI USA). The DNA was subjected to 35 cycles of amplification consisting of denaturation for 0.5 minutes at 94°C, annealing for 0.5 minutes at 60°C (for unmethylated-specific oligonucleotides) or 65°C (for wildtype- and methylated-specific oligonucleotides), and elongation for 0.5 minutes at 72°C, followed by a final elongation step of 10 minutes at 72°C. The PCR products were subjected to electrophoresis on a 2% agarose gel, stained with ethidium bromide and visualized with uv light.
Stable transfection of breast cancer cell lines with pUDH15-1 and pTET-SPLICE-p16 plasmids
Cells were grown in 35 mm plates until they reached 50-60% confluence. At this time, 2.4 μg of each of the plasmids, pUDH15-1 and pTET-SPLICE-p16 was mixed with a total of 12 μl of DharmaFect I lipid (Fisher Scientific, Pittsburgh, PA USA) per well in serum-free medium, and incubated at room temperature for 15 minutes. The cells were then washed with 1 ml of serum-free medium and overlaid with the DNA/lipid mixture. After 5 hours at 37°C, the DNA/lipid mixture was removed and replaced with medium supplemented with 10% serum, 500 μg/ml G418 and 500 μg/ml zeocin (antibiotics obtained from Sigma Aldrich, St. Louis, MO USA). The cells were grown for approximately two weeks after which individual G418/zeocin-resistant colonies were isolated using sterile cloning rings (Bellco Glass, Inc, Vineland, NJ USA), transferred to individual 25 cm2 flasks and cultured in the selection media (plus or minus 1μg/ml DOX obtained from Sigma Aldrich) both for PCR analysis (to confirm the presence of both plasmids) and western analysis (to determine the level of p16 protein expression in the presence and absence of DOX).
Identification of stable pUDH15-1/p16 TET transfectants by PCR analysis
An approximately 50% confluent 25 cm2 flask of each clone was vigorously shaken to detach cells undergoing mitosis. These cells were collected by centrifugation and lysed in a solution of 10 mM Tris HCL pH 8.7, 50 mM KCl, 1.2 mM MgCl2, 0.01% gelatin, 0.45% NP-40, 0.45% Tween-20 and 10 mg/ml proteinase K at 55°C for 60 minutes. Five microliters of each of the resulting cellular lysates were subjected to PCR analysis using oligonucleotide primers corresponding to pUDH15-1 or p16 (c5.1) in the following manner: A 20 μl reaction mixture contained 5 μl genomic DNA, 0.2 μM of each primer, PCR buffer (10 mM tris-HCL, pH 8.3; 5 mM KCl; 0.1% gelatin); 200 μM dNTP's; 1.5 mM MgCl2; and 0.06 units μl-1 Taq polymerase, all of which were supplied by Promega (Madison, WI USA). Following an initial denaturation period of 7 minutes at 95°C, the DNA was subjected to 30 cycles of amplification consisting of denaturation for 1 minute at 94°C, annealing for 1 minute at 52°C (for pUDH15-1 reactions) or 58°C (for p16 reactions), elongation for 1 minute at 72°C, and a final elongation step of 10 minutes at 72°C. The PCR products were subjected to electrophoresis on a 2 % agarose gel, stained with ethidium bromide and visualized with uv light.
Cell cycle analysis of stable pUDH15-1/p16 TET transfectants
MCF-7 and ZR75.1 TET transfectants were cultured in the presence or absence of 1μg/ml DOX for 5 days after which they were harvested, stained with propidium iodide (42) and analysed for cell cycle distribution at the University of Colorado Cancer Center Shared Resource.
Soft agar analysis
Five milliliters of a mixture containing 0.4% agar, 10% serum, MEM medium, 500 μg/ml G418, 500 μg/ml zeocin, plus or minus 1 μg/ml DOX (previously incubated at 45°C) was added to 60 mm plates, and allowed to harden at room temperature. The two MCF-7 transfectants (MCF-7/15-1/p16#69, and MCF-7/15-1/p16#13), two ZR75.1 transfectants, (ZR/15-1/p16#7, and ZR/15-1/p16#9), and MCF-7 and ZR75.1 cell lines transfected with the TET plasmids (but not expressing p16) were grown in the presence or absence of 1 μg/ml DOX in two 75 cm2 flasks each for 5 days then harvested and counted. The cells were resuspended in MEM medium supplemented with 10% serum to 1.25 X 104 cells/ml, and 8 ml of each suspension added to 12 ml of the above agar/medium mixture. After mixing, 2.5 ml of each cell-agar suspension was plated on to eight 60 mm plates containing a base layer of agar/medium. Fresh medium containing antibiotic selection plus or minus 1 μg/ml DOX was added to the plates twice per week. After 3 weeks, the total number of colonies per plate were counted (and averaged for the 8 replicate plates). The MCF-7 and ZR751. parent cell lines were also grown in the presence and absence of DOX for 5 days and subjected to soft agar analysis in the absence of G418 and zeocin selection.
Tumorigenicity assays in nude mice
The following protocol (#51801001(01)1E) was approved by the University of Colorado School of Medicine Institutional Animal Care and Use Committee (IACUC). The MCF-7 transfectants, MCF-7/15-1/p16#69 and MCF-7/15-1/p16#13, and the ZR75.1 transfectants, ZR/15-1/p16#7, and ZR/15-1/p16#9, were grown in the absence of 1 μg/ml DOX for 5 days then harvested and counted. Following resuspension in PBS, 5 X 106 cells of each transfectant were injected subcutaneously into the right shoulder of three female athymic nude mice. The same number of non-transfected MCF-7 and ZR75.1 cells were also injected into mice in triplicate. The growth of MCF-7 tumors in nude mice is estrogen-dependent. Thus, a 1.7 mg pellet of 17 ß-estradiol (Innovative Research of America, Sarasota, FL USA) that releases hormone over a 90-day period to give constant levels in the blood of 500-600 pg/ml was implanted into the mice prior to injection of the MCF-7 parent cell line and MCF-7 TET transfectants. The mice were monitored for the growth of tumors over a 12 week period after which the mice were euthanized and the tumors excised and weighed. The differences between groups were tested by one-way ANOVA followed by the Tukey's post-hoc HSD test.
Results
Loss of p16 expression in RB-positive MCF-7 and ZR75.1 cells
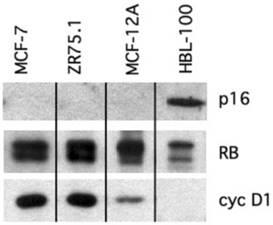
Breast cancers lose the ability to control the transition from G1 into S-phase of the cell cycle through the loss of p16 or, less frequently, RB expression (34, 43). Previous studies have shown that cells expressing functional RB but lacking endogenous p16 can be arrested in G1 phase by adenoviral-mediated overexpression of p16 (30-32,18). The aim of the current study was to determine the effects of regulated (rather than constitutively overexpressed) levels of p16 expression upon the transformed and tumorigenic properties of breast cancer cells. Using western blotting, we identified two breast cancer cell lines, MCF-7 and ZR75.1 that lacked detectable p16 protein but expressed RB (Figure 1). The presence of both hyperphosphorylated and hypophosphorylated forms of RB protein in the two cell lines indicated that the protein was functional and therefore capable of mediating a G1 growth arrest in response to the ectopic expression of functional p16 (Figure 1). We used the HBL-100 breast epithelial cell line as a positive control for p16 expression. These cells overexpress p16 because RB protein is inactivated by the presence of SV40 T antigen resulting in the loss of a futile negative feedback loop between RB and p16 (32,44). Both MCF-7 and ZR75.1 overexpressed cyclin D1 protein by approximately 10-fold relative to a non-transformed breast epithelial cell line, MCF-12A (Figure 1). Thus, the MCF-7 and ZR75.1 cell lines had overcome RB function through the complete loss of p16-mediated inhibition of G1 cyclin-dependent kinase activity.
Loss of p16 expression in RB-positive breast cancer cell lines. Protein isolated from logarithmic phase breast cancer cell lines, MCF-7 and ZR75.1, and two breast epithelial cell lines, MCF-12A (non-transformed, immortalized) and HBL-100 (SV40-transformed) was subjected to immunoblot analysis using antibodies to the p16, RB and cyclin D1 proteins. Both MCF-7 and ZR75.1 cells showed loss of p16 protein, expression of RB and overexpression of cyclin D1. RB protein in HBL-100 cells is inactive due to the presence of SV40 T antigen.
Loss of p16 expression through homozygous deletion and methylation of the gene in the MCF-7 and ZR75.1 cell lines, respectively
DNA from the MCF-7 and ZR75.1 breast cancer cell lines and the SV40 T antigen-transformed breast epithelial cell line, HBL-100, was subjected to duplex PCR analysis using oligonucleotides designed from intron 2 of the p16 gene, CDKN2A, (at chromosome 9p21), in combination with primers from the D9S199 locus (at 9p23). As the D9S199 locus in localized distal to the p16 gene and therefore unlikely to be within a homozygously deleted region on chromosome 9p21, we predicted that it would be amplified in each of the DNA samples assessed (and therefore acted as our positive control). HBL-100 and ZR75.1 DNA amplified both the p16 and D9S199 products. However, the MCF-7 cell line DNA underwent amplication with only the D9S199 primers, which indicated that it had undergone the homozygous deletion of the p16 gene (Figure 2A).
(A) The p16 gene is homozygously deleted in the MCF-7 breast cancer cell line. DNA derived from the MCF-7 and ZR75.1 breast cancer cell lines and the HBL-100 SV40-transformed breast epithelial cell line was subjected to PCR using primers to the p16 gene (c5.1) alone (first lane under each cell line), both p16 (c5.1) and D9S199 (control) (second lane under each cell line) or D9S199 alone (third lane under each cell line). Following amplification, the PCR products were subjected to electrophoresis on 3% NuSieve gels and the amplicons visualized with long-wave UV following ethidium bromide staining. (B) The p16 gene is both methylated and unmethylated in the ZR75.1 breast cancer cell line. Following sodium bisulfite modification, DNA from the indicated cell lines was subjected to PCR using primers specific for unmodified (WT), methylated (M) and unmethylated (U) p16. PCR products were subjected to electrophoresis on a 2% agarose gel and the amplicons visualized with uv following ethidium bromide staining.
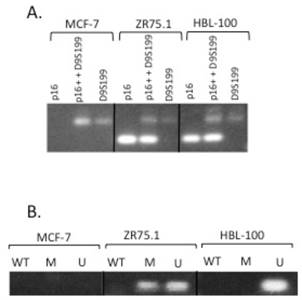
To determine the mechanism underlying the loss of p16 protein expression in the ZR75.1 cell line, DNA was subjected to methylation-specific PCR (MSP). Following modification of cell line DNA with sodium bisulfite (which converts all unmethylated cytosines to uracil), the resulting DNA was subjected to PCR using primers from the promoter of the p16 gene that were specific for unmodified (WT), methylated (M) and unmethylated (U) DNA (11). Notably, the ZR75.1 DNA underwent amplification with primers specific for both methylated and unmethylated DNA, which suggested that methylation of the gene was partially responsible for the lack of p16 expression but that there is likely another mechanism responsible for the complete loss of p16 protein expression in this cell line (Figure 2B).
Construction of stable MCF-7 and ZR75.1 transfectants demonstrating DOX-dependent expression of p16
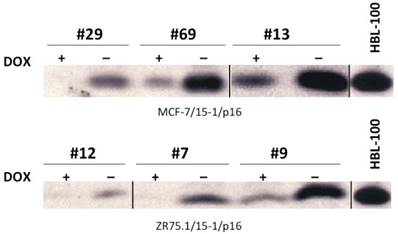
To determine the effect of expressing regulated levels of functional p16 on the anchorage dependence and tumorigenicity of the MCF-7 and ZR75.1 breast cancer cell lines (which lack endogenous p16 protein) we used the TET-OFF expression system. Following the co-transfection of the regulatory plasmid (pUDH15-1) and response plasmid (pTET-SPLICE-p16) into MCF-7 and ZR75.1 cells, we used PCR to identify multiple transfectants that contained both the pUDH15-1 and pTET-SPLICE-p16 plasmids. Subsequent immunoblot analysis allowed us to identify three transfectants derived from each of the two cell lines that expressed low, moderate and high levels of p16 protein in the absence of DOX (relative to the HBL-100 cell line, which overexpresses p16) (Figure 3 and Figure S3). Notably, those clones that expressed moderate to high levels of p16 in the absence of DOX, also expressed barely detectable to low levels of the p16 protein in the presence of DOX. This finding indicated a slight 'leakiness' in the TET-OFF system. In addition, all of the transfectants that demonstrated moderate to high expression of p16 in the absence of DOX showed a concomitant increase in the G1 phase population, the latter of which was more pronounced in the MCF-7 clones. Based on these data, we concluded that the ectopically expressed p16 was functional and able to contribute to cell cycle regulation in the cancer cells (Table 1) However, in contrast to the complete cessation of growth we previously reported in cells infected with Ad-p16 adenovirus (at an multiplicity of infection of 100 (32)), even those MCF-7 and ZR75.1 transfectants expressing the highest levels of p16 continued to proliferate, which made it possible for us to determine the effects of different levels of p16 upon the in vitro transformed and in vivo tumorigenic properties of the MCF-7 and ZR75.1 cells lines.
Inducible p16 expression in MCF-7 and ZR75.1 transfectants in the absence of DOX. MCF-7 and ZR75.1 cells stably transfected with both the regulatory (pUDH15-1) and response (pTET-SPLICE-p16) plasmids were cultured in the presence or absence of DOX for 5 days, harvested for protein isolation and subjected to immunoblot analysis to determine the degree of p16 induction. The HBL-100 SV40 T antigen-transformed cell line served as a control for overexpression of 16 protein.
Cell cycle analysis of MCF-7 and ZR75.1 TET transfectants grown in the presence or absence of DOX.
| Cell line/TET transfectant | % G1 | % S | % G2/M |
|---|---|---|---|
| MCF-7 | 72 | 8 | 20 |
| MCF-7/15-1/p16#69 (+ DOX) | 66 | 23 | 9 |
| MCF-7/15-1/p16#69 (- DOX) | 87 | 7 | 6 |
| MCF-7/15-1/p16#13 (+ DOX) | 63 | 22 | 15 |
| MCF-7/15-1/p16#13 (- DOX) | 87 | 7 | 6 |
| ZR75.1 | 63 | 31 | 6 |
| ZR75.1/15-1/p16#7 (+ DOX) | 80 | 11 | 9 |
| ZR75.1/15-1/p16#7 (- DOX) | 83 | 9 | 8 |
| ZR75.1/15-1/p16#9 (+ DOX) | 78 | 13 | 9 |
| ZR75.1/15-1/p16#9 (- DOX) | 82 | 10 | 8 |
DOX-dependent p16 expression suppresses the anchorage-independence of breast cancer cell lines
One of the hallmarks of tumor cells is their ability to grow in the absence of anchorage to a substrate. To assay the effect of p16 expression upon the anchorage-independence of MCF-7 and ZR75.1 cells, we subjected the MCF-7 and ZR75.1 TET transfectants expressing moderate (MCF-7/15-1/p16#69, ZR75.1/15-1/p16#7) and high (MCF-7/15-1/p16#13, ZR75.1/15-1/p16#9) levels of p16 to soft agar analysis. As negative controls, we also assessed the anchorage dependence of MCF-7 and ZR75.1 transfectants that contained both the pUDH15-1 and pTET-SPLICE-p16 plasmids but failed to express p16 protein and the non-transfected parent cell lines, MCF-7 and ZR75.1. All cells were grown in the presence or absence of 1 μg/ml DOX-containing medium for 5 days, after which 1.25 x 104 cells per cell line/transfectant were plated onto soft agar in 8 replicate 60 mm plates. Fresh medium containing antibiotic selection with or without DOX, was added to the plates twice per week. After 3 weeks, the total numbers of colonies per plate were counted.
The MCF-7 and ZR75.1 parent cells grew successfully both in the presence and absence of DOX forming large, healthy colonies with no evidence of growth suppression in the absence of anchorage (Table 2). In addition, the non-p16 expressing MCF-7 and ZR75.1 cells transfected with the pUDH15-1 and pTET-SPLICE-p16 plasmids also grew well with only a slight decrease in colony number relative to the non-transfected parent cell lines. In contrast, we observed a preponderance of inviable single cells and small clusters of 2-8 cells on the plates containing the MCF-7/15-1/p16#69 and MCF-7/15-1/p16#13 transfectants that were cultured in either the presence or absence of DOX. This finding suggested that even the low level of 'leaky' p16 expression exhibited by these two clones in the presence of DOX was sufficient to inhibit the growth of MCF-7 cells in soft agar. Consistent with these findings, both ZR75.1/15-1/p16#7 and ZR75.1/15-1/p16#9 transfectants also showed a marked restoration of anchorage dependence when plated onto soft agar in the absence of DOX. In addition, ZR75.1/15-1/p16#9 cells cultured in the presence of DOX (which showed low levels of 'leaky' p16 expression), were also suppressed for growth in soft agar. Notably, ZR75.1/15-1/p16#7 cells, which showed no detectable p16 expression in the presence of DOX grew to form multiple large, healthy colonies in soft agar, supporting the conclusion that the expression of p16 was responsible for the restoration of anchorage-dependence in the cells.
Growth of MCF-7 and ZR75.1 TET transfectants in soft agar.
| % Colony formation1 | ||
|---|---|---|
| Cell Line/transfectant | (+) | (-) |
| doxycycline | doxycycline | |
| MCF-7 | 100 | 100 |
| MCF-7/15-1/p16#69 | <0.1 | <0.1 |
| MCF-7/15-1/p16#13 | <0.1 | <0.1 |
| ZR75.1 | 100 | 100 |
| ZR75.1/15-1p16#7 | ~70-80 | <2 |
| ZR75.1/15-1/p16#9 | <2 | <2 |
| MCF-7/15-1/pTET2 | ~90 | ~90 |
| ZR75.1/15-1/pTET2 | ~90 | ~90 |
1The number of colonies that grew from each of the transfectants plated onto soft agar (in the presence or absence of doxycycline) was expressed as a percentage of the total number of colonies that arose from the corresponding parent cell line.
2MCF-7 and ZR75.1 control transfectants harbored both the regulator (pUDH15-1) and response (pTET-SPLICE-p16) plasmids, but did not express p16 protein.
DOX-dependent p16 expression suppresses the tumorigenicity of breast cancer cells in nude mice
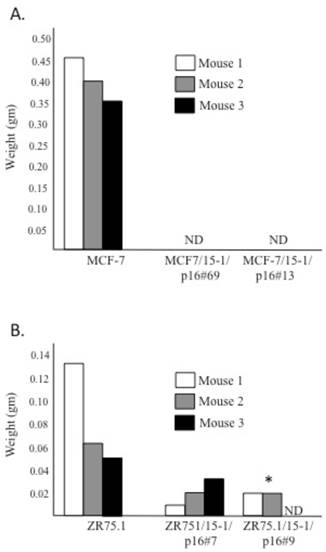
We performed nude mice assays to determine if the level of p16 in the MCF-7 and ZR75.1 TET transfectants was sufficient to suppress the tumorigencity of the parent cell lines. Both MCF-7 and ZR75.1 parent cell lines and each of the transfectants expressing moderate (MCF-7/15-1/p16#69, ZR75.1/15-1/p16#7) and high (MCF-7/15-1/p16#13, ZR75.1/15-1/p16#9) levels of p16 (relative to p16-overexpressing HBL-100 control cells) were grown in the absence of DOX to induce p16 expression for 5 days, after which 1 x 107 cells per cell line/transfectant were injected sub-cutaneously into the right shoulder of 3 female athymic nude mice. Tumors were measured biweekly for 12 weeks after which they were surgically resected and weighed. The tumorigenicity of both the MCF-7 and ZR75.1 transfectants was markedly suppressed relative to the parent cell lines as demonstrated by the complete lack of detectable tumors in mice injected with the MCF-7 transfectants and a statistically significant suppression of tumor growth (p<0.05) following injection of the high-p16 expressor, ZR75.1/15-1/p16#9 (Figure 4). From these data, we conclude that non-growth inhibitory levels of p16 protein contribute to the suppression of in vivo tumorigenicity of the MCF-7 and ZR75.1 breast cancer cell lines.
MCF-7 and ZR75.1 TET transfectants expressing p16 show decreased tumorigenicity. In addition to the non-transfected MCF-7 (Figure 4A) and ZR75.1 (Figure 4B) parent cell lines, transfectants expressing moderate (MCF-7/15-1/p16#69, ZR75.1/15-1/p16#7) and high (MCF-7/15-1/p16#13, ZR75.1/15-1/p16#9) levels of p16 (relative to the p16-overexpressing HBL-100 control cells) were grown in the absence of DOX for 5 days, after which 1 x 107 cells per cell line/transfectant were injected sub-cutaneously into the right shoulder of 3 female athymic nude mice. Tumors were resected and weighed after 12 weeks. ND indicates that no detectable tumor formed during the 12 week period of the experiment. *Significantly different as determined by One-way ANOVA followed post-hoc Tukey HSD test (p<0.05).
Discussion
The objectives of the current study were to assess the effect of p16 on both the in vitro and in vivo transformed properties of the breast cancer cell lines, MCF-7 and ZR75.1, both of which lacked p16 (due to the homozygous deletion and hemi-methylation of the gene, respectively) but expressed functional RB protein. To this end, we used the inducible TET-OFF expression system (39) to create transfectants that expressed non-growth inhibitory levels of p16 protein in the absence of DOX. Our desired outcome - to maintain cellular proliferation in the presence of ectopic p16 - contrasts with a previous study in which we used adenoviral-mediated overexpression of p16 to halt proliferation (in G1) in MCF-7 breast cancer cells (32).
We found that ZR75.1/15-1/p16#7 transfectants grown in the presence of DOX (no detectable p16 expression) retained the ability to grow in soft agar while those grown in the absence of DOX (moderate p16 expression) showed a marked reduction in anchorage independence. Further, the ZR75.1/15-1/p16#9 cells grown in the presence of DOX (low 'leaky' expression of p16) or absence of DOX (high p16 expression) were suppressed for growth in soft agar. Owing to the similarities of the cell cycle distribution profiles of each of these two transfectants, when cultured in the presence or absence of DOX, we concluded that it was the expression of p16 (and not changes in the growth rate of cells) that was responsible for restoration of anchorage-dependence in the ZR75.1 cells.
Whereas the MCF-7 transfectants cultured in the absence of DOX showed a more pronounced increase in G1 accumulation, the same cells cultured in the presence of DOX (which expressed a low 'leaky' level of p16) showed a cell cycle profile very similar to that of the non-transfected MCF-7 parent cell line. Analysis of the growth of MCF-7/15-1/p16#69 and MCF-7/15-1/p16#13 transfectants in soft agar showed that even those transfectants in which the low levels of p16 protein did not affect the cell cycle (in the presence of DOX), still showed loss of anchorage independence.
In comparison with the ZR75.1 parent cell line, the ZR75.1/15-1/p16#7 and ZR75.1/15-1/p16#9 transfectants induced the formation of smaller tumors in athymic nude mice (with statistically significant growth inhibition (p<0.05) in the case of ZR75.1/15-1/p16#9, which expressed the highest level of p16 in the absence of DOX). Given that the cell cycle distribution profiles of the ZR75.1/15-1/p16#9 transfectant grown in the presence or absence of DOX were similar to one another and to that of the ZR75.1/15-1/p16#7 cells grown in the presence of DOX (no detectable p16 expression), we again concluded that the p16-mediated suppression of tumorigenicity was independent of the growth rate of the cells.
Unlike the MCF-7 parent cell line, the MCF-7/15-1/p16#69 and MCF-7/15-1/p16#13 transfectants were unable to induce the formation of detectable tumors in athymic nude mice. Although the transfectants proliferated in culture, we recognized that the increase in G1 accumulation (in the absence of DOX) might influence the growth rate of tumors in mice. To mitigate this possibility, we monitored tumor development for an extended period of 12 weeks-post injection of MCF-7 transfectants. The subsequent absence of tumor formation suggested to us that the less tumorigenic phenotype of the MCF-7 transfectants was due, at least in part; to p16-mediated restoration of important cell cycle controls and possibly to p16's other anti-tumor effects. Indeed, Lu et al. (2012) (26) showed that AdRSVp16-mediated growth inhibition of MDA-MB-231 breast cancer cells is associated with increased apoptosis, decreased expression of the angiogenic factor, VEGF, and induction of senescence.
In summary, our study found that regulated, non-growth inhibitory levels of p16 are sufficient to restore anchorage-dependence and inhibit the tumorigenicity of breast cancer cells. We propose, therefore, that the constitutively high levels of p16 expression resulting from recombinant adenoviral infection are not necessarily critical to the success of p16-based gene therapeutic approaches. In contrast to the non-discriminating effects of adenoviral-mediated overexpression of p16 on both normal and tumor cells previously shown by our group (18), we propose that lower inducible levels of p16 expression may be sufficient to inhibit breast cancer without associated normal cell growth inhibition.
Supplementary Material
Figure S3.
Acknowledgements
This work was supported by a grant from the US Army Medical Research and Development Command No. DAMD17-94-J-4481, and by a University of Colorado Cancer Center support grant from the NCI No. 2P30 CA046934. We would like to thank Dr. Maggie Dalton and Ms. Wei Ming Chen for technical assistance and Dr. Maria Cuevas for help with the statistical analysis and preparation of the figures.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Key Statistics about Breast Cancer 2016. American Cancer Society. http://www.cancer.org/cancer/breastcancer/detailedguide/breast-cancer-key-statistics
2. Brenner AJ, Aldaz CM. Chromosome 9p allelic loss and p16/CDKN2 in breast cancer and evidence of p16 inactivation in immortal breast epithelial cells. Cancer Res. 1995;55:2892-2895
3. Liu Y, Heyman M, Wang Y, Falkmer U, Hising C, Szekely L, Einhorn S. Molecular analysis of the retinoblastoma gene in primary ovarian cancer cells. Int. J. Cancer. 1994;58:663-667
4. Quesnel B, Fenauz P, Philippe N, Fournier J, Bonneterre J, Preudhomme C, Peyrat JP. Analysis of p16 gene deletion and point mutation in breast carcinoma. Oncogene. 1995;10:351-353
5. Geradts J, Wilson PA. High frequency of aberrant p16 (INK4A) expression in human breast cancer. Am. J. Path. 1996;149:15-20
6. Xu L, Sgroi D, Sterner CJ, Beauchamp RL, Pinney DM, Keel S, Ueki K, Rutter JL, Buckler AJ, Louis DN, Gusella JF, Ramesh V. Mutational analysis of CDKN2 (MTS1/p16ink4) in human breast carcinomas. Cancer Res. 1994;54:5262-5264
7. Hunter T, Pines J. Cyclins and cancer. II: Cyclin D and CDK inhibitors come of age. Cell. 1994;79:573-82
8. Kamb A, Shattuck-Eidens D, Eeles R, Liu Q, Gruis NA, Ding W, Hussey C, Tran T, Miki Y, Weaver-Feldhaus J. Analysis of the p16 gene (CDKN2) as a candidate for the chromosome 9p melanoma susceptibility locus. Nat Genet. 1994;8:23-6
9. Nobori T, Miura K, Wu DJ, Lois A, Takabayashi K, Carson DA. Deletions of the cyclin-dependent kinase-4 inhibitor gene in multiple human cancers. Nature. 1994;368:753-756
10. Herman JG, Merlo A, Mao L, Lapidus RG, Issa J-P J, Davidson NE, Sidransky D. and Baylin SB. Inactivation of the CDKN2/p16/MTS1 gene is frequently associated with aberrant DNA methylation in all common human cancers. Cancer Res. 1995;55:4525-4530
11. Herman JG, Graff JR, Myohanen S, Nelkin BD, Baylin SB. Methylation-specific PCR: a novel PCR assay for methylation status of CpG islands. Proc. Natl. Acad. Sci. USA. 1996;93:9821-9826
12. Gonzalez-Zuleta M, Bender CM, Yang AS Nguyen T, Beart RW Van Tornout, JM Jones, PA. Methylation of the 5' CpG island in p16/CDKN2 tumor suppressor gene in normal and transformed tissues correlates with gene silencing. Cancer Res. 1995;55:4531-4535
13. Merlo A, Herman JG, Mao L, Lee DJ, Gabrielson E, Burger PC, Baylin SB, Sidransky D. 5' CpG island methylation is associated with transcriptional silencing of the tumour suppressor p16/CDKN2/MTS1 in human cancers. Nature Medicine. 1995;1:686-692
14. Cairns P, Mao L, Merlo A, Lee DJ, Schwab D, Eby Y, Tokino K, van der Riet P, Blaugrund JE, Sidransky D. Frequency of homozygous deletion at p16/CDKN2 in primary human tumours. Science. 1994;265:415-417
15. Sherr CJ. G1 phase progression: cycling on cue. Cell. 1994;73:1059-1065
16. Serrano M, Hannon GJ, Beach D. A new regulatory motif in cell cycle control causing specific inhibition of cyclin D/CDK4. Nature. 1993;366:704-707
17. McConnell BB, Gregory FJ, Stott FJ, Hara E, Peters G. Induced expression of p16(INK4a) inhibits both CDK4- and CDK2-associated kinase activity by reassortment of cyclin-CDK-inhibitor complexes. Mol. Cell. Biol. 1999;19:1981-1989
18. Grimison B, Langan TA, Sclafani RA. P16Ink4a tumor suppressor function in lung cancer cells involves cyclin-dependent kinase 2 inhibition by Cip/Kip protein redistribution. Cell Growth Differ. 2000;11:507-515
19. Malumbres M, Barbacid M. Cell cycle, CDKs and cancer: a changing paradigm. Nat Rev Cancer. 2009;9:153-166
20. Brehm A, Miska EA, Mccance DJ, Reid J, Bannister AJ, Kouzarides T. Retinoblastoma protein recruits histone deacetylase to repress transcription. Nature. 1998;391:597-601
21. Weintraub S, Prater CA, Dean DC. Retinoblastoma protein switches the E2F site from positive to negative element. Nature. 1992;358:259-261
22. Hara E, Smith R, Parry D, Tahara H, Stone S, Peters G. Regulation of p16CDKN2 expression and its implications for cell immortalization and senescence. Mol. Cell. Biol. 1996;16:859-867
23. Alcorta DA, Xiong Y, Phelps D, Hannon G, Beach D, Barrett JC. Involvement of the cyclin-dependent kinase inhibitor p16 (INK4a) in replicative senescence of normal human fibroblasts. Proc. Nat. Acad. Sci. USA. 1996;93:13742-13747
24. Loughran O, Malliri A, Owens D, Gallimore PH, Stanley MA, Ozanne B, Frame MC, Parkinson EK. Association of CDKN2A/p16INK4A with human head and neck keratinocyte replicative senescence: realtionship of dysfunction to immortality and neoplasia. Oncogene. 1996;13:561-568
25. Reznikoff CA, Yeager TR, Belair CD, Savelieva E, Puthenveettil JA, Stadler WM. Elevated p16 at senescence and loss of p16 at immortalization in human papillomacirus 16 E6, but not E7, transformed human uroepithelial cells. Cancer Res. 1996;56:2886-2890
26. Lu Y, Zhang X, Zhang J. Inhibition of breast tumor cell growth by ectopic expression of p16/INK4A via combined effects of cell cycle arrest, senescence and apoptotic induction, and angiogenesis inhibition. Journal of Cancer. 2012;3:333-344
27. Noble JR, Rogan EM, Neumann AA, Maclean K, Bryan TM, Reddel RR. Association of extended in vitro proliferative potential with loss of p16INK4 expression. Oncogene. 1996;13:1259-1268
28. Kiyono T, Foster SA. Koop JI, McDougal JK, Galloway DA and Klingelhutz, AJ. Both Rb/p16INK4a inactivation and telomerase activity are required to immortalize human epithelial cells. Nature. 1998;396:84-88
29. Wong DJ, Foster SA, Galloway DA, Reid BJ. Progressive region-specific de novo methylation of the p16 CpG island in primary human mammary epithelial cell strains during escape from M0 growth arrest. Mol. Cell. Biol. 1999;19:5642-5641
30. Jin X, Nguyen D, Zhang WW, Kyritsis AP, Roth JA. Cell cycle arrest and inhibition of tumor cell proliferation by the p16INK4 gene mediated by an adenovirus vector. Cancer Res. 1995;55:3250-3253
31. Craig C, Kim M, Ohri E, Wersto R, Katayosed D, Liz Z, Choi Y H, Mudahar B, Srivastava S, Seth P, Cowan K. Effects of adenovirus-mediated p16INK4A expression on cell cycle arrest are determined by endogenous p16 and Rb status in human cancer cells. Oncogene. 1998;16:265-272
32. Todd MC, Sclafani RA, Langan TA. Ovarian cancer cells that coexpress endogenous Rb and p16 are insensitive to overexpression of functional p16 protein. Oncogene. 2000;19:258-264
33. Perou CM, Sørlie T, Eisen MB, Van de Rijn M, Jeffrey SS, Rees CA, Pollack JR, Ross DT, Johnsen H, Akslen LA, Fluge O, Pergamenschikov A, Williams C, Zhu SX, Lønning PE, Børresen-Dale A-L, Brown PO, Botstein D. Molecular portraits of human breast tumours. Nature. 2000;406:747-752
34. Ma CX, Ellis MJ. The Cancer Genome Atlas: clinical applications for breast cancer. Oncology (Williston Park). 2013;27912:74-79
35. Herschkowitz JI, He X, Fan C, Perou CM. The functional loss of the retinoblastoma tumour suppressor is a common event in basal-like and luminal B breast carcinomas. Breast Cancer Res. 2008;10:R75
36. Subhawong AP, Subhawong T, Nassar H, Kouprina N, Begum S, Vang R, Westra WH, Argani P. Most basal-like breast carcinomas demonstrate the same Rb-/p16+ immunophenotype as the HPV-related poorly differentiated squamous cell carcinomas which they resemble morphologically. Am J Surg Pathol. 2009;33:163-175
37. Treré D, Brighenti E, Donati G, Ceccarelli C, Santini D, Taffurelli M, Montanaro L, Derenzini M. High prevalence of retinoblastoma protein loss in triple-negative breast cancers and its association with a good prognosis in patients treated with adjuvant chemotherapy. Ann Oncol. 2009;20:1818-1823
38. Bogina GS, Lunardi G, Marcolini L, Brunelli M, Bortesi L, Marconi M, Coati F, Valerio M, Guerriero M, Massocco A, Pegoraro MC, Giuseppe Zamboni. p16 but not retinoblastoma expression is related to clinical outcome in no-special-type triple-negative breast carcinomas. Modern Path. 2013;27:204-213
39. Gossen M, Freundlieb S, Bender G, Muller G, Hillen W, Bujard H. Transcriptional activation by tetracyclines in mammalian cells. Science. 1995;268:1766-1769
40. Hussussian CJ, Struewing JP, Goldstein AM, Higgins PA, Ally DS, Sheahan MD, Clark WH Jr, Tucker MA, Dracopoli NC. Germline p16 mutations in familial melanoma. Nat Genet. 1994;8:15-21
41. Laemmli UK. Cleavage of Structural Proteins during the Assembly of the Head of Bacteriophage T4. Nature. 1970;227:680-685
42. Krishan A. Rapid cytofluorometric analysis of mammalian cell cycle by propidium iodide staining. J. Cell Biol. 1975;66:188-193
43. Williams GH, Stoeber K. The cell cycle and cancer. J. Pathol. 2012;226:352-364
44. DeGregori J. Surprising dependency for retinoblastoma protein in ras-mediated tumorigenesis. Mol Cell Biol. 2006;26:1165-9
Author contact
![]() Corresponding author: Maria C Todd, Ph.D. Biology Department, Fondren Jones Science Building Room 143, Southwestern University, Georgetown, TX 78626, USA. Tel: (512) 863-1983; Fax: (512) 863-1696; e-mail: toddmedu.
Corresponding author: Maria C Todd, Ph.D. Biology Department, Fondren Jones Science Building Room 143, Southwestern University, Georgetown, TX 78626, USA. Tel: (512) 863-1983; Fax: (512) 863-1696; e-mail: toddmedu.