Impact Factor ISSN: 1837-9664

 Global reach, higher impact
Global reach, higher impactJ Cancer 2017; 8(5):894-902. doi:10.7150/jca.17064 This issue Cite
Research Paper
Increased TET1 Expression in Inflammatory Microenvironment of Hyperinsulinemia Enhances the Response of Endometrial Cancer to Estrogen by Epigenetic Modulation of GPER
Qiao-Ying Lv1*, Bing-Ying Xie1*, Bing-Yi Yang1, Cheng-Cheng Ning1, Wei-Wei Shan1, Chao Gu1, Xue-Zhen Luo1, Xiao-Jun Chen1 ![]() , Zhen-Bo Zhang2
, Zhen-Bo Zhang2 ![]() , You-Ji Feng2
, You-Ji Feng2
1. Obstetrics and Gynecology Hospital of Fudan University, Shanghai, 200011, China;
2. Department of Obstetrics and Gynecology, Shanghai General Hospital, Shanghai Jiao Tong University school of medicine, Shanghai, 201620, China.
* These authors contributed equally to this study.
Received 2016-8-1; Accepted 2016-12-9; Published 2017-3-12
Abstract
Background: Insulin resistance (IR) has been well studied in the initiation and development of endometrial endometrioid carcinoma (EEC). As yet, it has been largely neglected for estrogen sensitivity in local endometrium in hyperinsulinemia-induced systemic microenvironment. The aim of this study was to investigate the role of insulin in regulating estrogen sensitivity and explore the potential mechanisms in insulin-driven inflammatory microenvironment.
Methods: We first investigated the effect of insulin on estradiol-driven endometrial cancer cells proliferation in vitro to address the roles of insulin in modulating estrogen sensitivity. Then GPER, ERα and TET1 in EEC samples with or without insulin resistance were screened by immunohistochemistry to confirm whether insulin resistance regulates estrogen receptors. Further mechanism analysis was carried out to address whether TET1 was mediated epigenetic modulation of GPER in insulin-induced microenvironment.
Results: Insulin enhanced estradiol-driven endometrial cancer cells proliferation by up-regulating G-protein-coupled estrogen receptor (GPER) expression, but not ERα or ERβ. Immunohistochemistry of EEC tissues showed that GPER expression was greatly increased in endometrial tissues from EEC subjects with insulin resistance and was positively correlated with Ten-eleven-translocation 1 (TET1) expression. Mechanistically, insulin up-regulates TET1 expression, and the latter, an important DNA hydroxymethylase, could up-regulate GPER expression through epigenetic modulation.
Conclusion: This study identified TET1 as the upstream regulator of GPER expression and provides a possible mechanism that insulin-induced positive regulation of estrogen sensitivity in endometrial cancer cells. Increasing expression of GPER through TET1-mediated epigenetic modulation may emerge as the main regulator to enhance the response of endometrial cancer to estrogen in insulin-driven inflammatory microenvironment.
Keywords: Insulin resistance, Endometrial endometrioid cancer, estrogen sensitivity, GPER/GPR30, TET1.
Introduction
Sustained estrogenic stimulation without progesterone has been accepted to be the main pathogenesis of endometrial hyperplasia and endometrial endometrioid carcinoma (EEC) [1]. As yet, serum hormone analysis showed serum estradiol is not elevated in endometrial hyperplasia and EEC patients compared to control [2]. On the other hand, observational studies also reported there is no higher risk in postmenopausal women with unopposed 10 μg estradiol vaginal tablets for 1 year compared with those with no such exposure [3]. A 14-year observational follow-up in women initially randomised to receive one table of estrogen valerate (2mg) or placebo daily for 2 years showed that unopposed estrogen may be used safely by women with an intact uterus surviving a first myocardial infarction [4]. These clinical data and investigations suggested that it might be the estrogen sensitivity of local endometrial that drives endometrial carcinogenesis rather than increased circulating estradiol level.
The inflammatory microenvironment of endometrial cancer is the key to control estrogen sensitivity in local endometrium. Che et al. reported that IL-6 could up-regulate intratumoral aromatase level in stromal cells and increased aromatase promotes intratumoral 17β-estradiol (E2) biosynthesis in endometrial carcinoma microenvironment [5, 6]. Similarly, infiltrating tumor-associated macrophages (TAMs) microenvironment in prostate cancer showed elevated androgen receptor (AR) in stroma [7]. In our previous work, infiltrating macrophages increased the sensitivity of endometrial cancer cell to estrogen by inducing nuclear estrogen receptor α (ERα) expression in epithelium [2]. Thus, in inflammatory microenvironment, increased E2 biosynthesis or ERα expression could both up-regulate estrogen sensitivity of glandular epithelium.
Chronic, low-level inflammatory response is a well-known cause and also major characteristic of insulin resistance (IR) [8]. As a hallmark of insulin resistance, elevated circulating insulin levels could activate phosphoinositide3-kinase (PI3K), the mitogen-activated protein kinases (MAPK) or other pathways to affect endocrine status, glucose metabolic homeostasis and local inflammatory changes in endometrial microenvironment [9, 10, 11]. Previous studies showed that hyperinsulinaemic conditions could regulate gene methylation modification [12]. As estrogen receptors are master proteins that regulate cell proliferation, differentiation and homeostasis in endometrium [13], we asked whether inflammatory signaling in the microenvironment of insulin resistance promotes glandular epithelium growth by increasing estrogen receptor and thus increases estrogen sensitivity.
To address these questions, we investigated the effects of insulin on estradiol-driven endometrial cancer proliferation and expression of GPER and ERs. Then further analysis of GPER expression in endometrial tissues of EEC with and without IR was employed to clarify the relationship between hyperinsulinemia and GPER expression. Moreover, we explored the potential mechanism of insulin-induced GPER expression in endometrium. Our study will provide an important molecular mechanism of insulin sensitizing endometrial cancer cells to estrogen through epigenetic modulation of GPER without involvement of nuclear estrogen receptors.
Materials and Methods
Cell lines, cell cultures and drug treatment
The human endometrial adenocarcinoma cell line Ishikawa and HEC-1-A was kindly provided by Dr. Yu Yinhua (MD Anderson Cancer Center, Houston, TX) and Dr. Wei Lihui (Peking People Hospital, Beijing, China) respectively. Cells were cultured in DMEM/F12 (Gibco BRL, USA) supplemented with 10% fetal bovine serum (FBS; Gibco BRL, USA) and 100 U/ml penicillin & streptomycin (P&S, Life technology). These cells were maintained in a humidified incubator with 5 % CO2 at 37°C. Cells with approximately 80% confluence were treated with insulin (Sigma), 17β-estradiol (E2, Sigma), LY294002 (Selleck) in phenol-red free DMEM/F12 (Gibco BRL, USA) at indicated dose for indicated peroid.
siRNAs, plasmid construct, and transient transfection
The siRNAs for ERα, GPER and TET1 was designed by Dharmacon. The plasmid pPB-TET1 was kindly provided by Prof. Shi Yujiang (Harvard University, Cambridge, MA). The siRNAs and plasmids transfection was conducted using Lipofectamine 3000TM (Invitrogen) according to the manufacturer's protocol. The transfection efficiency was confirmed by western blot.
Cell proliferation analysis
The endometrial cancer cells were seeded in 96-well plates (2000 cells/well). After siRNAs transfection, these cells were incubated with insulin (100nM) and/or E2 (10nM) in phenol-red free and FBS free DMEM/F12 medium for 48h. Cell proliferation was measured by sulforhodamine-B (SRB) assay as previously described [14]. Cells were fixed with ice-cold 10% trichloroacetic acid (TCA) at 4°C for 1h, then washed five times in distilled water and allowed to dry in the air. 100μl SRB was added to each well and incubated at room temperature for 30 min. Unbound dye was then removed by 1% v/v acetic acid washing. Wells were placed on a shaker platform with 100μl unbuffered Tris Base (10 mM, pH 10.5) for 5 minutes and optical density (OD) at 492 nm was measured by Varioskan Flash (Thermo Scientific).
Protein extraction and Western blot analysis
Total cell protein was extracted using RIPA lysis buffer (Thermo Scientific, USA) with PMSF and Phosphatase Inhibitors Cocktail (Thermo Scientific, USA). Western blot analyses were carried out as previously described [15]. The primary antibodies to GAPDH, β-tublin, AKT, P-AKT were all purchased from Cell signaling technology company, ERα, TET1, GPER from Sigma Aldrich, ERβ and cyclinD1 from Abcam company. Each experiment was repeated at least three times.
Immunofluorescence analysis
Immunofluorescence analysis was described as before [16]. The primary antibodies to GPER and TET1 were purchased from Sigma Aldrich, 5-hmc from Active Motif and anti-rabbit IgG-FITC was purchased from Jackson. All these antibodies were used according to the manufacturer's instructions. DAPI was used for identifying cell nucleus. Fluorescence was detected by confocal microscopy (Leica TCS SP8 Configurable Confocal).
Total RNA extraction, Reverse Transcription and RT-PCR analysis
Total RNA extraction and reverse transcription were performed as before [17]. Real-time quantitative PCR (RT-PCR) analysis was performed with SYBR Premix Ex Taq (Takara, Dalian, China) according to the manufacturer's protocol. The RT-PCR primers used were synthesized by Sangon Biotech (Shanghai, China) and listed in Table S1.
Endometrial cancer sample collection, immunohistochemical staining and Semi-quantitative optical analysis
Each of 5 endometrial cancer cases with or without insulin resistance was collected. The pathological type of all these cases was adenocarcinoma Grade 1. Patients' information is provided in Table 1. Sections from endometrial cancer lesion were used for immunohistochemical staining of GPER, ERα and TET1. Primary antibodies to GPER and Ten-eleven translocation 1 (TET1) were purchased from Sigma aldrich, and ERα from Cell signaling technology. PBS was used as a negative control. All the clinical specimens in this study were approved by the Medical Ethics Committee of the Obstetrics and Gynecology Hospital of Fudan University (Shanghai, China).
Basic information Basic information of clinical samples for Immunohistochemistry.
| EEC(G1) | Total No. | Age (years) | FBG(mmol/L) | FINS(μU/L) | HOMA-IR | ||||
|---|---|---|---|---|---|---|---|---|---|
| Mean (SE) | P | Mean (SE) | P | Mean (SE) | P | Mean (SE) | P | ||
| EEC without IR | 5 | 48.8 (3.720) | 5.42 (0.162) | 7.148 (1.058) | 1.7314 (0.283) | ||||
| 0.582 | 0.179 | 0.003 | 0.003 | ||||||
| EEC with IR | 5 | 45.8 (3.680) | 5.88 (0.260) | 20.916 (3.180) | 5.4372 (0.829) | ||||
*p< 0.05, **p < 0.01 and ***p < 0.001
G1, well differentiated endometrial cancer; FBG, fasting blood glucose; FINS, fasting insulin; HOMA-IR, homeostasis model assessment-insulin resistance.
Semi-quantitative optical analysis of immunohistochemical staining of these markers was described as before [18]. This part was carried out by two independent blinded observers. Briefly, ten representative high-magnification fields (400×) per slide were selected for calculating positive cell percentage. According to staining intensity (0, negative; 1, weak; 2, moderate; and 3, strong), the multiplying product of positive cell percentage with staining index was calculated. Then, the product was divided by three, making the IHC score from 0 to 100. Pearson correlation test was used for correlation analysis of these markers.
Dot blot analysis
Genomic DNA extraction was performed according to the manufacturer's instructions (QIAGEN). Dot blotting analysis was carried out as previously described [2]. One μl DNA (0.5μg/μl) was dropped on nitrocellulose membranes and cross-linked by a UV lamp (2,000 w) for 5 min, then blocked with 10% milk for 1h and incubated with 5-hmC primary antibody (Active Motif) overnight at 4°C. Secondary antibody (Jackson) was applied to bind 5-hmC and ECL reagent to detect densitometry. Methylene blue (MB) staining was used as a loading control.
hMeDip assays
hMeDip assays were carried out as previously described[19]. 5-hmC antibody (Active Motif) was purchased from Active Motif, rabbit IgG was used as negative control. PCR was carried out using primers for GPER promoter, which is shown in Table S1.
Statistical analysis
SPSS 19.0 (IBM SPSS Software) was used for statistics. Unpaired t test, one-way ANOVA, Pearson correlation test were used for appropriate analyses. The P value of <0.05 (two-tailed) was considered to be significant.
Results
Insulin promotes estradiol-driven endometrial cancer cells proliferation by up-regulating GPER expression but not ERα or ERβ
Based on the findings that insulin resistance is a significant contributing factor to endometrial cancer risk [20], we hypothesized that insulin might play essential roles in up-regulating estrogen sensitivity in endometrium. We first analyzed the relative proliferative rates of Ishikawa and HEC-1A cells in response to estrogen with or without insulin treatment. Although estrogen or insulin alone could stimulate the proliferation of endometrial cancer cells, estrogen in combination of insulin had the highest pro-proliferation effect on endometrial cancer cells (Fig. 1A). This indicates that insulin can enhance estradiol-driven endometrial cancer cells proliferation. This result was in agreement with previous report showing that insulin and estrogen co-treatment could significantly up-regulate cyclinD1 and c-myc proteins in breast cancer cells compared to insulin or estrogen alone [21]. However, the precise mechanism is not completely defined.
Insulin enhances estradiol-driven endometrial cancer cells proliferation by up-regulating GPER expression but not ERα or ERβ. A. Insulin promotes estrogen-driven endometrial cancer cell proliferation. Endometrial cancer cell line Ishikawa and HEC-1A was treated with insulin (100nM) and/or E2 (10nM) for 48h. SRB assay was used to analyze cell proliferation. B. Insulin up-regulates GPER expression at a dose dependent manner but not ERα or ERβ. Endometrial cancer cells were treated with insulin for 48h at indicated dose. GPER, ERα and ERβ were analyzed by western blot. C. Insulin up-regulates GPER expression at a time dependent manner. Endometrial cancer cells were treated with 100nM insulin. GPER expression was analyzed by western blot at indicated time point. D. Insulin up-regulates GPER expression in cell cytoplasm. Endometrial cancer cells were treated with 100nM insulin for 48h Immunofluorescence staining shows increased GPER expression (green) in the cytoplasm of cells after insulin treatment. DAPI (blue) was used for nuclear staining. E. Silencing GPER compromised insulin-induced estrogen sensitivity in endometrial cancer cells. Ishikawa and HEC-1A cells transfected with siCON or siGPER were treated with insulin (100nM) and/or E2 (10nM) for 48h. SRB assay was used to analyze cell proliferation. Transfection efficiency was confirmed by western blot. F. Silencing ERα did not compromise insulin-induced estrogen sensitivity in endometrial cancer cells. HEC-1A and Ishikawa cells transfected with siCON or siERα were treated with insulin (100nM) and/or E2 (10nM) for 48h. SRB assay was used to analyze cell proliferation. Transfection efficiency was confirmed by western blot. n.s., no significant; * p < 0.05, **p < 0.01,***p < 0.001 compared to control.
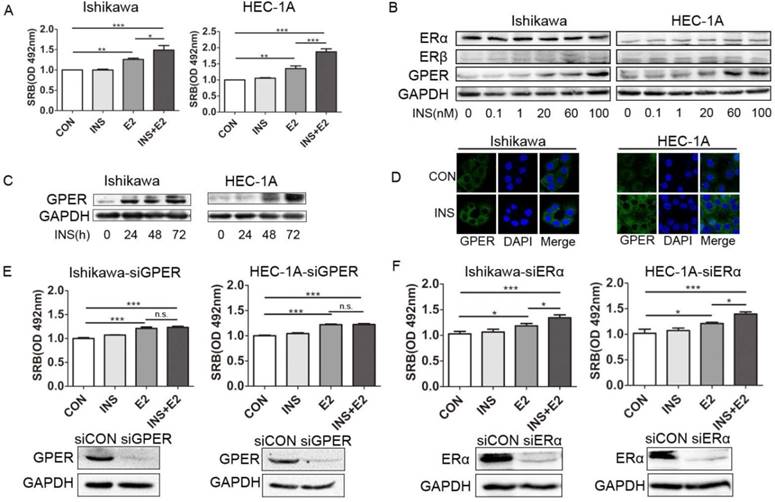
As estrogen receptors are the key factors mediating estrogen driven endometrial cancer cells proliferation [22], we first asked whether insulin promoted estrogen-driven endometrial cancer proliferation through up-regulating estrogen receptors. Western blot analysis showed that insulin could up-regulate membrane estrogen receptor GPER expression in endometrial cancer cells in dose-dependent manner, but had no effect on either ERα or ERβ which were nuclear estrogen receptors (Fig. 1B). Further study showed that insulin promoted GPER expression in a time-dependent manner (Fig. 1C). And Immunofluorescence assay showed that GPER staining intensity was prominently increased both in cell cytoplasm and cell membrane in response to insulin treatment (Fig. 1D). In this regard, we supposed that increased GPER expression induced by insulin might mediate increased estrogen sensitivity.
In order to testify our hypothesis that insulin promotes estradiol-driven endometrial cancer cell proliferation through up-regulating GPER instead of ERα, we silenced the expression of GPER or ERα expression respectively in endometrial cancer cells. Cell proliferation analysis showed that the pro-proliferation effect of insulin on estradiol-driven endometrial cancer proliferation disappeared after silencing GPER (Fig. 1E), but was not compromised when ERα expression was down-regulated (Fig. 1F). The findings confirmed that insulin enhanced estradiol-driven endometrial cancer cell proliferation through up-regulating GPER but not ERα.
GPER is strongly expressed in EEC with insulin resistance and positively correlates with TET1 expression
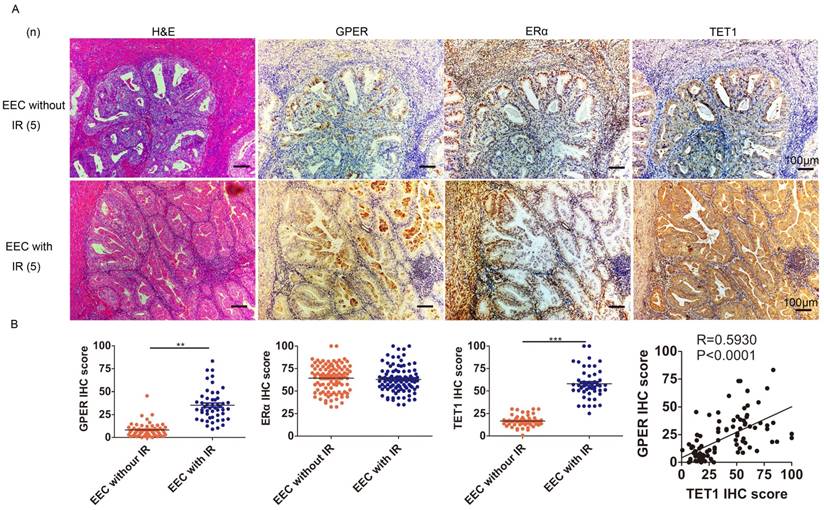
We next investigated the possible mechanism through which insulin up-regulated GPER. Epigenetic modulation is a key mechanism for the effect of insulin resistance on the body, and DNA hydroxymethylation is one of the most important epigenetic modulation pathways [23, 24], so we first investigated the relationship among insulin resistance, GPER and Ten-eleven-translocation 1 (TET1) in endometrial cancer lesions. TET1 is one of the primary components of the TET family, catalyzing the conversion of 5-methylcytosine (5-mC) to 5-hydroxymethylcytosine (5-hmC). Abnormal TET1 expression has been associated with the development of multiple types of cancer [25]. Our Immunohistochemical staining showed that GPER expression in endometrial cancer lesion was higher in patients complicated with insulin resistance compared to those without insulin resistance (Fig. 2A). No difference was found in ERα expression in endometrial cancer lesion between patients with or without insulin resistance. And TET1 expression was positively correlated with GPER but not ERα expression in endometrial cancer lesions (Fig. 2B). These results indicated that in patients with insulin resistance, increased TET1 might mediate GPER expression in endometrial cancer lesions.
GPER is strongly expressed in EEC with insulin resistance and positively correlates with TET1 expression. A. Immunohistochemical staining of GPER, ERα and TET1 expression in endometrial cancer lesion. GPER and TET1 expression were higher in patients suffering from insulin resistance (bottom panel) compared to those without insulin resistance (up panel). Number of cases (n) in each group is marked. Left column, H&E staining, original magnification 100×; right column, IHC staining with a monoclonal GPER, ERα and TET1 antibody, original magnification 100×. B. GPER, TET1 and ERα expression in endometrial cancer samples between groups with or without insulin resistance was compared by semi-quantitative optical analysis. Correlation of the IHC scores between GPER and TET1 was analyzed by pearson Correlation test. * p < 0.05, **p < 0.01,***p < 0.001 compared to control.
Increased TET1 expression and genomic DNA hydroxymethylation in insulin-driven microenvironment
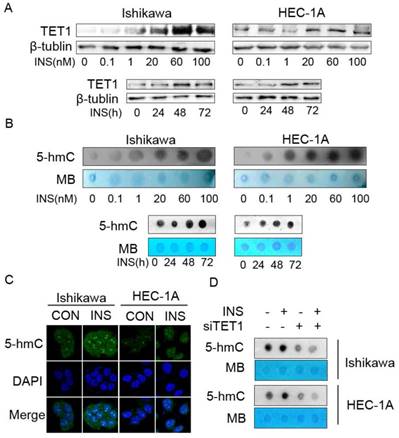
Because the clinical data showed that TET1 was highly expressed in endometrial cancer lesion with insulin resistance, we first investigated the effect of insulin on TET1 expression in endometrial cancer cells in vitro. Western blot analysis showed that insulin up-regulated TET1 expression in both a time- and dose- dependent manner (Fig. 3A). As the results showed that insulin up-regulated TET1 expression, and TET1 is the key enzyme catalyzing gene hydroxymethylation by converting 5-mC to 5-hmC, we then examined the global DNA hydroxymethylaiton after insulin treatment. Dot-blot analysis showed that insulin treatment increased 5-hmC level of global DNA in a dose- and time-dependent manner (Fig. 3B). Immunofluorescence staining assay also confirmed that insulin increased 5-hmC level in cell nuclear (Fig. 3C). These results revealed that insulin promoted DNA hydoxymethylation in endometrial cancer cells. Then we analyzed the 5-hmC level after silencing TET1 expression. As shown in Fig. 3D, additional insulin treatment could not rescue the attenuated 5-hmC level after silencing TET1 expression. These findings indicated that TET1 might mediate insulin-induced gene expression by promoting hydroxymethylation in endometrial cancer cells.
Insulin microenvironment shows increased TET1 expression and genomic DNA hydroxymethylation. A. Insulin promotes TET1 expression in endometrial cancer cells in a dose- and time-dependent manner. Cells were treated by insulin at indicated dose for 48h (up panel) or by 100nM insulin for indicated period (bottom panel). TET1 expression was analyzed by western blot. B. Insulin promotes genomic DNA hydroxymethylation. Endometrial cancer cells were treated with insulin at indicated dose for 48 h (up panel) or 100nM insulin for indicated period (bottom panel). 5-hmC levels were detected by dot-blot in total DNA. C. Insulin induces 5-hmC expression in cell nuclear. Confocal staining showed increasing 5-hmC expression (green) in the nuclei of endometrial cancer cells after insulin (100nM) treatment for 48h. DAPI (blue) was used as a DNA dye to indicate cell nuclei. D. TET1 specific siRNA abolished insulin-induced 5-hmC level of global DNA. Dot blot was used to analyze 5-hmC status. * p < 0.05, **p < 0.01,***p < 0.001 compared to control.
TET1 up-regulates GPER expression by promoting hydroxymethylation of GPER gene promoter in insulin-driven microenvironment
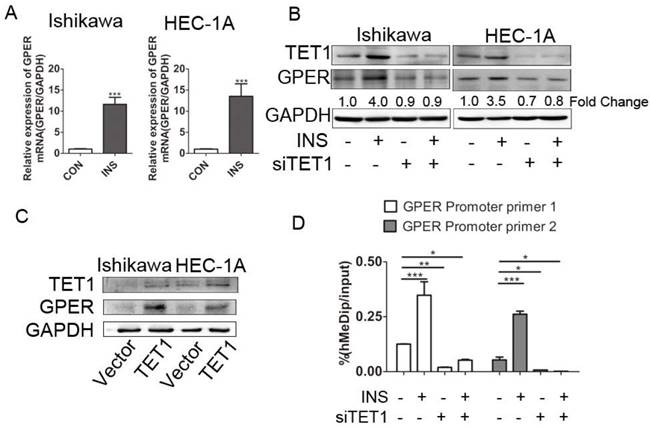
We further explored the possible mechanism involved in GPER up-regulation in insulin microenvironment. We first detected the transcriptional level of GPER after insulin treatment. The increased GPER mRNA level after insulin treatment documented that insulin promoted GPER transcription in endometrial cancer cells (Fig. 4A). Coupled to the previously described roles for TET1 in promoting gene expression [26], we assessed whether GPER up-regulation induced by insulin was directly attributable to TET1. We analyzed GPER expression in endometrial cancer cells after silencing or over-expressing TET1 by either siRNAs or cDNA plasmid. As shown in Fig. 4B, GPER expression was significantly down-regulated after silencing TET1, and additional insulin treatment could not rescue the attenuated GPER expression. On the other hand, overexpression of TET1 significantly enhanced GPER protein expression in endometrial cancer cells (Fig. 4C). Mechanistically, hMeDip assay showed showed that insulin treatment resulted in significant increase of 5-hmC in GPER gene promoter region in HEC-1A cells, whereas silencing TET1 by siRNA compromised GPER gene promoter hydroxymethylation (Fig. 4D). These findings indicated that TET1 is involved in insulin-driven GPER up-regulation through hydroxymethylation.
TET1 mediates GPER up-regulation by hydroxymethylation in insulin-driven microenvironment. A. Ishikawa and HEC-1A cells were treated with 100nM insulin for 24 h. GPER transcriptional level was analyzed by real time PCR. Data shown were normalized for GAPDH expression and then calculated as fold changes of relative mRNA expression. B. Insulin induced GPER expression through TET1. Endometrial cancer cells were transfected by siCON or siTET1. Cells with or without silencing TET1 were treated with 100nM insulin for 48h. The fold change of GPER relative to control group was calculated from densitometry. C. Increased TET1 expression up-regulates GPER expression. TET1 was overexpressed in endometrial cancer cells by tranfecting pPB-TET1 plasmid. GPER and TET1 expression were analyzed by western blot. D. TET1 affects hydroxymethylation of GPER gene promoter in insulin condition. HEC-1-A cells transfected with siTET1 or siCon were treated with 100nM insulin for 24h. 5-hmC level of GPER gene promoter fragment was detected by hMeDIP. * p < 0.05, **p < 0.01,***p < 0.001 compared to control.
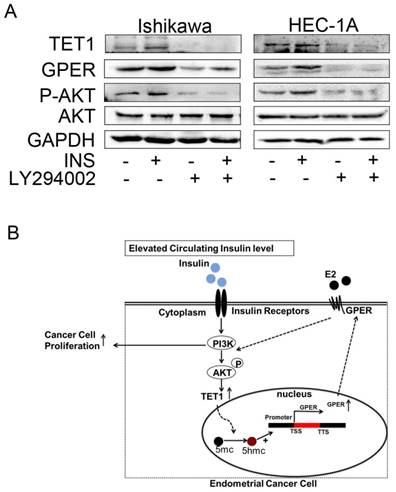
Insulin promotes TET1 expression through PI3K/AKT signaling pathway
It is well known that PI3K/AKT is the main pathway involved in insulin signaling [27]. But the relationship between PI3K/AKT and TET1 has not been reported before. Then we asked whether insulin promoted TET1 expression through PI3K/AKT pathway. To answer this question, LY294002, a specific PI3K/AKT inhibitor, was employed to inhibit PI3K/AKT signaling for investigating TET1 expression. As shown in Fig. 5A, blocking PI3K/AKT pathway compromised TET1 and subsequent GPER expression in endometrial cancer cells indicating that TET1 was up-regulated by insulin through PI3K/AKT pathway. Based on this finding, a proposed model was offered in Fig. 5B. The activated PI3K/AKT signaling induced by insulin up-regulates TET1 and GPER expression; the activated GPER signaling induced by estrogen promotes AKT activation, which causes further enhancement of GPER expression and cell proliferation. This positive loop in PI3K/AKT signaling accelerates estradiol-driven endometrial cancer cell growth by elevated GPER expression in insulin and estrogen co-treatment. Taken together, we can conclude that Insulin accelerates estradiol-driven endometrial cancer cells growth via a feed-forward PI3K/AKT signaling pathway.
Increased GPER signaling induced by Insulin establishes a feed-forward loop in PI3K/AKT signaling pathway. A. LY294002 abolished insulin-induced TET1 and GPER expression. The endometrial cancer cells were pre-treated by LY294002 (10uM) with or without insulin treatment for 48h. GPER and TET1 expression was analyzed by western blot. B. A proposed model that insulin induces GPER expression through TET1-mediated epigenetic mechanism to sensitize endometrial cancer cells to estrogen.
Discussion
Hyperinsulinemia is a significant contributing factor to endometrial cancer risk or even presents a causal association [28, 29]. In our previous study, we demonstrated that insulin resistance is present at early stages of endometrial hyperplasia; and elevated circulating insulin is positively correlated with disordered proliferative endometrium (DPE), endometrial hyperplasia (EH) with/without atypia and endometrial cancer [20]. But the precise mechanism of hyperinsulinemia in the process of carcinogenesis of endometrial cancer is still incompletely clarified. Elevated circulating insulin level induced by insulin resistance could contribute to the development of endometrial cancer by promoting cell proliferation through the PI3K/AKT and Ras/MAPK pathways and leading to changes in sex hormone levels in direct or indirect way [30]. Our latest work reported that inflammatory microenvironment might play roles in increased endometrial estrogen sensitivity, which is important in endometrial carcinogenesis [2]. The present study again showed that hyperinsulinemia, an important environmental factor, might increase local estrogen sensitivity in endometrium through up-regulating membrane estrogen receptor GPER but not ERα. This might be one of the important mechanisms hyperinsulinemia involved in carcinogenesis of endometrial cancer.
Epigenetic modulation is one of the important mechanisms of environmental factors regulating tumorigenesis [25]. Epigenetic changes might be caused by these environmental factors including hypoxia, inflammation, free radicals [31]. As a systemic environmental factor, hyperinsulinemia has been reported to regulate several genes methylation status [32]. Our results also showed that high level insulin treatment up-regulated the level of 5-hmC in genomic DNA, suggesting DNA hydroxymethylation regulation played an important role in insulin resistance-associated endometrial carcinogenesis.
Recent reports showed that the level of 5hmC and ten-eleven translocation (TET) family enzymes were altered in various types of cancers [33]. As a 5-hmC regulator, TET1 is a key epigenetic modulator in 5-mC to 5-hmC conversion. Hypoxia, chronic inflammation or other factors could up-regulate TET1 expression and trigger its downstream target gene transcription [26]. Mechanistical analysis showed that TET1 and 5-hmC are enriched at promoter regions of several pluripotency factors, including Nanog, Tcl1, and Esrrb [26, 34]. And also, TET1 regulated hypoxia-induced gene expression, such as INSIG1 (insulin induced gene 1), and epithelial-mesenchymal transition (EMT) [34]. Even in colorectal cancer, it was reported that IGF signaling could regulate Nanog via STAT3 signaling and promote EMT [35]. Besides, our latest study showed there was gradually increased TET1 expression in endometrial lesions with infiltrating macrophages [2]. In the present study, we found that TET1 was overexpressed in both insulin treatment and EEC samples with insulin resistance, suggesting TET1 might play roles in genes expression in endometrial cancer complicated with hyperinsulinmia. Accordingly, hydroxymethylation of GPER promoter region was altered when TET1 was up-regulated or silenced. This suggests that TET1 might be the up-stream regulator of GPER gene in the microenvironment of insulin resistance.
In this study, we also investigated the relationship between PI3K/AKT signaling and TET1 level. PI3K/AKT signaling is the classical pathway of insulin action on cells [27]. Inhibiting PI3K/AKT signaling blocked TET1 expression, suggesting that TET1 might be the down-stream molecular target of PI3K/AKT signaling.
The present study showed that insulin could promote endometrial cancer cells proliferation but not significantly. This result contradicts findings from Marco et al. [36] that IGF-Ⅰcould promote Ishikawa cell proliferation significantly. In our model, all SRB assays were carried out in FBS free medium. Conversely, their assays were incubated in medium containing 2.5% charcoal stripped FBS. The different pro-proliferative effect might be explained by the factors in charcoal-stripped serum.
In conclusion, our data provide compelling evidence that elevated GPER expression induced by insulin through TET1 mediated epigenetic modulation might be an important mechanism for increased estrogen sensitivity in endometrial cancer. This mechanism will help better understand the relationship between insulin resistance and endometrial cancer and provide potential therapeutic strategies for endometrial cancer with insulin resistance.
Supplementary Material
Figure S1 and Table S1.
Acknowledgements
This work was supported by the National Natural Science Foundation of China (Grant No. 81370688,81370074 and 81672562), Shanghai Science and Technology Development Funds (Grant No. 13JC1401300), Shanghai Science and Technology Development medical guide project (Grant No.134119a4500) and Shanghai Municipal Public Health Bureau (Grant No. XYQ2013119), and the "Chenxing Project" from Shanghai Jiao Tong University (Grant No. 15X100080023).
Competing Interests
The authors have declared that no competing interest exists.
References
1. R. Kaaks, A. Lukanova, M. S. Kurzer. Obesity, endogenous hormones, and endometrial cancer risk: a synthetic review[J]. Cancer Epidemiol Biomarkers Prev. 2002;11(12):1531-1543
2. C. Ning, B. Xie, L. Zhang, et al. Infiltrating Macrophages Induce ERalpha Expression through an IL17A-mediated Epigenetic Mechanism to Sensitize Endometrial Cancer Cells to Estrogen[J]. Cancer Res. 2016;76(6):1354-1366
3. J. Simon, L. Nachtigall, L. G. Ulrich, et al. Endometrial safety of ultra-low-dose estradiol vaginal tablets[J]. Obstet Gynecol. 2010;116(4):876-883
4. N. Cherry, R. McNamee, A. Heagerty, et al. Long-term safety of unopposed estrogen used by women surviving myocardial infarction: 14-year follow-up of the ESPRIT randomised controlled trial[J]. BJOG. 2014;121(6):700-705 705
5. Q. Che, B. Y. Liu, Y. Liao, et al. Activation of a positive feedback loop involving IL-6 and aromatase promotes intratumoral 17beta-estradiol biosynthesis in endometrial carcinoma microenvironment[J]. Int J Cancer. 2014;135(2):282-294
6. Q. Che, B. Y. Liu, F. Y. Wang, et al. Interleukin 6 promotes endometrial cancer growth through an autocrine feedback loop involving ERK-NF-kappaB signaling pathway[J]. Biochem Biophys Res Commun. 2014;446(1):167-172
7. X. Wang, W. J. Lin, K. Izumi, et al. Increased infiltrated macrophages in benign prostatic hyperplasia (BPH): role of stromal androgen receptor in macrophage-induced prostate stromal cell proliferation[J]. J Biol Chem. 2012;287(22):18376-18385
8. C. K. Glass, J. M. Olefsky. Inflammation and lipid signaling in the etiology of insulin resistance[J]. Cell Metab. 2012;15(5):635-645
9. P. M. Wairagu, A. N. Phan, M. K. Kim, et al. Insulin priming effect on estradiol-induced breast cancer metabolism and growth[J]. Cancer Biol Ther. 2015;16(3):484-492
10. S. Yang, B. Wang, F. Humphries, et al. The E3 ubiquitin ligase Pellino3 protects against obesity-induced inflammation and insulin resistance[J]. Immunity. 2014;41(6):973-987
11. Y. Wang, Y. Zhu, L. Zhang, et al. Insulin promotes proliferation, survival, and invasion in endometrial carcinoma by activating the MEK/ERK pathway[J]. Cancer Lett. 2012;322(2):223-231
12. A. B. Gaikwad, J. Gupta, K. Tikoo. Epigenetic changes and alteration of Fbn1 and Col3A1 gene expression under hyperglycaemic and hyperinsulinaemic conditions[J]. Biochem J. 2010;432(2):333-341
13. K. M. Yang, Y. Jung, J. M. Lee, et al. Loss of TBK1 induces epithelial-mesenchymal transition in the breast cancer cells by ERalpha downregulation[J]. Cancer Res. 2013;73(22):6679-6689
14. P. Houghton, R. Fang, I. Techatanawat, et al. The sulphorhodamine (SRB)assay and other approaches to testing plant extracts and derived compounds for activities related to reputed anticancer activity[J]. Methods. 2007;42(4):377-387
15. J. Chen, M. Bai, C. Ning, et al. Gankyrin facilitates follicle-stimulating hormone-driven ovarian cancer cell proliferation through the PI3K/AKT/HIF-1alpha/cyclin D1 pathway[J]. Oncogene. 2016;35(19):2506-2517
16. P. Rajbhandari, K. A. Schalper, N. M. Solodin, et al. Pin1 modulates ERalpha levels in breast cancer through inhibition of phosphorylation-dependent ubiquitination and degradation[J]. Oncogene. 2014;33(11):1438-1447
17. S. Liu, J. Sun, B. Cai, et al. NANOG regulates epithelial-mesenchymal transition and chemoresistance through activation of the STAT3 pathway in epithelial ovarian cancer[J]. Tumour Biol. 2016
18. X. Chen, A. Horiuchi, N. Kikuchi, et al. Hedgehog signal pathway is activated in ovarian carcinomas, correlating with cell proliferation: it's inhibition leads to growth suppression and apoptosis[J]. Cancer Sci. 2007;98(1):68-76
19. C. E. Nestor, R. Ottaviano, J. Reddington, et al. Tissue type is a major modifier of the 5-hydroxymethylcytosine content of human genes[J]. Genome Res. 2012;22(3):467-477
20. W. Shan, C. Ning, X. Luo, et al. Hyperinsulinemia is associated with endometrial hyperplasia and disordered proliferative endometrium: a prospective cross-sectional study[J]. Gynecol Oncol. 2014;132(3):606-610
21. E. Wik, M. B. Raeder, C. Krakstad, et al. Lack of estrogen receptor-alpha is associated with epithelial-mesenchymal transition and PI3K alterations in endometrial carcinoma[J]. Clin Cancer Res. 2013;19(5):1094-1105
22. M. Maggiolini, D. Picard. The unfolding stories of GPR30, a new membrane-bound estrogen receptor[J]. J Endocrinol. 2010;204(2):105-114
23. C. Ye, L. Li. 5-hydroxymethylcytosine: a new insight into epigenetics in cancer[J]. Cancer Biol Ther. 2014;15(1):10-15
24. H. Wu, Y. Zhang. Tet1 and 5-hydroxymethylation: a genome-wide view in mouse embryonic stem cells[J]. Cell Cycle. 2011;10(15):2428-2436
25. H. F. Chen, K. J. Wu. Epigenetics, TET proteins, and hypoxia in epithelial-mesenchymal transition and tumorigenesis[J]. Biomedicine (Taipei). 2016;6(1):1
26. S. Ito, A. C. D'Alessio, O. V. Taranova, et al. Role of Tet proteins in 5mC to 5hmC conversion, ES-cell self-renewal and inner cell mass specification[J]. Nature. 2010;466(7310):1129-1133
27. P. De Marco, F. Cirillo, A. Vivacqua, et al. Novel Aspects Concerning the Functional Cross-Talk between the Insulin/IGF-I System and Estrogen Signaling in Cancer Cells[J]. Front Endocrinol (Lausanne). 2015;6:30
28. A. V. Hernandez, V. Pasupuleti, V. A. Benites-Zapata, et al. Insulin resistance and endometrial cancer risk: A systematic review and meta-analysis[J]. Eur J Cancer. 2015;51(18):2747-2758
29. K. T. Nead, S. J. Sharp, D. J. Thompson, et al. Evidence of a Causal Association Between Insulinemia and Endometrial Cancer: A Mendelian Randomization Analysis[J]. J Natl Cancer Inst. 2015:107 (9)
30. N. Mu, Y. Zhu, Y. Wang, et al. Insulin resistance: a significant risk factor of endometrial cancer[J]. Gynecol Oncol. 2012;125(3):751-757
31. R. Madonna, R. De Caterina, Y. J. Geng. Epigenetic regulation of insulin-like growth factor signaling: A novel insight into the pathophysiology of neonatal pulmonary hypertension[J]. Vascul Pharmacol. 2015;73:4-7
32. A. B. Gaikwad, J. Gupta, K. Tikoo. Epigenetic changes and alteration of Fbn1 and Col3A1 gene expression under hyperglycaemic and hyperinsulinaemic conditions[J]. Biochem J. 2010;432(2):333-341
33. H. Wu, Y. Zhang. Tet1 and 5-hydroxymethylation: a genome-wide view in mouse embryonic stem cells[J]. Cell Cycle. 2011;10(15):2428-2436
34. Y. P. Tsai, H. F. Chen, S. Y. Chen, et al. TET1 regulates hypoxia-induced epithelial-mesenchymal transition by acting as a co-activator[J]. Genome Biol. 2014;15(12):513
35. C. Yao, L. Su, J. Shan, et al. IGF/STAT3/NANOG/Slug Signaling Axis Simultaneously Controls Epithelial-Mesenchymal Transition and Stemness Maintenance in Colorectal Cancer[J]. Stem Cells. 2016;34(4):820-831
36. P. De Marco, V. Bartella, A. Vivacqua, et al. Insulin-like growth factor-I regulates GPER expression and function in cancer cells[J]. Oncogene. 2013;32(6):678-688
Author contact
![]() Corresponding authors: Xiao-Jun Chen, Obstetrics and Gynecology Hospital of Fudan University, No. 419, Fangxie Road, Shanghai, 200011, P.R. China. Phone: 86-13601680784; Fax: 86-21-63455090; E-mail: xiaojunchen2013com; and Zhen-Bo Zhang, Department of Obstetrics and Gynecology, Shanghai General Hospital, Shanghai Jiao Tong University school of medicine, No. 650, Xinsongjiang Road, Shanghai, 201620, P.R. China. Phone: 86-15921516760; Fax: 86-21-63241377; E-mail: zhangzhenbozzbcom
Corresponding authors: Xiao-Jun Chen, Obstetrics and Gynecology Hospital of Fudan University, No. 419, Fangxie Road, Shanghai, 200011, P.R. China. Phone: 86-13601680784; Fax: 86-21-63455090; E-mail: xiaojunchen2013com; and Zhen-Bo Zhang, Department of Obstetrics and Gynecology, Shanghai General Hospital, Shanghai Jiao Tong University school of medicine, No. 650, Xinsongjiang Road, Shanghai, 201620, P.R. China. Phone: 86-15921516760; Fax: 86-21-63241377; E-mail: zhangzhenbozzbcom